

Abstract
Hypothetical hydroxide and proton migration along the linear water chain in Aquaporin GlpF from Escherichia coli are studied by ab initio Car-Parrinello molecular dynamics simulations. It is found that the protein stabilizes a bipolar single file of water. The single file features a contiguous set of water-water hydrogen bonds in which polarization of the water molecules vary with position along the channel axis. Deprotonation of the water chain promotes the reorientation of water molecules while the hydroxide ion rapidly migrates by sequentially accepting protons from the neighboring water molecules. The hydroxide ion is not attracted by a conserved, channel-lining arginine residue, but is immobilized at two centrally located, conserved Asparagine-Proline-Alanine motifs where fourfold coordination stabilizes the ion. Hydroxide transition from the channel vestibules into the channel lumen is strongly influenced by electrostatic coupling to two conserved oppositely aligned macrodipoles. This suggests that the macrodipole's negative poles play a role in preventing hydroxide ions from entering into the channel's inner vestibules. Water protonation within the lumen facilitates water reorientation and subsequent proton expelling occurs. In the periplasmic half-channel, expelling occurs via the Grotthuss mechanism. Protonation within the cytoplasmic half-channel implies wire-breakage at the Asn-Pro-Ala motifs. The proton is here diffusively rejected as (H5O2)+.
INTRODUCTION
All lifeforms need to accurately regulate cellular water contents through exchange of water with extracellular surroundings. Water regulation must be facilitated in a nondissipative manner with respect to the proton gradient, a prerequisite for proton-motive, force-driven ATP synthesis in any living organism. Accordingly, protons must be excluded from being conducted by the water-transporting device. This also applies to hydroxide ions, since transport of a proton in one direction is equivalent to transport of an hydroxide ion in the opposite direction. These tasks are ensured by specially designed water channels called Aquaporins (AQPs) (1–5). AQPs are homotetrameric, highly selective, water-transporting units—ubiquitously present in all living organisms exhibiting conduction rates of ∼109/s (3,6).
AQPs conserve two half-membrane spanning (re-entrant) loops that are one-half α-helical (denoted M3/M7 or HB/HE) and one-half coiled, respectively. The latter exposes carbonyl groups toward the channel lumen, thereby providing a conduction ladder for water (7–9). The α-helices terminate at the channel center at two conserved Asn-Pro-Ala (NPA) motifs (7–10). Multiple hydrogen bonds between the NPA motifs result in an NPA-configuration directing one amine hydrogen toward the pore, which also provides explicit hydrogen-bond donation for water molecules traversing the lumen (11–14). Moreover, a conserved, positively charged arginine (R) residue, consistently located in an extracellular (or periplasmic) channel entrance, together constitute, with an aromatic (ar; e.g., phenylalanine) and a polar residue (e.g., tryptophan or histidine, the latter eventually charged), the so-called AQP selectivity filter or ar/R constriction (7–10,12,15–17).
Medium-resolution models of human AQP1 (hAQP1) (7,9) and later high-resolution x-ray structures of the Escherichia coli glycerol uptake facilitator GlpF and bovine AQP1 (bAQP1) (8,10,18), and more recently x-ray structures of E. coli AqpZ (15) and of bovine and sheep AQP0 (16,17), provided structural insight into AQP selectivity by consistently revealing narrow water pores suggesting single-file transport. By extending the static, structural information into the dynamic domain, early molecular dynamics (MD) simulations of the hAQP1 model addressed this question, but also revealed that the low-resolution model structure was insufficient for reliable simulation studies of water permeation in AQPs (12,13,19). Single-file transport of water was, nonetheless, confirmed by MD simulations of the crystal structures of bAQP1 (10) and of GlpF (8,18) and of a refined hAQP1 model (11,12) in numerous MD studies (11,18,20–24). However, recent Monte Carlo (MC) studies of water transport in rigid bAQP1 and GlpF channels challenged the single-file picture of water transport (25).
A central question in the function of AQPs concerns the selectivity raised against protons and hydroxide ions. This question was indirectly addressed by classical MD simulations (11,18,22) and later by MC simulations (25). Selectivity against protons was from MD attributed to a bipolar water orientation (18,22,24–26,), observable in the GlpF lumen, and in lumens of the hAQP1 model (11) and of bAQP1 (see (23) and (26); and also Figs. 1 and 2, this article). It was argued that the bipolar orientation is stabilized by the electrostatic environment of the channel interior (18,22,27,28), which impedes formation of a so-called proton-wire (29), i.e., a monopolar orientation of the water molecules where protons are conducted by means of the Grotthuss mechanism (29–35), as illustrated in Fig. 1 c. However, neither the MD or MC simulations explicitly incorporated a proton (or a hydroxide ion) in the channel lumen. For the MD methodology this drawback was pointed out by Burykin and Warshel (36). A fundamentally different mechanism relying on spatial and not orientational disruption of the contiguous hydrogen (H)-bonded water network was proposed in Ren et al. (9) and supported by MD studies of the hAQP1 model finding disruption of the water file in either the channel center (19) or in the ar/R region (11). The latter finding was supported by MC studies (25) of the later resolved high-resolution bAQP1 structure (10) in rigid form.
FIGURE 1.

Schematic representation of bipolar (a) and monopolar (b) water wires and of Grotthuss transfer of a proton (c) and hydroxide ion (d) in monopolar water wires.
FIGURE 2.

(a) The QM/MM simulated system, consisting of one GlpF monomer with hydrating (capping) molecules. Nine (QM) water molecules, with the seven central ones within the constriction region, are displayed as solid van der Waals spheres. The remaining QM waters (six more in the cytoplasmic side and six more in the periplasmic side) are not represented. The selectivity filter is displayed with the W48 and F200 side chains in red-licorice representation (omitting H-atoms) and R206 is shown in standard colors. Depicted similarly are side chains of the conserved residues N68/N203 of the NPA motifs in the channel center. The direction of periplasm to cytoplasm is taken along increasing z. (b) AQP characteristic half-membrane spanning repeats with α-helical (69–79(M3), 206–217(M7)) and nonhelical (65–68, 199–205, the conduction pathway) parts rendered in brown and green, respectively. In licorice representation are displayed side chains of R206 and of N68/N203 of the NPA motifs. (c) Schematic hydrogen-bonding structure of the single-file waters and their hydrogen-bonding interactions with the polar groups of channel-lining residues.
Recently, simulation studies considered, explicitly, a proton in the GlpF or bAQP1 channel lumen. Consistently, one major barrier against proton transfer of electrostatic origin located in the center of the channel, i.e., at the position of the NPA motifs, was inferred (27,28,36–39). As recognized by most authors, the origin of AQP selectivity against protons should be found in the channel architecture (5,7,9–11,18,22,27,28,37,40). One expects that ionic (electrostatic) interactions with the conserved positively charged Arg residue in the selectivity filter (SF), e.g., R206 in GlpF, should play a role, but, seemingly, its influence is suppressed due to long-range (counter) electrostatic contributions from other charged (extraluminal) residues in the channel (22,25,27). The macrodipoles provided by the two conserved half-helices proved, in GlpF, to play an electrostatic role in dictating a bipolar water orientation that is not eligible for protonation (see (18) and (22); see also Figs. 1 and 2, this article). From analysis of the electrostatic forces acting on water inside the channel, it was also rationalized that a concerted reorientation of one-half of the bipolar, neutral single file, prerequisite to its protonation, was associated with a large barrier (22)—a suggestion later confirmed computationally (27). Another factor working against proton and hydroxide transfer in AQPs is an energetically enthalpic- and entropic-unfavorable dehydration energy associated with ion transfer from bulk water into a one-dimensional luminal confinement (36,39,41), which, in AQPs, offers the ion poor solvation, due to the narrow geometry and pronounced hydrophobicity of the channel lining. This is in clear contrast to proton-conducting channels (32,34,35,42).
To date, the origin of prevention of hydroxide ion migration across the AQP channel lumen has attracted little attention. So far only classical MD simulations have approached this question, and interestingly revealed luminal water-hydroxide co-diffusion leading to hydroxide immobilization at the NPA-motifs (37). The entrance of the hydroxide ion into the channel lumen was found obstructed by electrostatic barriers at the channel in-/outlets (37), presumably also a feature applying to the repelling of halide ions (25), although AQP6 conducts anions (see 43, and references therein).
Therefore, some divergence among conclusions regarding the nature of ion selectivity and ion transfer in AQPs prevails as recently reviewed (44). In this regard, it must be taken into account that rigid channel treatments (25,36,39) are potential sources of ambiguity (e.g., water transport is influenced by side-chain fluctuations at the SF region; see (24)). Moreover, discrepancies could well originate from differences in computational methodology; i.e., all charge transfer studies to date employ phenomenological (37) or empirical (25) or semi-empirical (27,28,36,38,39) descriptions of the charge transfer, not taking into account the electronic structure of the water wire.
We here report first-principles Car-Parrinello MD simulations (CPMD) of hydroxide and proton transfers in GlpF (8,18), fully representing the electronic structure of the water wire. CPMD applications to biological systems employing the Car-Parrinello method as well as its ab initio molecular dynamics/molecular mechanics (MD/MM) extension have been applied with success in investigations of biological problems (45–52) as well as in investigations of hydronium and hydroxide ion transfer in bulk water clusters (53,54).
METHODS
Below we describe modeling of GlpF when the channel embeds neutral and protonated/deprotonated water wires, respectively, followed by details of the simulations and analyses.
Initial structures, neutral wire
The starting configuration seen in Fig. 2 was derived by reducing an equilibrated configuration from classical MD simulation of the GlpF tetramer (8) embedded in a fully hydrated 16:0/18:1c9-palmitoyloleyl phosphatidylcholine (POPC) bilayer (24). Note that, apart from the lipid headgroup composition, the GlpF/POPC simulation and its results (24) were practically identical to those of GlpF embedded in a hydrated 16:0/18:1c9-palmitoyloleyl phosphatidylethanolamine bilayer (18,22). We selected a configuration in which the water wire was not disrupted, which is the most likely configuration (18,22,24). The appropriately reduced system (Fig. 2) included one GlpF monomer (3879 atoms); 3473 water molecules within an orthorhombic region measuring 49.7 by 55.4 by 79.6 Å3 in the x-, y-, and z-directions; and one Cl− anion, which ensures neutrality of the total system. In total, 14,262 atoms were retained.
Initial structures, charged wires
Since no constraints were imposed onto the proton in our simulation, migration of the proton within the channel lumen can, owing to electrostatic barriers (27,28,36–39), only be expected to be observable if the water wire initially is protonated inside the lumen, e.g., between the SF and Gly64 that define the lumen (see Fig. 2). Hydroxide transfer from the vestibule into the lumen is likely to occur as long as initial deprotonation is sufficiently close to the lumen. By resorting to this criterion for initial deprotonation, we prevented undesired hydroxide transfers within bulk/vestibule water molecules, where a three or fourfold-coordinated hydroxide ion is of lowest free energy (54,55). From the hybrid quantum-classical simulations (QM/MM) equilibrated neutral system we generated 2 × 2 configurations by protonating/deprotonating water molecules located within the lumen of the periplasmic and cytoplasmic half-channels. Specifically, we protonated/deprotonated the water molecules located at the extremes of the single file; i.e., W2 in the periplasmic half-channel and W8 in the cytoplasmic half-channel (see Fig. 2 c). These four starting configurations were employed to investigate intraluminal proton and hydroxide migration. All our simulations resorting to intraluminal deprotonation resulted in the hydroxide ion being immobilized at NPA motifs at the channel center. Consequently, we performed additional simulations in which the initial deprotonation was extraluminal. Deprotonation was either at the periplasmic channel entrance, or above this entrance, or in the periplasmic vestibule, albeit close to the channel—i.e., W1, W0, and W−1 in Fig. 2 c were deprotonated, respectively. To determine influence on hydroxide mobility from the electrostatic attraction/repulsion of the M3/M7 macrodipoles and from the NPA motifs, we repeated the deprotonated W1/W−1 simulations by turning off backbone charges of the M3/M7 half-helices and on the N68/N203 amino groups of the NPA motifs as described in Tajkhorshid et al. (18). Appropriate fixed 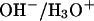 counterions were, in selected cases, added to the charged systems within the QM region (Fig. 2), opposing the initial position of protonation/deprotonation, to test whether system neutrality had impact on charge migration. However, within the short timescale of these simulations (∼1 ps), we found no significant differences among simulations either with or without counterions. Our charged-wire simulations are summarized in Table 1.
counterions were, in selected cases, added to the charged systems within the QM region (Fig. 2), opposing the initial position of protonation/deprotonation, to test whether system neutrality had impact on charge migration. However, within the short timescale of these simulations (∼1 ps), we found no significant differences among simulations either with or without counterions. Our charged-wire simulations are summarized in Table 1.
TABLE 1.
Summary and results of simulations
| Sim. | Ion | W | tsim | P | zi/zR/zN | Δz | NPA | R206 | M3/M7 |
 * *
|
nc | np |
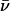 † †
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | OH− | W2 | 1286 | pin | 7.3/11.6/18.1 | 11.6 < z < 18.6 | on | on | on | 3 | 80 | 77 | 122 |
| 2‡ | OH− | W5 | 759 | N | 17.9/6.2/17.4 | 15.7 < z < 18.7 | off | on | on | 0 | 7 | 7 | 19 |
| 3‡ | OH− | W5 | 491 | N | 17.9/6.2/16.8 | 17.9 < z < 17.9 | off | off | on | 0 | 6 | 6 | 24 |
| 4 | OH− | W8 | 197 | C | 24.0/8.4/19.0 | 17.3 < z < 24.2 | on | on | on | −2 | 7 | 9 | 22 |
| 5 | H3O+ | W2 | 636 | pin | 11.6/7.3/18.1 | 5.1 < z < 11.6 | on | on | on | −3 | 11 | 14 | 37 |
| 6 | H3O+ | W2 | 187 | pin | 11.6/7.3/18.1 | 6.1 < z < 11.6 | off | on | on | −3 | 3 | 6 | 48 |
| 7a | H3O+ | W2 | 151 | pin | 6.8/7.2/17.5 | 11.5 < z < 11.5 | off | off | on | 0§ | 0 | 0 | 0 |
| 7b | H3O+ | W2 | 151 | pin | 6.8/7.2/17.5 | 11.5 < z < 11.5 | off | off | on | 0§ | 0 | 0 | 0 |
| 7c | H3O+ | W2 | 643 | pin | 6.8/7.2/17.5 | 11.5 < z < 19.1 | off | off | on | 1§ | 31 | 30 | 95 |
| 8¶ | H3O+ | W8 | 400 | C | 24.1/8.1/18.9 | 24.0 < z < 26.9 | on | on | on | 0 | 0 | 0 | 0 |
| 9 | OH− | W1 | 392 | P | 12.8/7.3/18.1 | 8.6 < z < 15.4 | on | on | on | 1 | 84 | 83 | 426 |
| 10 | OH− | W1 | 491 | P | 12.8/7.3/18.1 | 9.0 < z < 16.4 | on | on | off | 2 | 12 | 10 | 45 |
| 11 | OH− | W1 | 361 | P | 12.8/7.3/18.1 | 8.7 < z < 15.6 | off | on | off | 1 | 71 | 69 | 388 |
| 12 | OH− | W0 | 189 | P | 6.5/7.1/17.5 | 6.6 < z < 15.5 | on | on | on | 3 | 19 | 16 | 185 |
| 13 | OH− | W−1 | 983 | P | 13.4/15.2/25.8 | 12.3 < z < 18.9 | on | on | on | 4 | 91 | 87 | 181 |
| 14 | OH− | W−1 | 756 | P | 13.4/15.2/25.8 | 8.1 < z < 16.7 | on | on | off | −3 | 106 | 109 | 284 |
| 15 | OH− | W−1 | 418 | P | 13.4/15.2/25.8 | 9.3 < z < 14.6 | off | on | off | −1 | 41 | 42 | 201 |
Initial ion positions (zi in Å) were associated with a water molecule (W) positioned (P) in either cytoplasm (c), periplasm (p), inside periplasmic half-channel (pin) or at the NPA motifs (N). Simulation time (tsim) is given in femtoseconds. Positions of R206: Nη closest to the periplasm (zR) and of the midpoint between the Nδs of N68/N203 of the NPA motifs (zN) are given for reference in each simulation (these positions may vary among the simulations). The sampled z range of the ion during the simulation (Δz) is given in Å. Electrostatic interactions with side chains of NPA (i.e., N68/N203) and R206 were either retained (on) or eliminated (off).
The net number of ionic hops (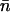 ) along the channel axis was computed as
) along the channel axis was computed as 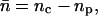 where nc and np denote the number of hops toward cytoplasm and periplasm, respectively, using bond and hop-cutoffs of 1.2 and 1.5 Å, respectively (see text for more details on the definition of a hop). Positive and negative values of
where nc and np denote the number of hops toward cytoplasm and periplasm, respectively, using bond and hop-cutoffs of 1.2 and 1.5 Å, respectively (see text for more details on the definition of a hop). Positive and negative values of 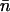 therefore indicates net ion movement toward the cytoplasm and periplasm, respectively (see Figs. 2, 7–9).
therefore indicates net ion movement toward the cytoplasm and periplasm, respectively (see Figs. 2, 7–9).
A mean hopping frequency 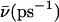 was computed as
was computed as 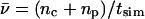 where tsim is the total simulation time varying for the different simulations.
where tsim is the total simulation time varying for the different simulations.
Restarted from the last configuration of Sim. 1 where OH− is immobilized at the NPA region.
Proton migration was only observed in the heavily constrained part of the simulation (643 fs out of 945 fs in total; see text).
Proton structural diffusion occurred (see text).
Hybrid quantum-classical simulations (QM/MM)
The QM/MM calculations were performed using the ab initio MD/MM approach (56), which combines the first-principles MD method of Car and Parrinello (CPMD) based on density functional theory (57,58) with a force-field MD methodology. In this approach, the system is partitioned into QM and MM fragments as illustrated in Fig. 2 where 21 water molecules (including nine single-file waters highlighted in Fig. 2) were QM-treated, whereas the reminder of the system was MM-treated. The QM fragment was enclosed in an isolated super-cell of dimensions a = b = 15 Å, c = 36 Å. For the simulations where W−1 initially was deprotonated, we used an extended QM region of 27 water molecules (20 waters capping the periplasmic vestibule plus seven single-file waters). The dynamics of the QM atoms depends on the electronic density computed with density functional theory, whereas that of the MM atoms is governed by an empirical force field. QM-MM electrostatic interactions are handled via a fully Hamiltonian coupling scheme (56), in which the short-range interactions were explicitly taken into account within a 6 Å sphere around every QM atom. An appropriately modified Coulomb potential was used to avoid unphysical escape of the electronic density from the QM to the MM region (56). For the QM region, electrostatic interactions with distant MM atoms are treated with a multipole expansion. Bonded and van der Waals interactions between the QM and the MM part are treated with a standard all-atom AMBER force field (59). Long-range MM electrostatic interactions were handled by a particle-particle-particle-mesh implementation (59) using a 64 × 64 × 64 mesh corresponding to a ∼1 Å grid spacing. For the neutral wire simulation, the QM subsystem is neutral, while it has a negative/positive charge for the ion migration simulations. The electronic wave functions were expanded in plane waves up to a kinetic energy cutoff of 70 Ry. We employed the Perdew-Burke-Ernzerhof generalized gradient-corrected approximation (60). Norm-conserving pseudo-potentials (61) were used to describe valence electrons. MD simulations used a time-step of 0.072 fs and the fictitious electronic mass of the Car-Parrinello Lagrangian was set to 900 a.u. The simulation of the neutral system was performed for 10 ps with an average temperature of 310 K. Charged-wire simulations departing from a selected snapshot of the equilibrated, neutral wire simulation were performed with a similar setup, except that no initial temperature was assigned. The temperature was then raised progressively, until it oscillated at ∼150 K for ∼0.5–1 ps. For proton transfer simulations, this time was sufficient for observing proton migration to the channel vestibules, i.e., unidirectional lumen vestibule transitions. For intraluminal hydroxide transfer simulations, this time was sufficient for observing ion immobilization at the NPA region, whereas bidirectional lumen
vestibule transitions. For intraluminal hydroxide transfer simulations, this time was sufficient for observing ion immobilization at the NPA region, whereas bidirectional lumen vestibule transitions were observed in the extraluminal hydroxide simulations.
vestibule transitions were observed in the extraluminal hydroxide simulations.
ANALYSIS OF OH− AND H3O+ MIGRATION
Below we outline our analysis of structural and dynamic quantities. Analysis was performed with VMD (62).
Dipole moments
Partial (dynamic) charges on the QM atoms were computed from the electrostatic potential (ESP) obtained from the simulations. Water dipole moments were computed from instantaneous positions and ESP charges. Individual water dipole moments were summed to yield the total wire dipole moment using, as origin, the center-of-mass of the water wire. Since the total dipole moment of the charged wires is origin-dependent, we only consider correlations among the three components of the unit-dipole moment accompanying charge transfers by means of the correlation coefficient 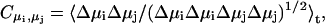 where
where 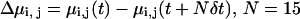 is the coordinate saving frequency, and δt = 0.072 fs is the time-step.
is the coordinate saving frequency, and δt = 0.072 fs is the time-step.
Ion localization
Hydroxide and proton positions were localized by searching for oxygen (O) atoms not having two (covalent) bonds. A covalent bond cutoff |rOH| ≤ 1.1 Å, based on the first peak in the radial distribution function ρ(|rO–H|) of the neutral wire, was applied. Typically, the excess negative charge is found in the wire as [HOH⋯OH]− or as delocalized between two O atoms in an extended (H5O3)− complex, i.e., [HOH⋯OH⋯⋯HOH]−, where one of the noncovalent O-H bonds usually will be longer than the other. The solvated proton also exists in the wire as a continuum of states between the fully localized hydronium ion H3O+ (the so-called Eigen-equivalent without a three-water solvation shell) and the delocalized Zundel proton (H5O2)+, i.e., [HOH⋯H⋯⋯HOH]+. We defined the proton (Zundel) position as that of the O atom to which the [HOH⋯H]+ distance is shortest and the hydroxide position as that of the O atom with only one covalent bond to H.
Ion translocation and mobility
The proton and hydroxide ion migrate via proton jumps involving the ion and its neighboring water molecule, i.e., a Grotthuss-type mechanism as illustrated in Fig. 1. For proton migration one proton is donated to the neighboring water (Fig. 1 c), whereas hydroxide migration implies that the ion accepts a proton from its neighboring water (Fig. 1 d). Another difference between proton and hydroxide migration is their respective geometric requirements. A proton (e.g., a hydronium ion) only migrates if the dipole of its neighboring water is oriented along the direction of migration. This condition can be fulfilled for a monopolar wire (Fig. 1 b) but not in any of the two strands in the bipolar wire opposing terminal protonation (Fig. 1 a). Hydroxide migration, i.e., the hopping of a proton hole, requires that the dipole of the neighboring water is oriented against the direction of migration (Fig. 1 d) a condition fulfilled in both strands of the bipolar wire (Fig. 1 a). Based on these geometric considerations, a proton (hydronium ion) penetrating into the AQP channel lumen from the bulk region is expected to be expelled, whereas the hydroxide ion could reach the channel center. We monitored the movement, referred to as translocation or hopping, of the ions in terms of their net number of hops 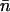 along the channel axis. A hop was registered if the displacement (Δz) satisfied
along the channel axis. A hop was registered if the displacement (Δz) satisfied 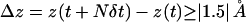 with δt and N defined above. Translocations toward cytoplasm (nc) and periplasm (np) imply positive and negative Δz values, respectively (see Fig. 2 b). From nc, np, and the total simulation time (tsim), a mean hopping frequency was computed as
with δt and N defined above. Translocations toward cytoplasm (nc) and periplasm (np) imply positive and negative Δz values, respectively (see Fig. 2 b). From nc, np, and the total simulation time (tsim), a mean hopping frequency was computed as 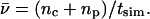
The value of 1.5 Å lies within the first water solvation shell of the O–O radial distribution function ρ(|rO–O|) obtained for the neutral wire. This distance is typically shorter between proton-donating and proton-accepting O atoms in [HOH⋯HOHH⋯HOH]+ and [HOH⋯OH⋯HOH]− configurations, which, however, elongates charged wires relative to the neutral wire overall. Although 1.5 Å is adopted as a somewhat arbitrary choice for hop criterion it was found to fully suffice for our purposes of consistently distinguishing between ion dynamics in protonated versus deprotonated wires, and for accessing changes in ion dynamics in charge-modified versus a fully charge-retained (“native”) channel.
RESULTS AND DISCUSSION
Neutral wire
Water structure
The simulation of the neutral wire shows that eight water molecules are equally spaced in a single file (Fig. 3, a and b), in accord with classical MD simulations (18,22,24). In CPMD simulations, the positions of the single-file water molecules (Fig. 3, a and b) fluctuate very little around their average position, indicative of the quasi-stationarity of the water wire on the CPMD timescale (as illustrated in Fig. 3 c, where the average positions within last part of our 10-ps trajectory of the neutral water wire are shown). Obviously, no water conduction occurs on this short timescale, which is to be expected upon comparison with classical MD results, finding that the mean hopping time for a successive water translocation by an amount equal to the average water-water spacing in the wire significantly (∼10-fold) exceeds a 10-ps timescale (21,22,24). In our simulations we find that the single-file water molecules remain H-bonded to each other and to the channel lining. Worth noting also is that the water molecules remain H-bonded across the SF region; i.e., no disruption of the H-bonded water network occurs on a short timescale. In contrast, water-water H-bond disruption was more frequently observed in MD simulations of the refined hAQP1 model (11,12) and in MC simulations of bAQP1 (25), where an adjacent histidine (H180/H182 in hAQP1/bAQP1), in addition to the arginine (R195/R197 in hAQP1/bAQP1), makes the ar/R region more polar and also competes for H-bonding with water at this constriction point (11,25). The present result for GlpF, however, agrees with classical MD simulations in which disruption at the SF is manifestly sparse (18,22,24). During our CPMD simulation, the single-file water strand exhibits and preserves the AQP characteristic bipolar orientation (Fig. 3 a). Of the eight single-file water molecules (W1–W8), W1–W4 have their dipoles oriented toward −z, whereas the dipoles of W6–W8 are oriented along +z. W5, oriented with its dipole perpendicular to z, acts as a bridge between the two wire fragments. Resorting to the concept of proton wires, it has been argued that this bipolar orientation is the main reason that protons cannot transfer across the channel (18,22).
FIGURE 3.

(a) Simulation snapshot of the channel displayed with the 21 QM water molecules rendered in CPK representation and selected MM water molecules located within the channel outlets displayed in licorice representation. F200/R206 selectivity filter side chains and N68/N203 side chains of the NPA motifs are displayed in standard colors, as are the exposed carbonyl groups of residues 64–66 and 199–202. (b) Trajectories (last 5 ps) of the center of mass of those water molecules occupying the constriction region (delineated by dashed lines). (c) Average positions and position fluctuations of water O and H atoms (H and H′) and of water center-of-mass (CM) of the same water molecules as in b. The constriction region (lumen) is delineated as in b.
The bipolar water orientation can be also inferred from the asymmetry of the dipole (μ) distributions in Fig. 4 a, where distributions of μ are compiled for the full channel and for each half-channel lumen during the last 5 ps of the neutral wire simulation. There are two clearly opposite mean orientations of μz corresponding, in which water molecules W1–W4 and W6–W8 align with their dipoles toward the periplasm (−z) and cytoplasm (+z), respectively, whereas μz(W5) ≅ 0 D indicates an average orientation perpendicular to the channel axis. Moreover, the μx and μy distributions reflect that the dipole of the periplasmic facing waters is slightly skewed toward +x,+y, whereas that of the cytoplasmic-facing waters is skewed along −x,−y—since the two conduction ladders of exposed carbonyl groups (C=O) in each half-channel essentially are orthogonally arranged (see Fig. 2 c).
FIGURE 4.

(a) Distributions of the three components of the dipole moment (in D) of the neutral water wire. The time-averaged distribution of the bipolarized wire is decomposed into distributions from the periplasmic and cytoplasmic halves of the channel using the NPA midpoint as channel axis origin. (a) The z-component, exhibiting a sign-change for the two half-channel distributions indicating bipolarity (b) x,y-components.
Polarization effects
Atomic charges of our QM waters 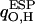 computed from the electrostatic potential (ESP) vary with time due to 1), dynamic polarization effects within the QM subsystem; and 2), QM/MM interactions. Moreover, the time-averaged atomic charge
computed from the electrostatic potential (ESP) vary with time due to 1), dynamic polarization effects within the QM subsystem; and 2), QM/MM interactions. Moreover, the time-averaged atomic charge 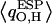 varies with position along the channel axis and exhibits substantial root mean-square (RMS) fluctuations, as shown in Fig. 5. The latter effect is not accounted for in simulations deploying nonpolarizable water models. Analysis of
varies with position along the channel axis and exhibits substantial root mean-square (RMS) fluctuations, as shown in Fig. 5. The latter effect is not accounted for in simulations deploying nonpolarizable water models. Analysis of 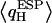 and corresponding RMS charge fluctuations reveal a significant degree of asymmetry of
and corresponding RMS charge fluctuations reveal a significant degree of asymmetry of 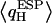 for H atoms of the same water (Fig. 5 b). One H atom is consistently more positively charged than the other, implying
for H atoms of the same water (Fig. 5 b). One H atom is consistently more positively charged than the other, implying 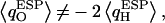 since one H atom H-bonds to a neighboring water molecule, whereas the other interacts with the exposed C=O groups of the (amphiphatic) channel lining. C=O⋯H interactions imply that
since one H atom H-bonds to a neighboring water molecule, whereas the other interacts with the exposed C=O groups of the (amphiphatic) channel lining. C=O⋯H interactions imply that 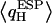 shifts toward positive value, whereas
shifts toward positive value, whereas  of the other H (water H-bonded) almost vanishes, since that H is symmetrically positioned between two water O atoms. The pronounced asymmetry in
of the other H (water H-bonded) almost vanishes, since that H is symmetrically positioned between two water O atoms. The pronounced asymmetry in 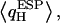 moreover, reflects that all luminal waters are QM-treated, whereas the channel lining is not. Hence, only hydrogens shared between two QM waters benefit from charge transfer/delocalization effects, whereas hydrogens H-bonded to MM-treated C=O groups do not.
moreover, reflects that all luminal waters are QM-treated, whereas the channel lining is not. Hence, only hydrogens shared between two QM waters benefit from charge transfer/delocalization effects, whereas hydrogens H-bonded to MM-treated C=O groups do not.
FIGURE 5.

Average charges and charge RMS fluctuations of the single-file water molecules during the last five picoseconds of the simulation. For comparison, the corresponding TIP3 point charges for O and H are delineated vertically. The constriction region (lumen) is delineated horizontally as in Fig. 3, b and c.
One discerns from Fig. 5 a that all O atoms of the single-file waters, except for W1 and W5, have similar  These O atoms either belong to water molecules having little interaction with the channel (W4, W6, and W7, as summarized in Fig. 2 c) or to waters H-bonding (C=O⋯HOH) to the channel lining (W2, W3, and W8; see Fig. 2 c). The O atoms of W1 and W5 exhibit very different, negatively shifted values of
These O atoms either belong to water molecules having little interaction with the channel (W4, W6, and W7, as summarized in Fig. 2 c) or to waters H-bonding (C=O⋯HOH) to the channel lining (W2, W3, and W8; see Fig. 2 c). The O atoms of W1 and W5 exhibit very different, negatively shifted values of 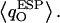 These O atoms, belonging to waters located in a very polar environment, interact with the R206:Hηs and with the Hδs of N68/N203 of the NPA motifs, respectively. Although
These O atoms, belonging to waters located in a very polar environment, interact with the R206:Hηs and with the Hδs of N68/N203 of the NPA motifs, respectively. Although 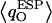 of W1 and W5 is negatively-shifted, the corresponding RMS fluctuations of
of W1 and W5 is negatively-shifted, the corresponding RMS fluctuations of 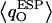 largely seem to be of uniform magnitude for all single-file water molecules, whereas RMS fluctuations of
largely seem to be of uniform magnitude for all single-file water molecules, whereas RMS fluctuations of 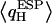 are significantly larger for W1 and W2 located in vicinity of the R206 guanidinium group. This reflects that water/channel electrostatic interactions in this region are particularly strong (22,25) yet insufficient to break the H-bonded network of the single file. Interestingly, e.g., the TIP3P water charges (63), commonly used in classical MD simulations and vertically delineated in Fig. 5, clearly deviate from
are significantly larger for W1 and W2 located in vicinity of the R206 guanidinium group. This reflects that water/channel electrostatic interactions in this region are particularly strong (22,25) yet insufficient to break the H-bonded network of the single file. Interestingly, e.g., the TIP3P water charges (63), commonly used in classical MD simulations and vertically delineated in Fig. 5, clearly deviate from 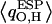 obtained in the heterogeneous channel lumen. Nevertheless, this merely influences the overall structure and persistence of the single file; its major structural properties are identical to those inferred from the classical MD simulations (11,18,20,22,24).
obtained in the heterogeneous channel lumen. Nevertheless, this merely influences the overall structure and persistence of the single file; its major structural properties are identical to those inferred from the classical MD simulations (11,18,20,22,24).
Charged wires
H-bonding structure
A radial distribution function ρ(|rO–H|) for the water molecules in the neutral, protonated, and deprotonated wires was computed for quantification of changes in the wire H-bonding structure upon introduction of an excess charge mimicking hypothetical luminal deprotonations or protonations (see Methods). Distributions were obtained with 0.1 Å resolution, followed by smoothening (64). The first peak in ρ(|rO–H|) shown in Fig. 6 is the intramolecular O–H distance (|rO−H| ≅ 1.0 Å), practically invariant among the wires. The second peak is the nearest-neighbor H-bonding distance (∼1.7 Å ≤ |rO−H| ≤ ∼2.0 Å). Deprotonation shifts first- and higher-order peaks to an ∼10% larger value, implying that H-bonds in the deprotonated wire on average are longer, and presumably less stable, relative to the neutral wire. This makes water molecules within the deprotonated wire more prone to reorientation relative to the neutral wire. In the protonated wire, second- and higher-order peaks become bimodal, reflecting the excess proton, while the second-order peak is shifted in a fashion similar to the deprotonated wire and with the same implications.
FIGURE 6.

Average hydrogen-bonded structure of the neutral, protonated, and deprotonated wires measured by the radial distribution function ρ(|rO–H|). Non-neutral wires were protonated and deprotonated, respectively, in the lumen of the periplasmic half-channel.
Ionic mobility
Due to favorable hydroxide-channel electrostatic interactions, one expects GlpF to favor luminal incorporation of hydroxide ions over protons if they were to enter (22,25,27,37). Electrostatic interactions between these ions and between the NPA motifs, R206, and the M3/M7 macrodipoles, should influence ionic mobility (22,25,27,37,38). Ionic mobility results, in terms of the mean hopping frequency 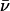 (see Methods), are collected in Table 1.
(see Methods), are collected in Table 1.
Proton and hydroxide mobility is indeed influenced by the luminal electrostatic environment. For the native channel the luminal hydroxide mobility is 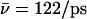 within the periplasmic half-channel (Simulation 1), noting that hydroxide translocation across the NPA region is always absent. Near the periplasmic outlet the mobility increases; i.e., for deprotonation of
within the periplasmic half-channel (Simulation 1), noting that hydroxide translocation across the NPA region is always absent. Near the periplasmic outlet the mobility increases; i.e., for deprotonation of 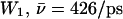 (Sim. 9); for deprotonation of
(Sim. 9); for deprotonation of 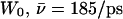 (Sim. 12); and for deprotonation of
(Sim. 12); and for deprotonation of 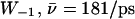 (Sim. 13; see Fig. 2 c). Within the periplasmic half-channel, elimination of the backbone charges of the M3/M7 α-helices (as in (18) and (22)) reduces mobility; for
(Sim. 13; see Fig. 2 c). Within the periplasmic half-channel, elimination of the backbone charges of the M3/M7 α-helices (as in (18) and (22)) reduces mobility; for  (Sim. 10), however, additional elimination of N68/N203 side-chain charges of the NPA motifs again increases mobility,
(Sim. 10), however, additional elimination of N68/N203 side-chain charges of the NPA motifs again increases mobility, 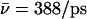 (W1, Sim. 11). Clearly, the hydroxide ion is well accommodated by the periplasmic half-channel, and readily oscillates back and forth between local binding sites within this region, whereas, in the cytoplasmic half-channel, this mobility is significantly lower, as
(W1, Sim. 11). Clearly, the hydroxide ion is well accommodated by the periplasmic half-channel, and readily oscillates back and forth between local binding sites within this region, whereas, in the cytoplasmic half-channel, this mobility is significantly lower, as 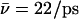 (Sim. 4). We attribute this difference to the more polar periplasmic region reinforcing water- and ion-channel electrostatic interactions relative to the rather hydrophobic cytoplasmic half-channel—in line with the fact that narrow hydrophilic confinements, such as gramicidin A, are better sustainers of ionic currents (32,34,65), than, e.g., hydrophobic carbon nanotubes (26,41). A drawback of the latter, in this respect, is that it offers poor solvation of the ion. For deprotonation of W−1 in the lumen-periplasmic vestibule boundary one finds (Sims. 13–15; see Fig. 2 c) that elimination of the M3/M7 backbone charges increases hydroxide mobility from
(Sim. 4). We attribute this difference to the more polar periplasmic region reinforcing water- and ion-channel electrostatic interactions relative to the rather hydrophobic cytoplasmic half-channel—in line with the fact that narrow hydrophilic confinements, such as gramicidin A, are better sustainers of ionic currents (32,34,65), than, e.g., hydrophobic carbon nanotubes (26,41). A drawback of the latter, in this respect, is that it offers poor solvation of the ion. For deprotonation of W−1 in the lumen-periplasmic vestibule boundary one finds (Sims. 13–15; see Fig. 2 c) that elimination of the M3/M7 backbone charges increases hydroxide mobility from 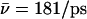 (Sim. 13) to
(Sim. 13) to 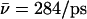 (Sim. 14), noting here that the M7 helix protrudes into the periplasmic vestibule (see Fig. 2). Additional elimination of the N68/N203 side-chain charges (Sim. 15) leads to a mobility close to that of the native channel (Sim. 13). In short, hydroxide mobility is higher in the periplasmic half-channel, while influence on mobility from the M3/M7 macrodipoles and from the NPA motifs is largely dependent on the initial position of the ion.
(Sim. 14), noting here that the M7 helix protrudes into the periplasmic vestibule (see Fig. 2). Additional elimination of the N68/N203 side-chain charges (Sim. 15) leads to a mobility close to that of the native channel (Sim. 13). In short, hydroxide mobility is higher in the periplasmic half-channel, while influence on mobility from the M3/M7 macrodipoles and from the NPA motifs is largely dependent on the initial position of the ion.
Luminal proton mobility is also largest for the native system when protonated in the periplasmic half-channel (see Sim. 5 versus Sim. 8). Relative to hydroxide mobility, that of the luminal proton is more unidirectional, since the proton is always outward-expelled and therefore, less “willing” to undergo transfers toward the channel center, which lowers 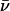 overall. For the proton, initially in the periplasmic lumen (Sim. 5),
overall. For the proton, initially in the periplasmic lumen (Sim. 5), 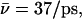 although elimination of N68/N203 side-chain charges increases this mobility,
although elimination of N68/N203 side-chain charges increases this mobility, 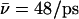 (Sim. 6), implying that GlpF accommodates the proton better in the absence of interactions with these side chains.
(Sim. 6), implying that GlpF accommodates the proton better in the absence of interactions with these side chains.
A residence correlation function c(t) = 〈θ(t, t′)〉t′, where t = t′Nδt (δ, N are defined in Methods) and θ(…) = 1 if z(t) = z(t′) and zero otherwise, was computed for short times (t ≪ 100 fs) for Sim. 1 and Sim. 5. We discretized the ionic coordinate z(t) using a bin-width of 1 Å. The c(t) (data not shown) suggests that the ionic residence correlation time is ∼50–100 fs. Long-term correlations cannot be accurately estimated, since both ionic species become immobilized (discussed below), implying that c(t→∞) approaches, asymptotically, a non-zero value. Taken together with the above mobility results, c(t) manifests that luminal translocation of both ionic species is a subpicosecond process in accord with related considerations (22,26,32,34,41,65). Consequently, even transient association of two disrupted wire fragments could allow for significant ionic current on a picosecond timescale, e.g., in GlpF, disruption of the H-bonded neutral wire occurs ∼30% of the time (18,22,24). These timescale considerations therefore conflict with proposed disruption of the neutral wire as a general mechanism of proton rejection in AQPs (9,11,25).
Intraluminal hydroxide migration
Hydroxide migration within the periplasmic half-channel lumen (Sim. 1) is represented in terms of simulation snapshots and the corresponding trajectory in Fig. 7. The position of initial deprotonation just below R206 is explicitly depicted by the first (t = 0) snapshot. As seen from Fig. 7, a and b, the hydroxide ion migrates as a proton hole, i.e., via proton relays from the neighboring water (see also Fig. 1 d). The hydroxide ion undergoes  net translocations (see Table 1) and becomes trapped at the NPA motifs in <1 ps, indicative of strong ion-channel attraction at the NPA region. This observation agrees with previous considerations regarding negative ions in the channel lumen (22,25,27,37). Consequently, the frequency histogram in Fig. 7 b depicts a monotonic increasing ion population toward the NPA region. Water molecules within the periplasmic half-channel exhibit sufficient orientational freedom to sustain a Grotthuss hop-and-turn translocation (a proton hole-type diffusion), i.e., the bipolar strands of the native (neutral) channel are not preserved in the deprotonated wire. As illustrated by all t ≠ 0 snapshots in Fig. 7 a, the waters anti-align sequentially upon the proton relay with respect to the native (neutral channel) bipolar orientation. Accordingly, intrawire and ion-channel center electrostatic interactions suffice for surmounting energetic barriers associated with water reorientation during hydroxide migration. Luminal deprotonation in the cytoplasmic half-channel, i.e., below the NPA motifs (Sim. 4, Table 1), leads in a similar fashion to
net translocations (see Table 1) and becomes trapped at the NPA motifs in <1 ps, indicative of strong ion-channel attraction at the NPA region. This observation agrees with previous considerations regarding negative ions in the channel lumen (22,25,27,37). Consequently, the frequency histogram in Fig. 7 b depicts a monotonic increasing ion population toward the NPA region. Water molecules within the periplasmic half-channel exhibit sufficient orientational freedom to sustain a Grotthuss hop-and-turn translocation (a proton hole-type diffusion), i.e., the bipolar strands of the native (neutral) channel are not preserved in the deprotonated wire. As illustrated by all t ≠ 0 snapshots in Fig. 7 a, the waters anti-align sequentially upon the proton relay with respect to the native (neutral channel) bipolar orientation. Accordingly, intrawire and ion-channel center electrostatic interactions suffice for surmounting energetic barriers associated with water reorientation during hydroxide migration. Luminal deprotonation in the cytoplasmic half-channel, i.e., below the NPA motifs (Sim. 4, Table 1), leads in a similar fashion to 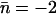 net translocations (Table 1) and within 120 fs again to immobilization and fourfold OH− coordination.
net translocations (Table 1) and within 120 fs again to immobilization and fourfold OH− coordination.
FIGURE 7.

(a) Simulation snapshots of the key part of GlpF taken from ion transfer simulation where the wire initially was deprotonated in the periplasmic lumen. For clarity only QM water molecules within the single file are displayed. The R206/N68/N203 side chains with their polar Hs, the exposed carbonyl groups of residues 65–66 and 199–201, are shown in licorice and CPK representations, respectively. Nonexposed carbonyl groups of residues L67 and M202, engaged in two hydrogen bonds to N203 and N68, respectively, are displayed along with the hydrogen bonds. The M3 and M7 half-helices are shown as solid cylinders/purple ribbons. (b) OH− trajectory and relative distributions of OH− along the channel axis. Trajectories of the water molecules explicitly involved in the ion transfer appear as gray lines for reference along with the center-of-mass trajectory of the N68/N203 side chains. (c) Schematic representation of a fourfold coordinated hydroxide ion trapped at the channel center between the N68/N203 side chains of the NPA motifs. (d) Corresponding electron density map calculated from the simulations and represented by the electron localizing function at contour level η = 0.9 (66).
The local structure around the centrally trapped hydroxide ion is schematically illustrated in Fig. 7 c and in more detail by means of the electron localizing function (66) in Fig. 7 d. Together this illustrates that N68/N203 and the two neighboring single-file water molecules coordinate the hydroxide ion, i.e., the δN–H bonds of N68/N203 and one O–H bond of each neighboring-file water molecule point toward the centroid electron density on the oxygen atom of the hydroxide ion. We recognize that this coordination implies that the two lower (cytoplasmic) water molecules in Fig. 7 c dipole-invert relative to their native orientation (see Fig. 2 c). A solvated transferring hydroxide ion is either four- or threefold coordinated (54,55), which underscores the particular OH− stability at the trapping position.
During hydroxide transfer, subtle concurrent changes in the dipole moment (μ) of the deprotonated wires occur (data not shown). However, these changes are within the RMS fluctuations of μ overall. Since GlpF is not ion-conducting, in reality we ascribe these small changes, in part, to the idea that the inherent electrostatic tuning of the lumen dictates a rigid charge distribution of the wire merely affected by the sequential water reorientation. In contrast, changes in μx,y,z are mutually strongly correlated. From the time-series of μx,y,z we obtain the correlation coefficients C(μx, μy) = 0.73, C(μx, μz) = 0.56, and C(μy, μz) = 0.68 (see Methods).
Influence from the NPA motifs on intraluminal hydroxide migration
Given the luminal hydroxide immobilization (Fig. 7), an obvious question is whether the N68/N203 side chains alone are responsible for this trapping. To address this question, we performed a simulation (Sim. 2, Table 1) where the ion initially was trapped between the NPA motifs, restarting from the last configuration of Sim. 1 and eliminating the charges of the N68/N203 side chains. To avoid possible structural artifacts caused by absence of charges we fixed the MM fragment. To gauge whether the potential trap could be thermally escaped, we performed the first part (343 fs) of this simulation at T = 150 K, after which the temperature was gradually elevated to reach T = 500 K. However, no net ion translocations resulted (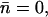 Table 1). Within the first time-interval, one net hydroxide transfer occurred toward the periplasm (np = 4, nc = 3), while during the thermally activated dynamics one net hydroxide transfer to the NPA motifs occurred (nc = 4, np = 3). Despite elimination of the N68/N203 charges, the hydroxide ion essentially remains immobilized, suggesting that other residues contribute in this trapping in accord with classical MC and MD results for transfer of hydroxide and halide ions within AQP lumens (25,37). Our above fixation of the MM fragment implies reduced channel-lining fluctuation, which tends to lower ion mobility and accordingly the probability for inducing and observing ion migration; e.g., for proton transfer in gramicidin A, charge translocation and channel-lining fluctuations are coupled (32,34). Therefore we performed an analogous simulation in which the MM fragment was free to move. Before an artificial breakdown of the crucial intrachannel H-bonding pattern at the NPA motifs (see Fig. 7 a), several ion transfers across the NPA motifs occurred. Note that beyond ∼0.55 ps, the simulation became unstable due to rotation of the N203 side chain. Upon rotation, the side chains moved apart by ∼2.5 Å and breakage of the H-bonded configuration of the NPA motifs resulted. Thereafter, OH− essentially freely crossed the NPA region, i.e., within the subsequent (after breakage) 0.25 ps. Ultimately, OH− escaped the channel through the cytoplasmic outlet. These findings caution against far-reaching conclusions based on simulations of rigid channels in general, but more importantly, confirm here that the N68/N203 side chains are indeed involved in trapping of the hydroxide ion.
Table 1). Within the first time-interval, one net hydroxide transfer occurred toward the periplasm (np = 4, nc = 3), while during the thermally activated dynamics one net hydroxide transfer to the NPA motifs occurred (nc = 4, np = 3). Despite elimination of the N68/N203 charges, the hydroxide ion essentially remains immobilized, suggesting that other residues contribute in this trapping in accord with classical MC and MD results for transfer of hydroxide and halide ions within AQP lumens (25,37). Our above fixation of the MM fragment implies reduced channel-lining fluctuation, which tends to lower ion mobility and accordingly the probability for inducing and observing ion migration; e.g., for proton transfer in gramicidin A, charge translocation and channel-lining fluctuations are coupled (32,34). Therefore we performed an analogous simulation in which the MM fragment was free to move. Before an artificial breakdown of the crucial intrachannel H-bonding pattern at the NPA motifs (see Fig. 7 a), several ion transfers across the NPA motifs occurred. Note that beyond ∼0.55 ps, the simulation became unstable due to rotation of the N203 side chain. Upon rotation, the side chains moved apart by ∼2.5 Å and breakage of the H-bonded configuration of the NPA motifs resulted. Thereafter, OH− essentially freely crossed the NPA region, i.e., within the subsequent (after breakage) 0.25 ps. Ultimately, OH− escaped the channel through the cytoplasmic outlet. These findings caution against far-reaching conclusions based on simulations of rigid channels in general, but more importantly, confirm here that the N68/N203 side chains are indeed involved in trapping of the hydroxide ion.
Influence from R206 on intraluminal hydroxide migration
To inspect R206's influence on hydroxide migration and trapping, we also conducted a simulation (Sim. 3, Table 1) in which electrostatic charges of the R206/N63/N203 side chains were simultaneously eliminated, starting again from the last (ion-immobilized) configuration of Sim. 1. The hydroxide ion again remains immobilized (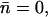 Table 1) within the entire simulation (0.5 ps). In the immobilized state we find that the hydroxide ion localizes toward the cytoplasmic side of the NPA motifs, i.e., N203 relative to the symmetric position of immobilization between the N68/N203 side chains found in Sim. 1 and Sim. 2. We attribute this shift in position to be the lack of electrostatic attraction toward the periplasm from the positively charged R206 guanidinium group. This lack of interactions merely affects ion mobility relative to Sim. 2, where only the N63/N203 side chains were eliminated;
Table 1) within the entire simulation (0.5 ps). In the immobilized state we find that the hydroxide ion localizes toward the cytoplasmic side of the NPA motifs, i.e., N203 relative to the symmetric position of immobilization between the N68/N203 side chains found in Sim. 1 and Sim. 2. We attribute this shift in position to be the lack of electrostatic attraction toward the periplasm from the positively charged R206 guanidinium group. This lack of interactions merely affects ion mobility relative to Sim. 2, where only the N63/N203 side chains were eliminated; 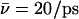 (Sim. 2, 343 ps of nonactivated dynamics) versus
(Sim. 2, 343 ps of nonactivated dynamics) versus 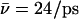 (Sim. 3).
(Sim. 3).
Extraluminal hydroxide migration
So far our results demonstrate that the hydroxide ion localizes at the channel center, thus blocking the channel once it reaches one of the two outward pointing termini of the bipolar wire. This does not happen at physiological conditions (nonalkaline pH). Accordingly, a mechanism preventing hydroxide ions from reaching the wire termini exists; e.g., hydroxide exclusion might be determined by two electrostatic barriers originating from the negative (vestibule) poles of the electrostatic M3/M7 macrodipoles (37). Desolvation energy penalties could also be a key factor preventing ions, regardless of charge, from penetrating into the channel lumen (36,39).
To get insight into these aspects, we performed simulations in which water molecules located with increasing distance from the channel center (W1,W0,W−1; see Fig. 2 c) were initially deprotonated. For deprotonation of W−1 located in the inner vestibule it was necessary to use an extended QM region (see Methods). Electrostatic influence from the M3/M7 macrodipoles was examined by switching off backbone atom charges as in Tajkhorshid et al. (18).
Results of the calculations are summarized in Table 1 and hydroxide trajectories for W1 and W−1 are displayed in Fig. 8. Deprotonation of W0 led to results similar to those of Sim. 1 and will not be discussed further. When W1 is deprotonated in the native channel, the hydroxide ion migrated to the channel lumen, despite frequent transfers between W0 and W1 (Fig. 8 a). Hydroxide-R206 electrostatic interactions do not trap the ion at the SF region; instead, the ion transfers to the inner lumen (W3–W4) in <0.5 ps. According to Fig. 8, b and c, suppression of the M3/M7 macrodipoles (Sim. 10) and, in addition, of the N68/N203 side chains (Sim. 11), does not change this qualitatively. Quantitatively (Table 1), the M3/M7 macrodipoles influence hydroxide mobility, i.e., 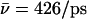 in the native channel (Sim. 9), whereas
in the native channel (Sim. 9), whereas 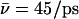 (Sim. 10) indicates suppression of the macrodipoles, since hydroxide attraction to the NPA motifs in the latter case dominate. Accordingly, mobility increases (
(Sim. 10) indicates suppression of the macrodipoles, since hydroxide attraction to the NPA motifs in the latter case dominate. Accordingly, mobility increases (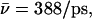 Sim. 11) if, in addition, the N68/N203 charges are eliminated. This scenario changes when W−1, located at the inner vestibule, is deprotonated (Fig. 2 c and Fig. 8, d–f). For the native channel (Sim. 13), the ion enters the lumen in <1 ps (Fig. 8 d;
Sim. 11) if, in addition, the N68/N203 charges are eliminated. This scenario changes when W−1, located at the inner vestibule, is deprotonated (Fig. 2 c and Fig. 8, d–f). For the native channel (Sim. 13), the ion enters the lumen in <1 ps (Fig. 8 d;  Table 1). Turning off the macrodipoles (Sim. 14) induces frequent hydroxide transfers along the W−1–W0–W1 fragment before escape into the vestibule (Fig. 8 e; n = −3, Table 1). Additional elimination of the N68/N203 side-chain charges (Sim. 15) implies that no intraluminal transfers are observed, and only transition into the inner vestibule region occur, possibly due to the short simulation time (Fig. 8 f). Clearly, the macrodipoles influence ion dynamics. Electrostatic interactions between the solvated ion and these dipoles facilitate ion penetration into the lumen, whereas the ion is readily repelled from the lumen suppressing the dipoles. This result might seem in conflict with the proposal that the macrodipoles have a role in preventing hydroxide ions from penetrating into the channel (37). However, the M7 helix protrudes far into the periplasm, and therefore is also distant from the position of W−1 (or R206). Consequently, as W−1 resides relatively far from the negative M7 pole, dipolar (ion-helix), interactions will, in the vicinity of W−1, reinforce lumen penetration instead of ion blocking. Hence, only hydroxide locations at or above the negative M7 pole, or, equivalently, below the negative M3 pole, would favor dipolar interactions that repel the negative ion. (A simulation starting with the ion in the bulk, i.e., at or near the negative M7 pole, is out of our current computational capabilities, since it requires a very large QM region to include a sufficient quantity of capping QM water molecules.) Consequently, we suggest that exclusion of negative ions must operate before the ion reaches the inner vestibule and relies partly on repulsion from the negative M3/M7 poles in accord with de Groot et al. (37). Moreover, our observed vestibule
Table 1). Turning off the macrodipoles (Sim. 14) induces frequent hydroxide transfers along the W−1–W0–W1 fragment before escape into the vestibule (Fig. 8 e; n = −3, Table 1). Additional elimination of the N68/N203 side-chain charges (Sim. 15) implies that no intraluminal transfers are observed, and only transition into the inner vestibule region occur, possibly due to the short simulation time (Fig. 8 f). Clearly, the macrodipoles influence ion dynamics. Electrostatic interactions between the solvated ion and these dipoles facilitate ion penetration into the lumen, whereas the ion is readily repelled from the lumen suppressing the dipoles. This result might seem in conflict with the proposal that the macrodipoles have a role in preventing hydroxide ions from penetrating into the channel (37). However, the M7 helix protrudes far into the periplasm, and therefore is also distant from the position of W−1 (or R206). Consequently, as W−1 resides relatively far from the negative M7 pole, dipolar (ion-helix), interactions will, in the vicinity of W−1, reinforce lumen penetration instead of ion blocking. Hence, only hydroxide locations at or above the negative M7 pole, or, equivalently, below the negative M3 pole, would favor dipolar interactions that repel the negative ion. (A simulation starting with the ion in the bulk, i.e., at or near the negative M7 pole, is out of our current computational capabilities, since it requires a very large QM region to include a sufficient quantity of capping QM water molecules.) Consequently, we suggest that exclusion of negative ions must operate before the ion reaches the inner vestibule and relies partly on repulsion from the negative M3/M7 poles in accord with de Groot et al. (37). Moreover, our observed vestibule lumen transition of the hydroxide ion in Fig. 8 d suggests that desolvation penalties at the vestibule-lumen boundary are of less importance, which is also claimed for proton transfer across this boundary (28). Given the strong negative ion-channel attraction, one expects that AQPs might sustain a weak OH− current at strong alkaline conditions.
lumen transition of the hydroxide ion in Fig. 8 d suggests that desolvation penalties at the vestibule-lumen boundary are of less importance, which is also claimed for proton transfer across this boundary (28). Given the strong negative ion-channel attraction, one expects that AQPs might sustain a weak OH− current at strong alkaline conditions.
FIGURE 8.

Hydroxide trajectories. Initial deprotonations were at water W1 (a–c) and at water W−1 (d–f); see Fig. 2 c. Trajectories of the fully interacting system are shown in a and d, trajectories where electrostatic interactions with backbone charges of the M3/M7 helices eliminated are presented in b and e, and trajectories resulting from additional elimination of the N68/N203 side-chain charges appear in c and f. The broken line represents the mean position of the R206:Nη pointing toward the periplasm. This position differs from a to c, and from d to f, solely due to different QM-box sizes.
Intraluminal proton migration
Our results from luminal proton transfer within the periplasmic half-channel are presented in Fig. 9. In this simulation (Sim. 5) the proton is expelled through the periplasmic outlet by means of the Grotthuss mechanism (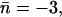 Table 1). From the snapshots in Fig. 9 a, one recognizes that the transferring proton predominantly is solvated as a Zundel complex (H5O2)+. Proton mobility in the R206 region is pronounced, as seen from the proton and R206 (guanidinium center-of-mass) trajectories in Fig. 9 b. The proton mobility is directed toward the periplasm, implying that the proton is more strongly repelled by the NPA region than by R206, in accord with the barrier against proton transport being the highest in that region (27,36–38)—and, again, agreeing with the relative ionic distributions in Fig. 9 b.
Table 1). From the snapshots in Fig. 9 a, one recognizes that the transferring proton predominantly is solvated as a Zundel complex (H5O2)+. Proton mobility in the R206 region is pronounced, as seen from the proton and R206 (guanidinium center-of-mass) trajectories in Fig. 9 b. The proton mobility is directed toward the periplasm, implying that the proton is more strongly repelled by the NPA region than by R206, in accord with the barrier against proton transport being the highest in that region (27,36–38)—and, again, agreeing with the relative ionic distributions in Fig. 9 b.
FIGURE 9.
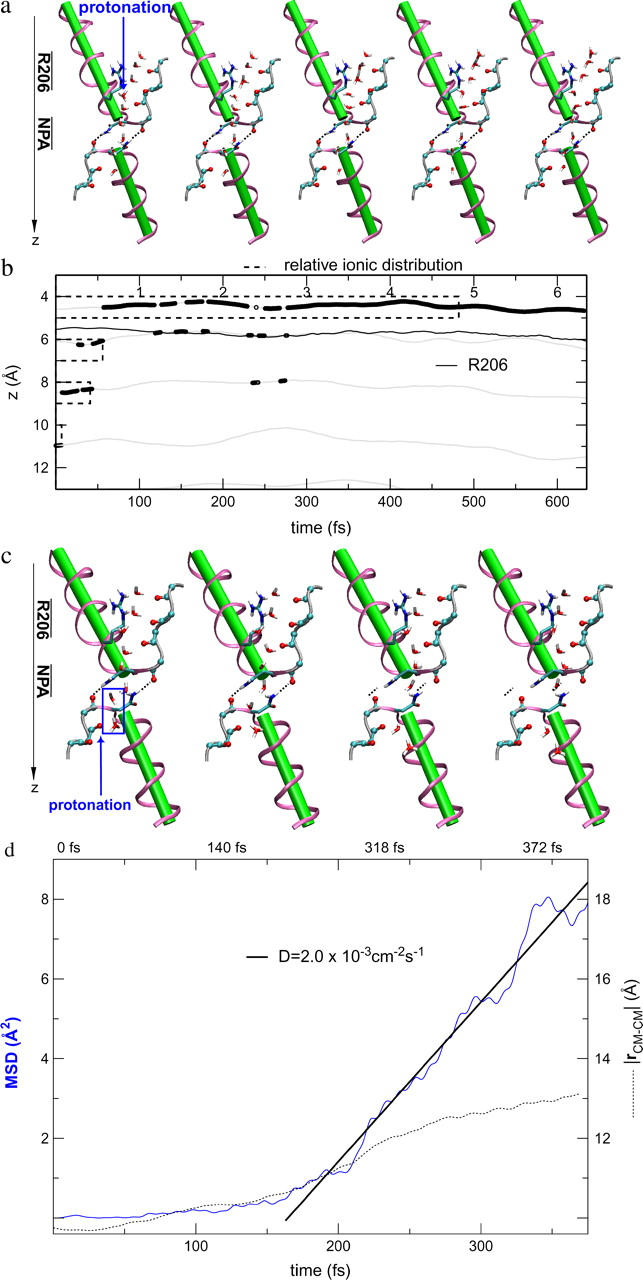
(a) Simulation snapshots taken from ion transfer simulation where the wire initially was protonated in the periplasmic lumen. (b) Complete H+ trajectory with the center-of-mass trajectory of the R206 side chain included for reference. See Fig. 7 legend for details. (c) Simulation snapshots taken from ion transfer simulation where the wire initially was protonated in the cytoplasmic lumen leading to breakage of the single-file below the cytoplasmic NPA motif. (d) Structural diffusion of the solvated proton (blue box of the wire in the t = 0 snapshot measured by 1), the center-of-mass distance between the two wire fragments (|rCM–CM|, dotted line); and 2), the MSD of the (H5O2)+ (Zundel) complex (blue line). An approximate diffusion constant D ≅ 2 × 10−3 cm2 s−1 was derived from the best fit to the MSD curve after its quasilinear onset.
As the proton moves toward the periplasmic vestibule, waters need—and are able—to reorient sequentially, which implies that at the end of this simulation, all waters in the periplasmic half-channel in Fig. 9 a are dipole-inverted relative to their native orientation (see also Fig. 1 c), and that a monopolar, proton-transferring wire, as in Fig. 1, b and c, is formed. Water reorientation is promoted by destabilizing the H-bonding network owing to protonation. Destabilization correlates with peak-shifts to larger |rO–H| in the radial distribution function for the protonated wire relative to the neutral wire in Fig. 6. Subtle changes in the dipole moment μ of the protonated wire also occur, albeit that changes in μ, concurrent with sporadic proton transfer, fall within RMS fluctuations of μ. Mutual changes in μx,y,z correlate with similar strength to that found for hydroxide migration: i.e., correlation coefficients C(μx, μy) = 0.63, C(μx, μz) = 0.60, and C(μy, μz) = 0.74 are obtained from the μx,y,z time-series.
Structural diffusion
We also investigated the effect of initially protonating the wire in the cytoplasmic half-channel (Sim. 8; results are shown in Fig. 9, c and d.) From simulation snapshots and results derived from the trajectory (Fig. 9, c and d, respectively), we find that the proton is expelled from the lumen to the inner cytoplasmic vestibule. Interestingly, the H-bonded, protonated wire starts to break below the cytoplasmic NPA motif at t ≅ 150 fs. Since  (Table 1), proton expelling does not occur by means of the Grotthuss mechanism—in contrast to expelling from the other half-channel lumen, discussed above. Rather, the proton co-diffuses with the two neighboring waters as (H5O2)+. (The diffusion is depicted by the snapshots in Fig. 9 c.) Breakage of the protonated wire causes a ∼3 Å increase in distance between the center-of-mass of the two wire fragments (|rCM–CM|) over ∼150 fs. A fairly linear mean-square-displacement (MSD) of the (H5O2)+ center-of-mass sets “on” after a 150–200-fs lag period, which, accordingly, is the approximate time required to break the wire. The mean-square displacement implies a somewhat high diffusion constant of ∼2.0 × 10−3 cm2 s−1, which is two orders-of-magnitude larger than the channel's water diffusion coefficient of ∼3.0 × 10−4 cm2 s−1 to ∼4.0 × 10−4 cm2 s−1 (18,22,24), presumably owing to downhill diffusion on an underlying potential originating from the electrostatic repulsion from the NPA region (22,25,27,36–38).
(Table 1), proton expelling does not occur by means of the Grotthuss mechanism—in contrast to expelling from the other half-channel lumen, discussed above. Rather, the proton co-diffuses with the two neighboring waters as (H5O2)+. (The diffusion is depicted by the snapshots in Fig. 9 c.) Breakage of the protonated wire causes a ∼3 Å increase in distance between the center-of-mass of the two wire fragments (|rCM–CM|) over ∼150 fs. A fairly linear mean-square-displacement (MSD) of the (H5O2)+ center-of-mass sets “on” after a 150–200-fs lag period, which, accordingly, is the approximate time required to break the wire. The mean-square displacement implies a somewhat high diffusion constant of ∼2.0 × 10−3 cm2 s−1, which is two orders-of-magnitude larger than the channel's water diffusion coefficient of ∼3.0 × 10−4 cm2 s−1 to ∼4.0 × 10−4 cm2 s−1 (18,22,24), presumably owing to downhill diffusion on an underlying potential originating from the electrostatic repulsion from the NPA region (22,25,27,36–38).
Obviously, structural diffusion is a slower mechanism of ion rejection compared to the Grotthuss transfer and our short trajectory implies that computing a diffusion coefficient is associated with large uncertainty. Although likely, the actual significance of the breakage of the protonated wire in the vicinity of the cytoplasmic NPA motif therefore remains questionable. It could (in part) be ascribed to the fact that the water molecule at the position of breakage cannot H-bond to the carbonyl group of the adjacent channel-lining residue (L67)—which serves to facilitate fast dipole inversion in the neutral channel and ensure structural preservation by means of an appropriate H-bonded configuration of the N68/N203 residues of the NPA motifs (8,13,14,18,22). M202 is the counterpart of L67; the L67:O⋯Hδ:203, M202:O⋯Hδ:68 H-bonds can be discerned from Fig. 9 c. This implies that the H-bonded network of the protonated wire at this position is too weak to persist against strong ion-channel electrostatic repulsion originating from the NPA region. For comparison, classical MD simulations lead to a disruption frequency of the neutral wire of 30% at this position, coinciding with the overall maximum of H-bond disruption for GlpF (18,22).
Influence from the NPA motifs and R206 on intraluminal proton migration
To determine whether ion-channel electrostatic interactions preventing proton conduction across the NPA motifs could be suppressed, we carried out two simulations of proton transfer within the periplasmic lumen, first eliminating charges of the N63/N203 side chain (Sim. 6) and then eliminating charges of the R206 guanidinium group (Sim. 7, see Table 1). The excess proton was introduced in the lumen of the periplasmic half-channel. In Sim. 6, the proton readily crossed the R206/SF region several times, whereas no migration across the NPA region occurred. Relative to the native channel simulation (Sim. 5), the proton mobility was merely affected (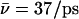 in Sim. 5 vs.
in Sim. 5 vs. 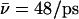 in Sim. 6). The water molecules again reorient upon charge relay, and the proton is expelled by the channel to the periplasmic vestibule, following the Grotthuss mechanism. The same was found for Sim. 7; i.e., additional elimination of R206 side-chain charges had no effect. After manually reorienting the first and second water molecules (W0 and W1 in Fig. 2 c) such that they initially anti-aligned with respect to their native orientation (see Fig. 2), we resumed the simulation. The imposed reorientation was intended to serve to obstruct the second water molecule from acting as a proton acceptor upon proton relay from the water molecule below. However, the obstructing water molecule did reorient to take its native orientation, enabling proton acceptance from the neighboring water and, accordingly, proton expelling. Hence, in GlpF, electrostatic interactions governing proton rejection are so robust that the (artificial) barriers against initial dipolar reorientation are easily surmounted in a protonated wire. Only by fixing the second, initially anti-aligned water molecule, did we find, upon resuming the simulation, that the proton indeed can be forced to transfer toward and across the NPA region (
in Sim. 6). The water molecules again reorient upon charge relay, and the proton is expelled by the channel to the periplasmic vestibule, following the Grotthuss mechanism. The same was found for Sim. 7; i.e., additional elimination of R206 side-chain charges had no effect. After manually reorienting the first and second water molecules (W0 and W1 in Fig. 2 c) such that they initially anti-aligned with respect to their native orientation (see Fig. 2), we resumed the simulation. The imposed reorientation was intended to serve to obstruct the second water molecule from acting as a proton acceptor upon proton relay from the water molecule below. However, the obstructing water molecule did reorient to take its native orientation, enabling proton acceptance from the neighboring water and, accordingly, proton expelling. Hence, in GlpF, electrostatic interactions governing proton rejection are so robust that the (artificial) barriers against initial dipolar reorientation are easily surmounted in a protonated wire. Only by fixing the second, initially anti-aligned water molecule, did we find, upon resuming the simulation, that the proton indeed can be forced to transfer toward and across the NPA region (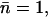 Sim. 7c, Table 1). The observed difficulties for the proton, in overcoming major electrostatic interactions preventing proton transfer across the NPA region, agree with related studies (25,27,28,36–39).
Sim. 7c, Table 1). The observed difficulties for the proton, in overcoming major electrostatic interactions preventing proton transfer across the NPA region, agree with related studies (25,27,28,36–39).
CONCLUSION
We have reported here, for the first time to our knowledge, ab initio MD simulations of an AQP water channel (E. coli GlpF) that fully include the electronic structure of the water wire featured in the channel. Along this level of description we also model hypothetical Grotthuss relays of protons and hydroxide ions (proton holes), along charged wires inside and outside the channel lumen, permitting us to follow details of the charge transfer processes.
We find that GlpF features two contiguous bipolarized strands of eight water molecules in single file, in agreement with classical MD simulations (11,18,22,24) and with x-ray data—taking into account that thermal differences rationalize discrepancies in water occupancy (18). Such a network is unable to sustain the Grotthuss relay of protons as well as of hydroxide ions. In the neutral wire, water molecules exhibit strong local polarization effects, reflecting, in part, their location inside the channel.
In qualitative agreement with related classical MD studies of hydroxide diffusion (37), our simulations of intraluminal hydroxide ion transfer demonstrated that, independent of deprotonation site, the hydroxide ion is immobilized at the NPA motifs, adopting here a particularly stable fourfold coordination as borne out in the electron density of the water wire. Eliminating electrostatic interactions with the N63/N203 side chains and with the R206 side chain merely permits the hydroxide ion to escape its trap at short timescales. This also holds the temperature is elevated. We find that hydroxide transfer occurs by propagation of a proton hole, i.e., by means of a hop-and-turn Grotthuss mechanism parallel to proton transfer in water wires (30–32,65). The Grotthuss mechanism implies sequential water reorientation, a feature we find promoted by charging the wire.
Our results clearly suggest that electrostatic attractions driving the hydroxide ion toward the channel center also involve contribution from the conserved α-helical M3/M7 macrodipoles. The M3/M7 influence on hydroxide migration is clearly demonstrated by deprotonating a water molecule adjacent to the M7 helix in the inner periplasmic vestibule. The M7 macrodipole readily facilitates hydroxide penetration into the lumen, since absence of the M7 dipole permits the ion to migrate to the outer periplasmic vestibule, rendering desolvation effects at the vestibule-lumen boundary secondary in this respect. The latter finding also applies to proton blocking according to recent results (28). Prevention of hydroxide ion transport in AQPs must therefore predominantly rely on ion rejection in the vestibule regions adjacent to the negative poles of the M3/M7 helices in accord with de Groot et al. (37).
The dominant barrier to proton blocking in AQPs has been claimed (22) and demonstrated (27,36–38) to lie in the electrostatic interior of the water pore, or more precisely, in the channel center where the largest barrier is met. This is confirmed by our ab initio QM/MM simulations of water wires carrying an excess proton, demonstrating that the positive charge (regardless of position of initial protonation) consistently becomes expelled from the channel center while translocating in the periplasm, according to the hop-and-turn Grotthuss mechanism (30–32). The excess proton readily crosses the region of the positively charged R206, regardless of whether electrostatic interactions with this residue are retained in the simulation or not. Therefore R206 does not individually act as a proton filter. Its absence would, however, eventually cause luminal proton attraction since mutual cancellation of electrostatic contributions from R206 and from negatively charged residues in its vicinity leads to neutrality overall (22). Protonation in the cytoplasmic lumen led to breakage of the wire into two fragments. Wire breakage at this point is likely, since water-channel interactions are modest, whereas electrostatic repulsion from the NPA region in contrast is maximal. Albeit slower than Grotthuss transfer, the proton consequently diffuses out of the channel as a Zundel ((H5O2)+) complex.
Acknowledgments
This work was supported by grants No. 2001SGR-00044 (Generalitat de Catalunya, Spain) and No. BQU2001-04587-CO-02 (Ministry of Science and Technology, Spain, MCYT) and via grants given to the MEMPHYS-Center for Biomembrane Physics, University of Southern Denmark, and to the Quantum Protein Center, Technical University of Denmark from the Danish National Research Foundation. The computer resources were provided by the CEPBA-IBM Research Institute of Barcelona. C.R. thanks the Ramon y Cajal program of the MCYT and the ICREA Foundation for financial support. M.Ø.J. acknowledges financial support from the ACCESS Mobility Program, Center for Parallelism of Barcelona (CEPBA, Spain).
References
- 1.Preston, G. M., P. Piazza-Carroll, W. B. Guggino, and P. Agre. 1992. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 water channel. Science. 256:385–387. [DOI] [PubMed] [Google Scholar]
- 2.Agre, P. 1998. The Aquaporins, blueprints for cellular plumbing systems. J. Biol. Chem. 273:14659–14662. [DOI] [PubMed] [Google Scholar]
- 3.Borgnia, M., S. Nielsen, A. Engel, and P. Agre. 1999. Cellular and molecular biology of the Aquaporin water channels. Annu. Rev. Biochem. 68:425–458. [DOI] [PubMed] [Google Scholar]
- 4.Yasui, M., T. H. Kwon, M. A. Knepper, S. Nielsen, and P. Agre. 1999. Aquaporin-6: an intracellular vesicle water channel protein in renal epithelia. Proc. Natl. Acad. Sci. USA. 96:5808–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozono, D., M. Yasui, L. S. King, and P. Agre. 2002. Aquaporin water channels: atomic structure and molecular dynamics meet clinical medicine. J. Clin. Invest. 109:1395–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agre, P., and D. Kozono. 2003. Aquaporin water channels: molecular mechanisms for human diseases. FEBS Lett. 555:72–78. [DOI] [PubMed] [Google Scholar]
- 7.Murata, K., K. Mitsuoka, T. Hirai, T. Walz, P. Agre, J. B. Heymann, A. Engel, and Y. Fujiyoshi. 2000. Structural determinants of water permeation through Aquaporin-1. Nature. 407:599–605. [DOI] [PubMed] [Google Scholar]
- 8.Fu, D., A. Libson, L. J. W. Miercke, C. Weitzman, P. Nollert, J. Krucinski, and R. M. Stroud. 2000. Structure of a glycerol conducting channel and the basis for its selectivity. Science. 290:481–486. [DOI] [PubMed] [Google Scholar]
- 9.Ren, G., V. S. Reddy, A. Cheng, and A. K. Mitra. 2001. Visualization of a water-selective pore by electron crystallography in vitreous ice. Proc. Natl. Acad. Sci. USA. 98:1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sui, H., B.-G. Han, J. K. Lee, P. Wallan, and B. K. Jap. 2001. Structural basis of water-specific transport through the AQP1 water channel. Nature. 414:872–877. [DOI] [PubMed] [Google Scholar]
- 11.de Groot, B. L., and H. Grubmüller. 2001. Water permeation across biological membranes: mechanism and dynamics of Aquaporin-1 and GlpF. Science. 294:2353–2357. [DOI] [PubMed] [Google Scholar]
- 12.de Groot, B. L., A. Engel, and H. Grubmüller. 2001. A refined structure of human Aquaporin-1. FEBS Lett. 504:206–211. [DOI] [PubMed] [Google Scholar]
- 13.Zhu, F., E. Tajkhorshid, and K. Schulten. 2001. Molecular dynamics study of Aquaporin-1 water channel in a lipid bilayer. FEBS Lett. 504:212–218. [DOI] [PubMed] [Google Scholar]
- 14.Jensen, M. Ø., E. Tajkhorshid, and K. Schulten. 2001. The mechanism of glycerol conduction in aquaglyceroporins. Structure. 9:1083–1093. [DOI] [PubMed] [Google Scholar]
- 15.Savage, D. F., P. F. Egea, Y. Robles-Colmenares, J. D. O'Connell, and R. M. Stroud. 2003. Architecture and selectivity in Aquaporins: 2.5 Å resolution x-ray structure of Aquaporin Z. PLoS Biol. 1:334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harries, W. E. C., D. Akhavan, S. Miercke, L. J. W. Khademi, and R. M. Stroud. 2004. The channel architecture of Aquaporin 0 at a 2.2-Å resolution. Proc. Natl. Acad. Sci. USA. 101:14045–14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonen, T., P. Sliz, J. Kistler, Y. Cheng, and T. Walz. 2004. Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature. 429:193–196. [DOI] [PubMed] [Google Scholar]
- 18.Tajkhorshid, E., P. Nollert, M. Ø. Jensen, L. J. W. Miercke, J. O'Connel, R. M. Stroud, and K. Schulten. 2002. Global orientational tuning controls the selectivity of the AQP water channel family. Science. 296:525–529. [DOI] [PubMed] [Google Scholar]
- 19.Kong, Y., and J. Ma. 2001. Dynamics mechanisms of the membrane water channel Aquaporin-1 (AQP1). Proc. Natl. Acad. Sci. USA. 98:14345–14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu, F., E. Tajkhorshid, and K. Schulten. 2002. Pressure-induced water transport in membrane channels studied by molecular dynamics. Biophys. J. 83:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu, F., E. Tajkhorshid, and K. Schulten. 2004. Theory and simulation of water simulation in Aquaporin-1. Biophys. J. 86:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen, M. Ø., E. Tajkhorshid, and K. Schulten. 2003. Electrostatic control of permeation and selectivity in Aquaporin water channels. Biophys. J. 85:2884–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vidossich, P., M. Cascella, and P. Carloni. 2004. Dynamics and energetics of water permeation through the Aquaporin channel. Proteins Struct. Funct. Bioinform. 55:924–931. [DOI] [PubMed] [Google Scholar]
- 24.Jensen, M. Ø., and O. G. Mouritsen. 2004. Lipids do influence protein function—the hydrophobic matching hypothesis revisited. Biochim. Biophys. Acta. 1666:205–226. [DOI] [PubMed] [Google Scholar]
- 25.Miloshevsky, G. V., and P. C. Jordan. 2004. Water and ion permeation in bAQP1 and GlpF channels: a kinetic Monte Carlo study. Biophys. J. 87:3690–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu, F., and K. Schulten. 2003. Water and proton conduction through carbon nanotubes as models for biological channels. Biophys. J. 85:236–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakrabarti, N., E. Tajkhorshid, B. Roux, and R. Pomes. 2004. Molecular basis of proton blockage in Aquaporins. Structure. 12:65–74. [DOI] [PubMed] [Google Scholar]
- 28.Chakrabarti, N., B. Roux, and R. Pomes. 2004. Structural determinants of proton blockage in Aquaporins. J. Mol. Biol. 343:493–510. [DOI] [PubMed] [Google Scholar]
- 29.Nagle, J. F., and S. Tristram-Nagle. 1983. Hydrogen-bonded chain mechanism for proton conduction and proton pumping. J. Membr. Biol. 74:1–14. [DOI] [PubMed] [Google Scholar]
- 30.de Grotthuss, C. J. T. 1806. On the decomposition of water and their solution species using galvanic electricity. Ann. Chim. LVIII:54–74. [Google Scholar]
- 31.Agmon, N. 1995. The Grotthuss mechanism. Chem. Phys. Lett. 224:456–462. [Google Scholar]
- 32.Pomes, R., and B. Roux. 1996. Structure and dynamics of a proton wire: a theoretical study of H+ translocation along the single-file water chain in the gramicidin A channel. Biophys. J. 71:19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brewer, M. L., U. W. Schmitt, and G. A. Voth. 1999. The formation and dynamics of proton wires in channel environments. Biophys. J. 80:1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pomes, R., and B. Roux. 2002. Molecular mechanism of H+ conduction in the single-file water chain of the gramicidin channel. Biophys. J. 82:2304–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smondyrev, A. M., and G. A. Voth. 2002. Molecular dynamics simulation of proton transport through the Influenza-A virus M2 channel. Biophys. J. 85:1987–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burykin, A., and A. Warshel. 2003. What really prevents proton transport through Aquaporin? Charge self-energy versus proton wire proposals. Biophys. J. 85:3696–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Groot, B. L., T. Frigato, V. Helms, and H. Grubmuller. 2003. The mechanism of proton exclusion in the Aquaporin-1 water channel. J. Mol. Biol. 333:279–293. [DOI] [PubMed] [Google Scholar]
- 38.Ilan, B., E. Tajkhorshid, K. Schulten, and G. A. Voth. 2004. The mechanism of proton exclusion in Aquaporin channels. Proteins Struct. Funct. Bioinform. 55:223–228. [DOI] [PubMed] [Google Scholar]
- 39.Burykin, A., and A. Warshel. 2004. On the origin of the electrostatic barrier for proton transport in Aquaporin. FEBS Lett. 570:41–46. [DOI] [PubMed] [Google Scholar]
- 40.Fujiyoshi, Y., K. Mitsuoka, B. L. de Groot, A. Philippsen, H. Grubmller, P. Agre, and A. Engel. 2002. Structure and function of water channels. Curr. Opin. Struct. Biol. 12:509–515. [DOI] [PubMed] [Google Scholar]
- 41.Dellago, C., M. M. Naor, and G. Hummer. 2003. Proton transport through water-filled carbon nanotubes. Phys. Rev. Lett. 90:105902. [DOI] [PubMed] [Google Scholar]
- 42.Wu, Y., and G. A. Voth. 2003. A computer simulation study of the hydrated proton in a synthetic proton channel. Biophys. J. 85:864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu, K., D. Kozono, Y. Kato, P. Agre, A. Hazama, and M. Yasui. 2005. Conversion of Aquaporin-6 from an anion channel to a water-selective channel by a single amino acid substitution. Proc. Natl. Acad. Sci. USA. 102:2192–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Groot, B. L., and H. Grubmüller. 2005. Dynamics and energetics of water permeation and proton exclusion in Aquaporins. Curr. Opin. Struct. Biol. 15:176–183. [DOI] [PubMed] [Google Scholar]
- 45.Hünenberger, P. 2000. Optimal charge-shaping functions for the particle-particle-particle-mesh (P3M) method for computing electrostatic interactions in molecular simulations. J. Chem. Phys. 113:10464–10476. [Google Scholar]
- 46.Carloni, P., and U. Röthlisberger. 2001. Theoretical and Computational Chemistry. Elsevier, New York. 215.
- 47.Carloni, P., U. Röthlisberger, and M. Parrinello. 2002. The role and perspective of ab initio molecular dynamics in the study of biological systems. Acc. Chem. Res. 35:455–464. [DOI] [PubMed] [Google Scholar]
- 48.Magistrato, A., W. F. DeGrado, A. Laio, U. Röthlisberger, J. Van de Vondele, and M. L. Klein. 2003. Characterization of the dizinc analogue of the synthetic diiron protein DF1 using ab initio and hybrid quantum/classical molecular dynamics simulations. J. Phys. Chem. B. 107:4182–4188. [Google Scholar]
- 49.Rovira, C., B. Schulze, M. Eichinger, J. Evanseck, and M. Parrinello. 2001. Influence of the heme pocket conformation on the structure and vibrations of the Fe-CO bond in myoglobin: a QM/MM density functional study. Biophys. J. 81:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rousseau, R., U. W. Kleinschmidt, U. Schmitt, and D. Marx. 2004. Modeling protonated water networks in bacteriorhodopsin. Phys. Chem. Chem. Phys. 6:1848–1859. [Google Scholar]
- 51.Rousseau, R., U. W. Kleinschmidt, and D. Marx. 2004. Assigning protonation patterns in water networks in bacteriorhodopsin based on computed IR spectra. Angew. Chem. Intl. Ed. Eng. 43:4804–4807. [DOI] [PubMed] [Google Scholar]
- 52.Piana, S., D. Bucher, P. Carloni, and U. Röthlisberger. 2004. Reaction mechanism of HIV-1 protease by hybrid Car-Parrinello/classical MD simulations. J. Phys. Chem. B. 108:11139–11149. [Google Scholar]
- 53.Marx, D., M. E. Tuckerman, J. Hutter, and M. Parrinello. 1999. The nature of the hydrated excess proton in water. Nature. 397:601–604. [Google Scholar]
- 54.Tuckerman, M. E., D. Marx, and M. Parrinello. 2002. The nature and transport mechanism of hydrated hydroxide ions in aqueous solution. Nature. 417:925–929. [DOI] [PubMed] [Google Scholar]
- 55.Asthagiri, D., L. R. Pratt, J. D. Kress, and M. A. Gomez. 2005. Hydration and mobility of HO−(aq). Proc. Natl. Acad. Sci. USA. 101:7229–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laio, A., J. Van de Vondele, and U. J. Röthlisberger. 2001. A Hamiltonian electrostatic coupling scheme for hybrid Car-Parrinello molecular dynamics simulations. J. Chem. Phys. 116:6941–6947. [Google Scholar]
- 57.Car, R., and M. Parrinello. 1985. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 55:2471–2474. [DOI] [PubMed] [Google Scholar]
- 58.Marx, D., and J. Hutter. 2000. Ab initio molecular dynamics: theory and implementation. In Modern Methods and Algorithms of Quantum Chemistry. J. Grotendorst, editor. John von Neumann Institute for Computing, Julich, Germany. 301–409.
- 59.Pearlman, D. A., D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, S. Debolt, D. Ferguson, G. Seibel, and P. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91:1–41. [Google Scholar]
- 60.Perdew, J. P., K. Burke, and M. Ernzerhof. 1996. Generalized gradient approximation made simple. Phys. Rev. Lett. 77:3865–3868. [DOI] [PubMed] [Google Scholar]
- 61.Troullier, N., and J. L. Martins. 1991. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B. 43:1993–2006. [DOI] [PubMed] [Google Scholar]
- 62.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 63.Jorgensen, W. L., J. Chandrasekhar, J. D. Medura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential models for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 64.Allen, M. P., and D. J. Tildesley. 1988. Computer Simulation of Liquids. Clarendon, Oxford.
- 65.Pomes, R., and B. Roux. 1998. Free energy profiles for H+ conduction along hydrogen-bonded chains of water molecules. Biophys. J. 75:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Becke, A. D., and K. E. Edgecombe. 1990. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 92:5397–5403. [Google Scholar]