

Abstract
The x-ray structure of the Escherichia coli chloride/proton antiporter ClC-ec1 provides a structural paradigm for the widespread and diverse ClC family of chloride channels and transporters. To maximize the usefulness of this paradigm, it is important to directly relate structure to function via studies of ClC-ec1 itself; however, few functional studies of this protein have been performed. In an endeavor to develop new tools for functional analysis of ClC-ec1, we have discovered that this transporter is inhibited by the stilbenedisulfonate 4,4-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS). In planar lipid bilayers, DIDS inhibits ClC-ec1 activity reversibly, with an apparent affinity in the micromolar range. Since ClC-ec1 is randomly oriented in the bilayers, ascertaining whether DIDS inhibits from the intracellular or extracellular side required an indirect approach. Using the ClC-ec1 structure as a guide, we designed a strategy in which modification of Y445C was monitored in conjunction with inhibition by DIDS. We found that DIDS inhibits transporters specifically from the intracellular side. Transporters with their extracellular side exposed to DIDS function normally, maintaining stoichiometric proton/chloride antiport over a wide range of proton and chloride concentrations. The side-dependent nature of DIDS inhibition will be useful for generating “functionally oriented” preparations of ClC-ec1, in which DIDS is used to silence transporters in one orientation but not the other.
INTRODUCTION
ClC family members move chloride across membranes in all cell types, and their physiological roles are diverse (for reviews, see Pusch and Jentsch (1) and Jentsch et al. (2)). They are critical for skeletal muscle excitability, neuronal chloride distribution, endocytosis, epithelial ion transport, and extreme acid tolerance (3–10). A great advance in the molecular understanding of the ClC family came with the crystal structures of two prokaryotic ClC homologs from Salmonella typhimurium and Escherichia coli (11,12). These prokaryotic homologs are homodimers, and chloride ions are seen bound in each subunit. A similar molecular arrangement, with each subunit forming its own chloride-permeation pathway, occurs in at least three eukaryotic channels, as indicated by functional studies on ClC-0, ClC-1, and ClC-2 (13–16). Although the sequence identity between prokaryotic and eukaryotic ClC members is only 15–20%, the sequences surrounding the core of the structure, including the chloride-binding sites, are highly conserved. In addition, functional studies of several eukaryotic ClC channels suggest that the overall fold among ClCs is most likely conserved (12,17–21).
In light of the likely structural similarity between the prokaryotic and eukaryotic ClC homologs, electrophysiological recordings revealed an unexpected functional dissimilarity: the E. coli homolog ClC-ec1 is a chloride/proton antiporter, not a chloride channel (22). Although an antiport mechanism might seem strikingly different from a channel mechanism, there are compelling functional similarities between the ClC-ec1 transporter and the eukaryotic ClC channels. For example, the eukaryotic ClC channels have “transporter-like” gating properties. Conformational changes in transporters are typically promoted by increases in substrate concentrations and are sensitive to transmembrane gradients of substrate (23). In the eukaryotic ClC channels that have been studied, gating is regulated by both chloride and protons (the ClC antiporter substrates) (24–27) and also by the transmembrane gradient for chloride (28). Another similarity between antiporter and channel homologs is seen with mutation of a conserved glutamate residue, which results in loss of proton sensitivity in both ClC-ec1 and ClC-0 (12,22). In addition, in ClC-ec1, this mutation eliminates proton transport, thus transforming the chloride-proton antiporter to a purely chloride-selective uniporter or channel (22). The similarities between the ClC transporter ClC-ec1 and eukaryotic ClC channels suggest that the structural and functional boundaries separating ClC channels from transporters may be quite subtle. To understand how subtle structural differences relate to changes in function, we must first learn more about the mechanism of ClC-ec1, currently the only ClC member for which both structure and function can be studied.
Inhibitors are classic experimental tools for studying mechanism in channels and transporters. However, there is a dearth of inhibitors for the ClCs in general and, until now, no known inhibitor of ClC-ec1. The stilbenedisulfonate DIDS is a widely used anion-transport inhibitor (for reviews, see Salhany (29), Romero et al. (30), and Jentsch et al. (31)).
Although DIDS inhibits some anion transport proteins rather nonspecifically via reaction with lysine residues (32), we show here that inhibition of ClC-ec1 is reversible and hence does not rely on covalent modification. Identification of the binding site will be useful for probing mechanism and possibly for designing additional pharmacological modulators of ClC family members. As a first step toward identifying the binding site, we sought to determine whether DIDS inhibits ClC-ec1 from the intracellular side, the extracellular side, or both. One difficulty in this endeavor is that ClC-ec1 is randomly oriented in the reconstituted vesicles used for functional studies: some of the transporters have their intracellular side facing inside the vesicles, whereas others are oriented in the opposite direction, with their extracellular side facing inside the vesicles. Indeed, this situation is a major obstacle to many studies, as it precludes detailed determination of chloride, proton, and voltage dependence.
To determine whether DIDS inhibits ClC-ec1 from the intracellular or extracellular side, we designed a strategy that uses the known structure of ClC-ec1 combined with cysteine modification. Using this strategy, we show that DIDS inhibits ClC-ec1 from the intracellular side and has no functional effect when applied to the extracellular side. The side-specific nature of inhibition by DIDS will allow for functional orientation of ClC-ec1.
MATERIALS AND METHODS
Protein purification
ClC-ec1 containing a C-terminal polyhistidine (His) tag was overexpressed in E. coli and purified essentially as described (33). Briefly, cells were disrupted using sonication (Branson Ultrasonics, Danbury, CT) or a French press (SLM Instruments, Rochester, NY), and the protein was extracted for 2 h at room temperature in the presence of 50 mM decylmaltoside (Anatrace, Maumee, OH). The extract was centrifuged at 20,000 × g for 45 min, and the protein was then purified over a Co2+ column (BD Biosciences, San Jose, CA). The His-tag was cleaved off of the purified protein with Endoproteinase Lys-C (Roche, Basel, Switzerland), and the protein was then further purified over a Poros HQ anion exchange column (Applied Biosystems, Foster City, CA). For planar lipid bilayer experiments, only the aliquots corresponding to the rising phase of the elution peak were used. For experiments involving cysteine modification, dithiothreitol (DTT) (Invitrogen, Carlsbad, CA) was present throughout all steps of the purification to prevent oxidation of the cysteines. For these preparations, the Co2+ beads were replaced by Ni2+ beads (Amersham Biosciences, Buckinghamshire, England) that could tolerate high concentrations of DTT. A total of 5 mM DTT was used for all steps throughout the Ni2+-column purification, and 1 mM DTT was used for the anion exchange column and reconstitution.
Reconstitution
ClC-ec1 reconstitution was similar to that described (33). E. coli polar lipids (Avanti Polar Lipids, Alabaster, AL) were dried under argon or by rotary evaporation under vacuum, washed twice with equal volumes of pentane, and then suspended to a final concentration of 20 mg lipid/mL in reconstitution buffer (for bilayers, either 150 mM KCl, 15 mM citrate, 15 mM phosphate pH 7.0 with KOH or 450 mM KCl, 25 mM citrate, 25 mM phosphate pH 7.0 with KOH; for flux assays, as described below). Octylglucoside (Anatrace) was added (4–5% final concentration), and the suspension was sonicated to clarity using a cylindrical bath sonicator (Laboratory Supplies, Hicksville, NY). Solubilized ClC-ec1 was added at 50 μg protein per mg lipid for bilayer studies and 10 μg protein per mg lipid for proton flux assays, and the mixtures were diluted to 10 mg lipid/mL with reconstitution buffer. Detergent was removed by dialysis over 24 h with three 1-L buffer changes. The resulting liposomes were freeze-thawed 2–5 times in an acetone/dry ice bath and frozen at −80°C until the day of use.
Flux assays
Proton flux assays were performed as described (22) except at symmetric pH 5.7. Briefly, vesicles were reconstituted in 350 mM KCl, 50 mM citrate, 20 mM phosphate pH 7.0 with KOH. Vesicles were adjusted to pH 5.7 with phosphoric acid, and then intra- and extravesicular solutions were equalized by freezing and thawing the samples 3–4 times. Either 1% dimethyl sulfoxide (DMSO) (no-DIDS control) or 2.5 mM DIDS was then added to the vesicles and equilibrated across the membranes by a series of freeze-thaws. Small unilamellar vesicles were formed by bath sonication for 5–10 s. Vesicles (100 μL) were then spun through Sephadex G-50 (Amersham) columns equilibrated in low-chloride solution (3 mM KCl, 300 mM K2SO4, 2 mM glutamic acid pH 5.7 with KOH). This decreases the extracellular chloride concentration, brings the vesicles into a weakly buffered external solution, and removes unbound DIDS. The effluent from the spin column (∼150 μL), containing the buffer-exchanged vesicles, was immediately added to 1.2 mL of the low-chloride solution. Inward proton movement, driven by outward chloride flux, was initiated 30–40 s after dilution of the vesicles by the addition of 1.8 μL valinomycin (Fluka, Buchs, Switzerland, 1.1 mg/mL in ethanol). After an additional 45–70 s, 1 μL carbonyl cyanide p-(trifluoro-methoxy)phenyl hydrazone (FCCP) (Sigma, St. Louis, MO, 2 mg/mL in ethanol) was added to collapse the proton gradient. The extravesicular proton concentration was monitored using a pH electrode (Beckman Coulter, Fullerton, CA) connected to an Orion 710A pH meter (Thermo Electron, Beverly, MA) and interfaced with a PC computer running HyperTerminal software (Microsoft, Redmond, WA). The voltage output from the pH meter was recorded every 5 s. The voltage output was converted to external proton concentration using a standard curve generated on the day of experiments from solutions of known pH (pH 4.0, 7.0, and 10.0). All flux assays were performed at room temperature (∼22°C). The initial rate of change in [H+]ext (Fig. 1 C) was determined over the 10 s after the addition of valinomycin. The nonspecific “leak” change in [H+]ext (Fig. 1 C) was determined from the 10–30 s immediately preceding the addition of valinomycin. Vesicles in which this leak was >8 nM/s were excluded from analysis.
FIGURE 1.
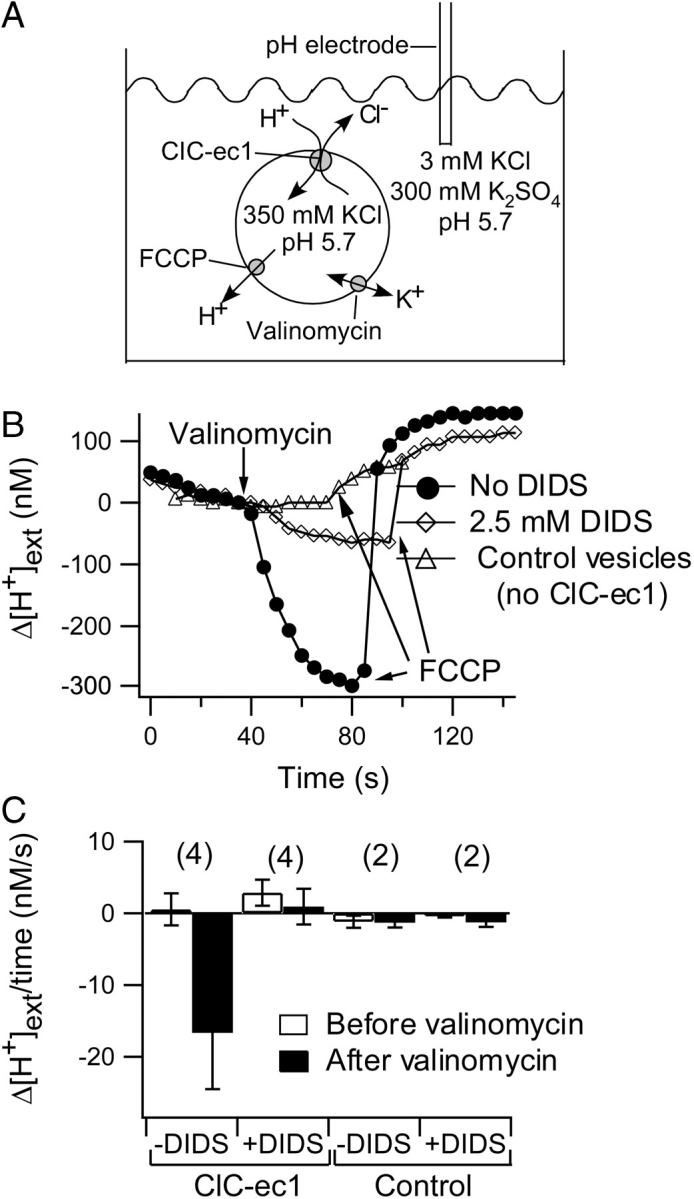
DIDS inhibition of ClC-ec1. (A) Cartoon illustrating ion movement in the proton flux assays. An outwardly directed Cl− gradient drives H+ uptake through ClC-ec1. The chloride and proton conditions listed are initial conditions before the potassium-selective ionophore valinomycin was added to allow bulk movement of Cl− and H+ through ClC-ec1. FCCP is added at the end of each experiment to collapse the proton gradient and release accumulated protons. (B) Inhibition of chloride-driven proton flux. Vesicles reconstituted with ClC-ec1 were treated with 1% DMSO (no-DIDS control, •) or 2.5 mM DIDS (⋄). Control vesicles lacking ClC-ec1 were treated with 1% DMSO (▵). To align the traces for visual comparison, the change in proton concentration (Δ[H+]ext), rather than the absolute proton concentration is plotted. The change is in reference to the time point immediately before addition of valinomycin. Traces were aligned along the x axis such that valinomycin was added at the same time point. Straight lines connect the data points in all figures unless otherwise stated. (C) The rate of Δ[H+]ext is summarized for several flux assays under each condition (mean ± SE, or ± range for n = 2). Negative values on the y axis represent a decrease in [H+]ext over time, and positive values represent an increase in [H+]ext. The rates measured before (open bars) and after (solid bars) the application of valinomycin differ significantly only in the case of ClC-ec1-containing vesicles in the absence of DIDS. The numbers in parentheses represent the number of flux assays performed under each condition.
Bilayer recordings
Bilayer recordings were performed with a horizontal bilayer chamber as described (34). The trans chamber is grounded and is equivalent to the “outside” by standard electrophysiological convention: negative (inward) current corresponds to net positive ion flow from the trans to the cis chamber. Bilayers were formed from 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) (Avanti Polar Lipids). A 3:1 mixture of POPE/POPG stored in chloroform was dried, washed 1–2 times with pentane, dried again, and resuspended in n-decane to 5 or 10 mg lipid/mL. Formation of bilayers was monitored by measuring capacitance using an Axopatch 200B amplifier interfaced with a Digidata 1322A acquisition board and pClamp software (Axon Instruments, Union City, CA). Solutions used for each experiment are described in the figure legends. No functional differences were observed between the two types of buffers used (5 mM citrate-5 mM phosphate or 5 mM citrate-40 mM HEPES). Salt bridges containing 300 mM KCl or 300 mM Na-glutamate (to allow for the use of low-chloride solutions) and 2% agar or agarose were used in all experiments. A salt gradient (300 mM KCl cis, 40 mM KCl trans) was used to promote vesicle fusion. Additionally, 2–4 μL of 3 M KCl were added immediately above the bilayer to further promote fusion (increasing the final KCl concentration in the cis chamber by <6%). Currents in response to various voltages were recorded after fusions had stopped and the current at −100 mV was constant over a period of ∼5 min. Recordings were made at room temperature (22°C).
DIDS
Stock solutions of 250 mM DIDS (Molecular Probes, Eugene, OR) were made in DMSO. They were kept dark and frozen at −20°C or −80°C until immediately before use, when they were diluted to a final concentration of 6.25 μM − 2.5 mM.
Cysteine modification
[2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET) was obtained from Toronto Research Chemicals (Toronto, Ontario, Canada). Stock solutions (0.1–1.0 M) were made in distilled water, aliquoted, and stored at −80°C until immediately before use. A total of 1–4 mM MTSET was added to bilayers in which both the cis and the trans chambers contained 300 mM KCl, 5 mM citrate, 40 mM HEPES pH 6.0. For this experiment, a pH of 6.0 was used as a happy medium for obtaining both a sufficient amount of current (the activity of ClC-ec1 increases as the pH is lowered) and a sufficiently fast rate of reactivity of MTSET (which generally decreases as the pH is lowered). Recordings were made at least 5 min after addition of the MTSET, when the effect of MTSET had reached steady state.
Determining reversibility and apparent affinity of DIDS in ClC-ec1
To determine the reversibility of inhibition by DIDS, 250 μM DIDS was applied to the trans chamber for ∼5 min before washing away the unbound DIDS. After washout, currents were measured only every 2–5 min to minimize the probability of the bilayer breaking. Approximate rates of reversibility were determined by fitting the current decay to a single exponential. Since only a few time points were used, and since perfusion took ∼2 min, these rates are estimates.
To determine the apparent affinity of DIDS in ClC-ec1, currents were measured before and after addition of DIDS to the trans chamber. Each bilayer received 1–3 consecutive additions of DIDS. The data for inhibition by 6–250 μM DIDS (Fig. 3 C) were obtained from eight separate bilayers, and the data for inhibition by 2.5 mM DIDS (Fig. 3 C) were obtained from 20 separate bilayers. The concentrated stock of DIDS used in these experiments was made fresh or stored at −20°C or −80°C for <1 day before use.
FIGURE 3.

Reversibility of DIDS inhibition of ClC-ec1. (A) Initial currents through wild-type ClC-ec1 (○) were blocked ∼40% by addition of 250 μM DIDS added to the trans chamber (□). Complete reversal of inhibition was observed ∼40 min after washing out the DIDS (⋄). The cis chamber contained 300 mM KCl, 5 mM citrate, and 40 mM HEPES pH 5.0, and the trans chamber contained 40 mM KCl, 5 mM citrate, and 40 mM HEPES pH 5.0. (B) Identical to A, except that the trans chamber contained solution at pH 7.0. Initial currents (○) were blocked ∼35% by addition of 250 μM DIDS to the trans chamber (□). Complete reversal was observed ∼15 min after washing out the DIDS (⋄). (C) Concentration dependence of inhibition by DIDS. In these experiments, the cis chamber contained 300 mM KCl, 5 mM citrate, and 40 mM HEPES pH 5.0; the trans chamber contained 40 mM KCl, 5 mM citrate, and 40 mM HEPES pH 5.7. Data are from several bilayers (see Materials and Methods) in which DIDS was added to the trans chamber only, such that maximal inhibition is ∼50%. The inhibition was measured at +100 mV (⋄) or −100 mV (○). The solid line is a fit of the data to a rectangular hyperbola: F = Fmax/(1 + (KM/[DIDS])), where F is the percentage current inhibition, Fmax is the maximal inhibition (48%), and KM is the apparent affinity for DIDS (26 μM).
Determination of reversal potentials in “oriented samples”
DIDS was added to either the cis or the trans chamber in 2–5 bilayers under each condition listed in Table 3. Additional perfusions were then made to alter the chloride and/or proton conditions on the opposite side of the bilayer: for example, if DIDS was added to the cis chamber, the chloride or proton concentration of the trans solution was changed by perfusion. Reversal potentials were determined from current-voltage curves.
TABLE 3.
Application of DIDS to the extracellular side of ClC-ec1 does not affect proton-chloride coupling
| [Cl−] (mM) (cis/trans) | pH (cis/trans) | Vrev (mV) “unoriented” | Vrev (mV) “oriented” | ECl− (mV) | EH+ (mV) |
|---|---|---|---|---|---|
| 300/40 | 5/5 | 28.1 ± 2.1 (15) | 28.9 ± 5.3 (7) | 46.8 | 0 |
| 300/40 | 5/7 | −8.8 ± 2.6 (3) | −9.1 ± 1.4 (4) | 46.8 | −117.1 |
| 300/40 | 5/5.7 | 19.6 ± 4.3 (6) | 17.7 ± 5.5 (6) | 46.8 | −41.0 |
| 300/40 | 5.7/5 | 46.7 ± 2.3 (3) | 47.0 ± 3.6 (3) | 46.8 | 41.0 |
| 300/40 | 5/4 | 47.7 ± 2.6 (3) | 48.1 ± 2.4 (4) | 46.8 | 58.6 |
| 300/40 | 4/5 | 11.2 ± 1.4 (3) | 9.2 ± 4.1 (3) | 46.8 | −58.6 |
| 300/40 | 4/7 | −25.2 ± 0.8 (3) | −24.1 ± 0.9 (4) | 46.8 | −175.7 |
| 300/3 | 5/5 | 66 ± 4.4 (8) | 59.2 ± 11.0 (7) | 109.2 | 0 |
| 300/300 | 4/4 | 2.3 ± 0.96 (4) | 1.5 ± 1.5 (4) | 0 | 0 |
Reversal potentials under a wide range of chloride and proton concentrations and gradients. Solutions were buffered with 5 mM citrate-5 mM phosphate. Vrev “unoriented” represents the mean reversal potential ± SD (No. of bilayers) in the absence of DIDS. Vrev “oriented” represents the mean reversal potential ± SD (No. of bilayers) when 2.5 mM DIDS was added to either the cis or the trans chamber. ECl− and EH+ represent the calculated reversal potentials for Cl− and H+, respectively. Activity coefficients of 0.688, 0.818, and 0.940 were used for 300 mM, 40 mM, and 3 mM Cl−, respectively (45).
RESULTS
DIDS inhibits ClC-ec1
To test whether DIDS inhibits ClC-ec1, we expressed and purified ClC-ec1 and reconstituted the protein into lipid vesicles. Chloride-driven proton flux through ClC-ec1 was measured using a pH electrode in a weakly buffered extravesicular solution (22). At symmetric pH, an outwardly directed chloride gradient favors movement of chloride out of the vesicles and concomitant movement of protons into the vesicles through ClC-ec1 (Fig. 1 A). Bulk chloride efflux and proton influx through ClC-ec1 do not occur until the potassium ionophore valinomycin is added to dissipate the transmembrane electrical potential. With ClC-ec1-containing vesicles, the external proton concentration ([H+]ext) decreased after valinomycin was added (Fig. 1 B, circles). The proton ionophore FCCP was added at the end of the experiment to release intravesicular protons and confirm that the change in [H+]ext was in fact due to proton accumulation. Addition of 2.5 mM DIDS to both sides of the ClC-ec1 vesicles (see Materials and Methods) inhibited this chloride-driven proton influx (diamonds). With control vesicles lacking ClC-ec1, no valinomycin-induced proton influx was observed (triangles). In each experiment, the slight change in [H+]ext that occurred before the addition of valinomycin likely resulted from a combination of drift in the electrode and proton leak (via permeation of HCl or CO2) across the vesicle membrane (35). This background Δ[H+]ext varied in magnitude and direction from experiment to experiment and is summarized for each condition in Fig. 1 C. Δ[H+]ext is significantly different before and after addition of valinomycin only in the case of ClC-ec1-containing vesicles in the absence of DIDS.
DIDS inhibits ClC-ec1 in a side-dependent manner
To examine inhibition by DIDS more closely, we used bilayer recordings, which allow direct control of the solutions on both sides of the membrane and the voltage across the membrane. ClC-ec1 currents in response to various test voltages are shown in Fig. 2 A (left), and the current-voltage relationship is plotted in Fig. 2 B (circles). In these recordings, some of the transporters have their intracellular side facing the trans chamber, whereas the remaining transporters have their intracellular side facing the cis chamber (Fig. 2 D, left). This random orientation occurs because dispersion in detergent renders the proteins free to insert in either direction. Such dispersion is necessary for purification of ClC-ec1 away from ion channels that overwhelm and mask the ClC-ec1 currents (33). Although it is theoretically possible that the proteins reconstitute in a preferred orientation, this is not likely given that both orientations are observed when ClC-ec1 is reconstituted to form two-dimensional crystals (36), and our results in Figs. 4 and 5 additionally refute this possibility.
FIGURE 2.

Side-dependent inhibition of ClC-ec1 by DIDS. (A) ClC-ec1 currents in response to the voltage protocol shown below the center current trace. This voltage protocol was used for all experiments unless otherwise stated. (Left) Initial current family. (Center) After addition of 2.5 mM DIDS to the cis chamber. (Right) After subsequent addition of 2.5 mM DIDS to the trans chamber. The cis chamber contained 300 mM KCl, 5 mM citrate, and 40 mM HEPES pH 5.0, and the trans chamber contained 40 mM KCl, 5 mM citrate, and 40 mM HEPES pH 5.7. (B) Current-voltage plots, where currents were measured at the end of the 1.5-s voltage pulses shown in A. (C) Current-voltage plots from a separate bilayer before and after addition of 1% DMSO to the cis chamber and subsequently to the trans chamber. Solutions were identical to those used in A and B. (D) Cartoon depicting a model in which DIDS inhibits ClC-ec1 specifically from one side. Transporters insert into the bilayers with random orientation; some have their intracellular side facing the trans chamber, whereas others have their intracellular side facing the cis chamber. In this model, DIDS binds to one face of ClC-ec1, inhibiting all of the transporters in one orientation when added to the cis chamber and all of the transporters in the opposite orientation when added to the trans chamber. For simplicity, one DIDS molecule is shown binding to each ClC-ec1 molecule.
FIGURE 4.

Side-specific potentiation of Y445C by MTSET. (A) Structure of ClC-ec1 (Protein Data Bank 1OTS) (12). Y445 is shown spacefilled in red. The chloride ions (two per subunit) are spacefilled in green. (B) Wild-type ClC-ec1 currents were recorded in response to 3-s voltage pulses from −120 mV to +150 mV in 30 mV steps, with a holding voltage of 0 mV. Currents plotted were from the end of the 3-s pulse: initial current (black circles); after addition of 2 mM MTSET to the cis chamber (red triangles); after subsequent addition of 2 mM MTSET to the trans chamber (blue squares). The cis and trans chambers both contained 300 mM KCl, 5 mM citrate, and 40 mM HEPES pH 6.0. (C) Addition of 2 mM MTSET causes side-specific potentiation of Y445C-ClC-ec1. Currents were recorded in response to 3-s voltage pulses from −150 mV to +150 mV in 30 mV steps, with a holding voltage of 0 mV. Initial current (black circles); after addition of 2 mM MTSET to the trans chamber (blue squares); after subsequent addition of 2 mM MTSET to the cis chamber (red triangles). Solutions were as described in B. (D) Cartoon depicting a model in which MTSET modifies Y445C specifically from the intracellular side. For simplicity, one MTSET molecule is shown binding to each ClC-ec1 molecule.
FIGURE 5.

DIDS and MTSET act from the same side. Each panel shows Y445C-ClC-ecl currents before and after indicated solution changes. Conditions were as described in Fig. 4 B. In each cartoon, the transporter depicted on the left has its intracellular side facing the trans chamber, and the transporter on the right has its intracellular side facing the cis chamber. For simplicity, only one DIDS and one MTSET are shown bound to each ClC-ec1 transporter. (A) Initial current (black circles); after addition of 2.5 mM DIDS to the cis chamber (green diamonds); after subsequent addition of 4 mM MTSET to the cis chamber (red triangles); after subsequent addition of 2 mM MTSET to the trans chamber (blue squares); and after subsequent addition of 2.5 mM DIDS to the trans chamber (purple crosses). (B) Initial current (black circles); after addition of 1 mM MTSET to the cis chamber (red triangles); after subsequent addition of 1 mM DIDS to the cis chamber (green diamonds); and after subsequent addition of 2 mM MTSET to the trans chamber (blue squares).
Addition of saturating concentrations of DIDS (2.5 mM) to the cis side inhibited ∼40% of the current (Fig. 2 A, center, and Fig. 2 B, diamonds). Subsequent addition of 2.5 mM DIDS to the trans chamber inhibited an additional ∼50% of the current (88% total block, Fig. 2 A, right, and Fig. 2 B, crosses). DMSO alone had no significant effect on ClC-ec1 currents (Fig. 2 C). Table 1 summarizes results from several experiments. These data are consistent with a model in which DIDS inhibits the ClC-ec1 activity in a side-dependent manner (Fig. 2 D).
TABLE 1.
Inhibition of wild-type ClC-ec1 by DIDS
| % Inhibition at −100 mV | % Inhibition at +100 mV | Vrev (mV) | |
|---|---|---|---|
| No DIDS | 17.5 ± 3.4 (23) | ||
| DIDS cis only | 38 ± 7 (11) | 40 ± 10 (10) | 16.7 ± 4.4 (9) |
| DIDS trans only | 50 ± 8 (12) | 50 ± 10 (12) | 17.9 ± 3.0 (12) |
| DIDS both sides | 74 ± 10 (13) | 73 ± 10 (12) | 13.5 ± 5.6 (13) |
DIDS inhibits ClC-ec1 in a side-dependent manner. Values represent mean ± SD. Numbers in parentheses represent the number of bilayers tested. Conditions were as in Fig. 2, except in some experiments solutions were buffered with 5 mM phosphate in place of 40 mM HEPES. In each case, 2.5 mM DIDS was added to the cis chamber (300 mM KCl pH 5.0), the trans chamber (40 mM KCl pH 5.7), or both. The order of addition did not affect the amount of inhibition. The fact that the reversal potential (Vrev) does not change significantly upon addition of DIDS suggests that much of the residual current after DIDS inhibition is due to currents through ClC-ec1.
We expect that some variability in the percentage inhibition might occur because of variation in the fraction of transporters in each orientation. Although on average the bilayers will contain 50% of the transporters in each orientation, some bilayers will have more transporters oriented in one direction than in the other. Indeed, we saw anywhere from 27–55% inhibition from DIDS added only to the cis chamber and 42–75% inhibition from DIDS added only to the trans chamber. If all of this variation was due to random variability in the orientation of transporters, then we should have observed significantly less variation when DIDS was added to both sides of the membrane. However, we observed a large variation in inhibition (60–88%) even when DIDS was added to both sides of the membrane. Potential sources of variability include leak in the bilayers and instability of DIDS solutions (see Discussion). Although we are not yet certain of the reason for this variability, DIDS consistently inhibits the majority of ClC-ec1 activity both in flux assays and lipid bilayer recordings.
DIDS inhibits ClC-ec1 reversibly
DIDS is known to modulate anion channel and transporter activity by both irreversible and reversible mechanisms (32). We found that ClC-ec1 inhibition by DIDS reverses completely (Fig. 3, A and B), with a time constant of 9 ± 3 min at pH 5.0 and of 2 ± 1 min at pH 7.0 (mean ± SD, n = 3 bilayers for each condition). The apparent affinity of DIDS for ClC-ec1 is ∼30 μM under the conditions described in Fig. 3 C. Although changes in chloride or proton concentrations could alter this value (by affecting the distribution of transporter conformations or the occupancy of ions in the permeation pathway), 30 μM is of the same magnitude observed for reversible inhibition of other chloride transport proteins, including ClC-Ka (∼100 μM) (37) and the endogenous volume regulated anion channels in T-lymphocytes and HEK293 cells (∼25 μM) (38,39).
DIDS acts from the intracellular side
To determine whether DIDS inhibits ClC-ec1 specifically from the intracellular face or the extracellular face, we used the known structure of ClC-ec1 together with a cysteine-modification approach. In the ClC-ec1 structure, Y445 coordinates one of the bound chloride ions near the intracellular side (Fig. 4 A). In ClC-0, a cysteine at the homologous position (Y512) can be modified by the membrane-impermeant reagent MTSET applied to the intracellular side (18). In wild-type (WT) ClC-ec1, addition of MTSET had no functional effect (Fig. 4 B). In contrast, in transporters containing the Y445C mutation (Y445C-ClC-ec1), addition of MTSET to the trans chamber increased the current at positive voltages (Fig. 4 C, blue squares), and subsequent addition of MTSET to the cis chamber increased the current at negative voltages (red triangles). Similar results were observed in several bilayers (Table 2), and this voltage-dependent potentiation did not depend on whether MTSET was added to the cis chamber or trans chamber first. Potentiation by MTSET was completely reversible by DTT. These results are consistent with a model in which MTSET modification of Y445C occurs specifically from one side of ClC-ec1 (Fig. 4 D). In contrast, our data are not consistent with a model in which MTSET is able to modify Y445C from either the intracellular or extracellular side of ClC-ec1; this model would predict that addition of MTSET to either the cis or the trans chamber would be sufficient for complete modification of Y445C, and subsequent addition to the opposite side of the bilayer should have no further functional effect. Hence, potentiation of the Y445C mutant is due to MTSET reacting specifically with Y445C from one side of the protein. Based on the structure of ClC-ec1 and the experimental observation that the homologous position in ClC-0 (Y512C) is modified from the intracellular side (18,21), we conclude that in ClC-ec1, MTSET modifies Y445C specifically from the intracellular side.
TABLE 2.
Potentiation of Y445C-ClC-ec1 by MTSET
| Potentiation at −120 mV | Potentiation at +120 mV | |
|---|---|---|
| MTSET cis | 2.60 ± 0.39 (9) | 1.18 ± 0.11 (8) |
| MTSET trans | 1.15 ± 0.10 (4) | 1.72 ± 0.32 (3) |
| MTSET cis if DIDS previously added cis | 0.77 ± 0.13 (4) | 0.89 ± 0.09 (4) |
| MTSET trans if DIDS previously added cis | 0.95 ± 0.14 (4) | 1.99 ± 0.34 (4) |
Potentiation is defined as the current after addition of MTSET divided by the current before addition of MTSET (mean ± SD). Numbers in parentheses represent the number of bilayers tested. Conditions were as in Fig. 4.
We reasoned that we could use the sidedness of this MTSET effect on Y445C transporters to determine whether DIDS inhibits from the intracellular or extracellular side. Although the shape of the current-voltage relationship for Y445C transporters differs somewhat from that observed with WT transporters (Fig. 4 C versus B), the Y445C currents are inhibited by DIDS in a side-specific manner as observed with WT, so it is unlikely that the structure of the DIDS-binding site is altered significantly by this point mutation. To determine whether DIDS acts from the intracellular side (like MTSET) or the extracellular side, we performed an experiment consisting of four consecutive solution changes (Fig. 5 A). In this experiment, addition of a saturating concentration of DIDS to the cis chamber blocked ∼35% of the current (black circles without DIDS, green diamonds with DIDS, states 1 and 2 in the cartoon). If DIDS inhibits ClC-ec1 from the intracellular side, subsequent application of MTSET to the same (cis) chamber should have no effect, since DIDS has already inhibited the transporters with their intracellular side facing the cis chamber (and since DIDS may additionally prevent MTSET from reacting). If, however, DIDS inhibits from the extracellular side, the MTSET-sensitive transporters should still be available and normal potentiation should occur when MTSET is added to the same chamber as DIDS. We observed that subsequent addition of MTSET to the cis chamber had no effect on current (Fig. 5 A, red triangles, state 3 in the cartoon). Thus, DIDS either inhibited all the MTSET-reacted transporters or prevented MTSET from reacting. Either way, these results suggest that DIDS and MTSET act on the same (intracellular) side. If this interpretation is correct, then transporters oriented in the opposite direction (with their intracellular side facing the trans chamber) should be potentiated when MTSET is subsequently added to the trans chamber. Indeed, subsequent addition of MTSET to the trans chamber potentiated the current at positive voltages (Fig. 5 A, blue squares, state 4 in the cartoon), exactly as seen in the absence of any cis additions (Fig. 4 C, blue squares). Thus, DIDS has no significant effect from the extracellular side of ClC-ec1. A final addition of DIDS to the trans chamber inhibited nearly all of the remaining current (Fig. 5 A, purple crosses, state 5 in cartoon). This demonstrates that DIDS can still bind and inhibit from the intracellular side of ClC-ec1 after MTSET has reacted.
To determine whether the order of DIDS and MTSET additions would affect our conclusion, we varied the order of the perfusions. In the example shown in Fig. 5 B, MTSET was first added to the cis chamber, which potentiates the current at negative voltages with little effect at positive voltages (black circles, before MTSET; red triangles, after addition of MTSET to the cis chamber, states 1 and 2 in cartoon). Subsequent addition of DIDS to the cis chamber inhibited the MTSET-potentiated current (green diamonds, state 3 in cartoon), as expected if DIDS and MTSET are acting on the same transporters. Further confirmation comes from our observation that the usual potentiation occurs when MTSET is next added to the trans chamber (blue squares, state 4 in cartoon). Similar results were observed in several bilayers (Table 2). These data together with the structure of ClC-ec1 strongly support the conclusion that DIDS inhibits ClC-ec1 specifically from the intracellular side.
The side specificity of DIDS inhibition suggests that it will be useful for “functional orientation” of ClC-ec1 preparations. To functionally orient ClC-ec1, DIDS can be added to one side of the bilayer, silencing transporters with their intracellular side exposed to DIDS and allowing transporters with their extracellular side exposed to DIDS to be studied in isolation. For DIDS to be useful for this purpose, it is critical that transporters with their extracellular side exposed to DIDS function normally. For example, one could imagine a scenario in which DIDS inhibits transporters only when exposed to the intracellular side but also binds somewhere on the extracellular side and modulates chloride-proton coupling. To verify that extracellular exposure of DIDS does not alter the chloride-proton coupling of ClC-ec1, we measured the reversal potential before and after addition of DIDS to one side of the bilayer (“unoriented” and “oriented” conditions, respectively) under several chloride and proton conditions (Fig. 6, Table 3). In all cases, reversal-potential data were well described by the thermodynamic relationship for 2:1 stoichiometric antiport, in which two chloride ions are transported in one direction for every proton transported in the opposite direction (22):
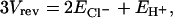 |
(1) |
where Vrev is the experimentally measured reversal potential and ECl− and EH+ are the calculated reversal potentials for chloride and protons, respectively, as explained in Table 3. Hence, even when the extracellular side of ClC-ec1 is exposed to millimolar concentrations of DIDS, ClC-ec1 still acts to transport two chloride ions across the membrane for every proton countertransported.
FIGURE 6.

Normal proton-chloride coupling is observed in “oriented” transporters. Reversal potential data (Table 3) are plotted according to Eq. 1. The dashed line represents the expected curve for a transporter that stoichiometrically couples the movement of two chloride ions in one direction to one proton in the opposite direction. Vrev is the experimentally measured reversal potential; ECl− and EH+ are the calculated reversal potentials for chloride and protons, respectively, as explained in Table 3. “Unoriented” refers to measurements made in the absence of DIDS; “oriented” refers to measurements made in the presence of 2.5 mM DIDS in either the cis or the trans chamber. In all cases, DIDS inhibited approximately half (35–67%) of the current, consistent with the idea that DIDS inhibits specifically from one side under all of these conditions.
DISCUSSION
Inhibition of ClC-ec1 by DIDS
ClC-ec1 is inhibited by DIDS in flux assays and bilayer recordings. We used the mutant Y445C, which is potentiated in a side-specific manner by MTSET (Fig. 4), to identify the side-specificity of inhibition by DIDS. Since DIDS and MTSET act from the same side of Y445C-ClC-ec1 (Fig. 5), we conclude that DIDS acts from the intracellular side. Thus, addition of DIDS to the cis chamber of the bilayer results in a “functionally oriented” preparation in which most of the active transporters are oriented with their extracellular face toward the cis chamber.
One complication of using DIDS for functional orientation is the variability in the magnitude of inhibition (60–88% when added to both sides of the bilayer). We do not have a satisfactory explanation for all of this variability, but it likely results at least in part from variability in bilayer leak and in efficacy of the DIDS solution. Variability in the efficacy of DIDS inhibition in ClC-ec1 could arise from a combination of several known properties of DIDS: 1), DIDS will react with accessible deprotonated primary amines. The lipids used for vesicles and bilayers contain substantial amounts of phosphatidylethanolamine, which could serve as a DIDS sink. 2), Exposure to light varies the ratio of the trans and cis forms of DIDS, and these isomers could have different efficacies for ClC-ec1. The trans isomer is known to inhibit other chloride transport proteins by several orders of magnitude more efficiently than the cis isomer (32). 3), DIDS is not stable in aqueous solution. We stored the DIDS solutions frozen in DMSO, and it is possible that small amounts of water in these stock solutions caused variability in the purity of the DIDS. Despite these sources of variability, we found that DIDS consistently inhibited the majority of the ClC-ec1 activity in a side-dependent manner.
A second complication in using DIDS for functional orientation is that maximal inhibition is not 100%. The fact that the reversal potential was not significantly altered after saturating concentrations of DIDS were added to both sides (Table 1) suggests that much of the residual current was due to activity through ClC-ec1 rather than leak in the bilayers. To use DIDS for functional orientation, it will be best to use a subtraction protocol, which would overcome complications from the fact that maximal inhibition by DIDS is <100% and from the aforementioned variability in maximal inhibition by DIDS. For example, in bilayers oriented with DIDS in the cis chamber, the “DIDS-sensitive current” would be obtained by subtracting current observed after addition of DIDS to the trans chamber. An important point is that we have not yet demonstrated that inhibition by DIDS is side specific under all possible combinations of chloride and proton concentrations and that this would need to be verified for each condition. Nevertheless, under all conditions tested in Table 3 (ranging 3–300 mM Cl− and pH 4–pH 7), application of DIDS to one side of the bilayer inhibited 35–67% of the current, consistent with the idea that DIDS inhibits transporters specifically from the intracellular side under a variety of conditions.
MTSET modification of Y445C-ClC-ec1
Modification of Y445C by the positively charged reagent MTSET causes voltage-dependent potentiation of Y445C-ClC-ec1. In ClC-0, the homologous mutation, Y512C, is inhibited nearly completely by MTSET (18) and also by the negatively charged reagent sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES) (18,21). Surprisingly, in Y445C-ClC-ec1, addition of MTSES had little functional effect, inhibiting the currents by <20% when applied to both sides of the membrane. MTSES was able to react with all of the Y445C sites, as demonstrated by the fact that application of MTSES prevented MTSET from potentiating Y445C-ClC-ec1 current (K. Matulef and M. Maduke, unpublished data). Thus, these different effects of MTSET and MTSES likely arise from the difference in charge on these reagents. The charge at residue 445 could affect currents by altering the intrinsic voltage dependence of ClC-ec1 or by altering the local concentrations of chloride and protons. Further work is necessary to elucidate the mechanism of potentiation by MTSET.
Comparison to inhibition of other ClCs by DIDS
ClC-Ka and ClC-K1 are inhibited by DIDS from the extracellular side (37). Two residues near the outer mouth of the pore are necessary for potent inhibition of these channels by DIDS (37), and these residues are not conserved in ClC-ec1, ClC-0, or other homologs that are insensitive to extracellular DIDS: ClC-2, ClC-3, and ClC-5 (40–42). Thus, it is not surprising that DIDS does not inhibit ClC-ec1 or ClC-0 from the extracellular side.
Although DIDS is a widely used chloride-transport inhibitor, the only other ClC homolog for which intracellular sensitivity to DIDS has been reported is ClC-0 (43,44). Like ClC-ec1, DIDS inhibits ClC-0 specifically from the intracellular side, but unlike ClC-ec1, inhibition of ClC-0 is irreversible (43). Irreversible inhibition usually occurs as a result of reaction of one or both of the isothiocyanate groups with susceptible unprotonated lysines. The reversible inhibition of ClC-ec1 by DIDS suggests that either the DIDS-binding site in ClC-ec1 is not close to an unprotonated lysine residue or that DIDS is rigidly bound, such that its isothiocyanate groups cannot become suitably oriented for reaction with a nearby lysine. We cannot rule out the possibility that DIDS can slowly inhibit ClC-ec1 irreversibly, but if so, it must do so on a timescale much slower than is observed with ClC-0. In ClC-0, 12 μM DIDS inhibits irreversibly within 40 s (43); in these experiments with ClC-ec1 (Fig. 3, A and B), 250 μM DIDS applied for ∼5 min was completely reversible.
It remains to be seen whether DIDS inhibits other ClC homologs from the intracellular side and whether DIDS binds at homologous sites in ClC-0 and ClC-ec1. Since good pharmacological tools are lacking for ClC members, identifying the binding site for DIDS in ClC-ec1, which is amenable to crystallization, may aid in designing better inhibitors for these critical proteins.
Acknowledgments
We thank Mila Gadzinski for technical assistance and Alessio Accardi and Christopher Miller for graciously donating the Y445C-ClC-ec1 construct and their adventitiously generated variant of BL21-DE3 cells found to contain low levels of contaminating porin channels. We also thank Richard Aldrich, Robert Blaustein, Anita Engh, Anthony Fodor, Weiyan Li, Gilbert Martinez, Joseph Mindell, and Richard Reimer for comments on the manuscript.
This work was supported by the Mathers Foundation and by the Esther Ehrman Lazard Faculty Scholar Award (M.M.). K.M. was supported by the Katherine McCormick Fellowship, the Stanford University School of Medicine Dean's Postdoctoral Award, and the Ruth L. Kirchstein National Research Service Award.
References
- 1.Pusch, M., and T. J. Jentsch. 2005. Unique structure and function of chloride transporting CLC proteins. IEEE Trans Nanobioscience. 4:49–57. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch, T. J., M. Poet, J. C. Fuhrmann, and A. A. Zdebik. 2005. Physiological functions of CLC Cl− channels gleaned from human genetic disease and mouse models. Annu. Rev. Physiol. 67:779–807. [DOI] [PubMed] [Google Scholar]
- 3.Jentsch, T. J., C. Lorenz, M. Pusch, and K. Steinmeyer. 1995. Myotonias due to CLC-1 chloride channel mutations. Soc. Gen. Physiol. Ser. 50:149–159. [PubMed] [Google Scholar]
- 4.Staley, K., R. Smith, J. Schaack, C. Wilcox, and T. J. Jentsch. 1996. Alteration of GABAA receptor function following gene transfer of the CLC-2 chloride channel. Neuron. 17:543–551. [DOI] [PubMed] [Google Scholar]
- 5.Piwon, N., W. Gunther, M. Schwake, M. R. Bosl, and T. J. Jentsch. 2000. ClC-5 Cl− -channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature. 408:369–373. [DOI] [PubMed] [Google Scholar]
- 6.Bosl, M. R., V. Stein, C. Hubner, A. A. Zdebik, S. E. Jordt, A. K. Mukhopadhyay, M. S. Davidoff, A. F. Holstein, and T. J. Jentsch. 2001. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl(−) channel disruption. EMBO J. 20:1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipecka, J., M. Bali, A. Thomas, P. Fanen, A. Edelman, and J. Fritsch. 2002. Distribution of ClC-2 chloride channel in rat and human epithelial tissues. Am. J. Physiol. Cell Physiol. 282:C805–C816. [DOI] [PubMed] [Google Scholar]
- 8.Wills, N. K., and P. Fong. 2001. ClC chloride channels in epithelia: recent progress and remaining puzzles. News Physiol. Sci. 16:161–166. [DOI] [PubMed] [Google Scholar]
- 9.Jentsch, T. J. 2005. Chloride transport in the kidney: lessons from human disease and knockout mice. J. Am. Soc. Nephrol. 16:1549–1561. [DOI] [PubMed] [Google Scholar]
- 10.Iyer, R., T. M. Iverson, A. Accardi, and C. Miller. 2002. A biological role for prokaryotic ClC chloride channels. Nature. 419:715–718. [DOI] [PubMed] [Google Scholar]
- 11.Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- 12.Dutzler, R., E. B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- 13.Ludewig, U., M. Pusch, and T. J. Jentsch. 1996. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature. 383:340–343. [DOI] [PubMed] [Google Scholar]
- 14.Middleton, R. E., D. J. Pheasant, and C. Miller. 1996. Homodimeric architecture of a ClC-type chloride ion channel. Nature. 383:337–340. [DOI] [PubMed] [Google Scholar]
- 15.Saviane, C., F. Conti, and M. Pusch. 1999. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 113:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinreich, F., and T. J. Jentsch. 2001. Pores formed by single subunits in mixed dimers of different CLC chloride channels. J. Biol. Chem. 276:2347–2353. [DOI] [PubMed] [Google Scholar]
- 17.Estevez, R., B. C. Schroeder, A. Accardi, T. J. Jentsch, and M. Pusch. 2003. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 38:47–59. [DOI] [PubMed] [Google Scholar]
- 18.Lin, C. W., and T. Y. Chen. 2003. Probing the pore of ClC-0 by substituted cysteine accessibility method using methane thiosulfonate reagents. J. Gen. Physiol. 122:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Traverso, S., L. Elia, and M. Pusch. 2003. Gating competence of constitutively open CLC-0 mutants revealed by the interaction with a small organic inhibitor. J. Gen. Physiol. 122:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen, M. F., and T. Y. Chen. 2003. Side-chain charge effects and conductance determinants in the pore of ClC-0 chloride channels. J. Gen. Physiol. 122:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engh, A. M., and M. Maduke. 2005. Cysteine accessibility in ClC-0 supports conservation of the ClC intracellular vestibule. J. Gen. Physiol. 125:601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Accardi, A., and C. Miller. 2004. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 427:803–807. [DOI] [PubMed] [Google Scholar]
- 23.Stein, W. D. 1990. Channels, Carriers, and Pumps. Academic Press, Inc., San Diego, CA.
- 24.Hanke, W., and C. Miller. 1983. Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 82:25–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen, T. Y., and C. Miller. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pusch, M., U. Ludewig, A. Rehfeldt, and T. J. Jentsch. 1995. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature. 373:527–531. [DOI] [PubMed] [Google Scholar]
- 27.Pusch, M. 2004. Structural insights into chloride and proton-mediated gating of CLC chloride channels. Biochemistry. 43:1135–1144. [DOI] [PubMed] [Google Scholar]
- 28.Richard, E. A., and C. Miller. 1990. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 247:1208–1210. [DOI] [PubMed] [Google Scholar]
- 29.Salhany, J. M. 1996. Allosteric effects in stilbenedisulfonate binding to band 3 protein (AE1). Cell Mol. Biol. (Noisy-le-grand). 42:1065–1096. [PubMed] [Google Scholar]
-
30.Romero, M. F., C. M. Fulton, and W. F. Boron. 2004. The SLC4 family of
 transporters. Pflugers Arch. 447:495–509. [DOI] [PubMed] [Google Scholar]
transporters. Pflugers Arch. 447:495–509. [DOI] [PubMed] [Google Scholar] - 31.Jentsch, T. J., V. Stein, F. Weinreich, and A. A. Zdebik. 2002. Molecular structure and physiological function of chloride channels. Physiol. Rev. 82:503–568. [DOI] [PubMed] [Google Scholar]
- 32.Cabantchik, Z. I., and R. Greger. 1992. Chemical probes for anion transporters of mammalian cell membranes. Am. J. Physiol. 262:C803–C827. [DOI] [PubMed] [Google Scholar]
- 33.Accardi, A., L. Kolmakova-Partensky, C. Williams, and C. Miller. 2004. Ionic currents mediated by a prokaryotic homologue of CLC Cl− channels. J. Gen. Physiol. 123:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heginbotham, L., L. Kolmakova-Partensky, and C. Miller. 1998. Functional reconstitution of a prokaryotic K+ channel. J. Gen. Physiol. 111:741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nozaki, Y., and C. Tanford. 1981. Proton and hydroxide ion permeability of phospholipid vesicles. Proc. Natl. Acad. Sci. USA. 78:4324–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mindell, J. A., M. Maduke, C. Miller, and N. Grigorieff. 2001. Projection structure of a ClC-type chloride channel at 6.5 Å resolution. Nature. 409:219–223. [DOI] [PubMed] [Google Scholar]
- 37.Picollo, A., A. Liantonio, M. P. Didonna, L. Elia, D. C. Camerino, and M. Pusch. 2004. Molecular determinants of differential pore blocking of kidney CLC-K chloride channels. EMBO Rep. 5:584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis, R. S., P. E. Ross, and M. D. Cahalan. 1993. Chloride channels activated by osmotic stress in T lymphocytes. J. Gen. Physiol. 101:801–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helix, N., D. Strobaek, B. H. Dahl, and P. Christophersen. 2003. Inhibition of the endogenous volume-regulated anion channel (VRAC) in HEK293 cells by acidic di-aryl-ureas. J. Membr. Biol. 196:83–94. [DOI] [PubMed] [Google Scholar]
- 40.Thiemann, A., S. Grunder, M. Pusch, and T. J. Jentsch. 1992. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 356:57–60. [DOI] [PubMed] [Google Scholar]
- 41.Li, X., K. Shimada, L. A. Showalter, and S. A. Weinman. 2000. Biophysical properties of ClC-3 differentiate it from swelling-activated chloride channels in Chinese hamster ovary-K1 cells. J. Biol. Chem. 275:35994–35998. [DOI] [PubMed] [Google Scholar]
- 42.Mo, L., H. L. Hellmich, P. Fong, T. Wood, J. Embesi, and N. K. Wills. 1999. Comparison of amphibian and human ClC-5: similarity of functional properties and inhibition by external pH. J. Membr. Biol. 168:253–264. [DOI] [PubMed] [Google Scholar]
- 43.Miller, C., and M. M. White. 1980. A voltage-dependent chloride conductance channel from Torpedo electroplax membrane. Ann. N. Y. Acad. Sci. 341:534–551. [DOI] [PubMed] [Google Scholar]
- 44.Miller, C., and M. M. White. 1984. Dimeric structure of single chloride channels from Torpedo electroplax. Proc. Natl. Acad. Sci. USA. 81:2772–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson, R. A., and R. H. Stokes. 1965. Electrolyte Solutions. Butterworths, London.