

Abstract
Structural rearrangements of the myosin upper-50 kD subdomain are thought to play a key role in coordinating actin binding with nucleotide hydrolysis during the myosin ATPase cycle. Such rearrangements could open and close the active site in opposition to the actin-binding cleft, helping explain the opposing affinities of myosin for actin and nucleotide. To directly examine conformational changes across the active site during the ATPase cycle we have genetically engineered a mutant of chicken smooth-muscle myosin, F344W motor domain essential light chain, which contains a single tryptophan (344W) located on a short loop between two alpha helixes that traverse the upper-50 kD subdomain in front of the active site. Fluorescence resonance energy transfer was examined between the 344W donor probe and 2′(3′)-O-(N-methylanthraniloyl) (mant)-nucleotide acceptor probes in the active site of this construct. The observed fluorescence resonance energy transfer efficiencies were 6.4% in the presence of mant ADP and 23.8% in the presence of mant ATP, corresponding to distances of 33.4 Å and 24.9 Å, respectively. Our results are consistent with structural rearrangements in which there is an 8.5-Å closure between the 344W residue and the mant moiety during the transition from the strongly (ADP) to weakly (ATP) actin-bound states of the myosin ATPase cycle.
INTRODUCTION
Muscle contraction is driven by cyclical interactions between the myosin II motor protein and filamentous actin. To fulfill its motor function myosin must hydrolyze nucleotide and then undergo a conformational change, the power stroke, to release the stored energy to create force and movement. Myosin coordinates its power stroke with binding to and releasing from actin, a process thought to be mediated by the occupancy of its active site. Thus the ATPase cycle acts as an internal clock timing myosin's rearrangements such that the energy released by hydrolysis is harnessed as directed motion. A simple version of the myosin ATPase cycle is shown below (Scheme 1),
 |
where A = actin, M = myosin, AM = the acto-myosin complex, and Pi = inorganic phosphate (1).
From the strongly actin-bound AM rigor complex, myosin binds tightly to ATP causing a decrease in its affinity for actin by >4 orders of magnitude, triggering actin release. After ATP hydrolysis, Pi release is thought to result in a conformational change in which myosin increases its affinity for actin from micromolar to nanomolar and initiates its power stroke. Myosin remains strongly bound to actin after ADP release, until a new molecule of ATP binds at the active site. To avoid confusion, we will refer to myosin as being strongly bound to actin but tightly bound to ATP throughout the text. Myosin has a high affinity for actin in the apo and ADP states (even in the absence of actin) and thus we will refer to these as strong-binding states throughout the text.
A wealth of myosin II crystal structures, solved in the absence of nucleotide and in the presence of nucleotides and nucleotide analogs (2–7), have allowed assignment of force production, actin binding, and nucleotide hydrolysis to specific structural domains (Fig. 1). The myosin force-generation function is thought to be provided by a lever-arm domain (residues 781–820), which has been observed in numerous positions in crystal structures of myosin. It has been proposed that the various positions all correlate to one of two conformational classes, either a pre-power stroke state in which the lever arm is tilted up relative to the core domain or a post-power stroke “near-rigor” state in which the lever arm is pointed down (8).
FIGURE 1.

Structure of F344W-MDE with the mant nucleotide (cyan) and the 344W residue shown as a ball-and-stick representation (green). Also highlighted are the essential light chain (ELC, blue) and the switch I, switch II, and P-loop (red). The chicken smooth-muscle myosin MDE structure (1BR4.pdb (2)) was superimposed with mant ADP coordinates (1LVK.pdb (4)), and a distance of 22.4 Å was measured between 344W and the mant moiety. This figure was prepared in Swiss PDB Viewer (GlaxoSmithKline).
The actin-binding site of myosin is defined by a large cleft that divides the catalytic domain into the so-called upper- and lower-50-kD subdomains. Limited movement of the actin-binding cleft has been observed by comparing myosin crystal structures in various states of the ATPase cycle, although the cleft does actually close somewhat in the recent myosin V apo structure (9) and undergoes some rearrangements in the recent myosin II apo structure of Reubold et al. (8). As myosin V binding to actin is diffusion-limited, it has been suggested that the myosin V actin-binding cleft is already closed in the apo state in the absence of actin, making myosin V an instructive special case. Fitting skeletal and smooth-muscle myosin crystal structures into cryoelectron microscopy (cryo-EM) density maps of S1-decorated actin filaments predicts a greater closing of the actin-binding cleft in the posthydrolysis state than is seen in any of the crystal structures (10,11). In fact, rotation of the upper- and lower-50-kD subdomains is seen in the recent myosin apo structures. For chicken smooth-muscle myosin II, a solution study using an intrinsic tryptophan probe located in the 50-kD cleft showed that actin-binding and closure of the actin-binding cleft occur at nearly identical rates (12). Fluorescence data from this study suggested cleft closure in strongly actin-bound states even in the absence of actin. This data supports a model of the transition from the weakly to the strongly actin-bound state in which the sum of many small attractions between myosin and actin produces movement of the upper-50-kD domain, culminating in cleft closure. Similarly, a study of Dictyostelium myosin II using extrinsic probes demonstrated that cleft movement is linked to actin binding (13). Thus, a growing body of evidence suggests that the actin-binding cleft begins to close in strong-binding states of the ATPase cycle even in the absence of actin, and closes to a greater degree upon actin binding.
Although domain movements of the lever arm and actin-binding cleft are now well established in myosin, conformational changes at the active site have proven difficult to observe by crystallography. The active site would be expected to open in the strongly actin-bound states to allow for the exchange of ADP and ATP, and close upon ATP binding to provide a catalytically competent conformation for hydrolysis. Rayment et al. predicted from their original myosin crystal structure that opening of the active site could be triggered by actin-binding cleft closure (14). Further, pivoting the upper-50-kD subdomain around a hinge located near the active site allows for greatly improved fitting of the skeletal myosin crystal structure to cryo-EM reconstructions of the acto-myosin complex (11). Such a pivot would close the actin-binding cleft while opening the active site, as predicted by Rayment et al. (14).
The myosin active site, considered from a perspective closer to the γ-phosphate binding pocket, contains several loop motifs in common with the members of the kinesin and G-protein superfamilies of nucleotide triphosphate-binding proteins (15). Three loops, termed the phosphate-binding loop (P-loop), switch I, and switch II, are thought to play coordinating roles during the hydrolysis cycle. However, aside from these similarities, large structural differences exist between the active sites of G-proteins, kinesins, and myosins. The switch I and switch II loops are exposed in G-proteins and have been shown to participate directly in binding to G-protein targets (16–18); e.g., switch II of the α-subunit of heterotrimeric G-proteins interacts directly with its βγ subunits and switch I of the Rap-1 G-protein interacts with its target Raf. In contrast, in the much larger and more complex myosins, the switches are buried and are not involved in interactions with actin but instead act to coordinate the distal domains that carry out myosin function. Although crystal structures have been obtained for a large variety of G-proteins in all stages of the hydrolysis cycle, in myosin the structural picture is incomplete, as only the weakly actin-bound states have been captured. This may be a consequence of the complexity of myosin active sites relative to those of G-proteins. Even comparing myosins to the closely related kinesins, elements of the active site are thought to act by somewhat different mechanisms, e.g., strong binding of myosin to actin and kinesin to microtubules don't occur in identical nucleotide states. Further, whereas switch I has been shown to close on microtubule binding for kinesins (16), for myosins there is reason to believe that the opposite occurs, as actin binding may trigger opening of switch I (8,11). Thus, despite information about hydrolysis mechanisms in G-proteins and kinesins, the question of how the myosin active site is altered during the ATPase cycle remains open and controversial. Of particular importance is the question of whether structural events at the myosin active site required for enzymatic function can be accommodated solely by rearrangements of the P-loop, switch I, and switch II, or must include more global changes.
In this study, fluorescence resonance energy transfer (FRET) is used to examine distance changes across the active site in nucleotide states that correspond to both strongly and weakly actin-bound states. A chicken smooth-muscle myosin fragment, F344W motor domain essential light chain (MDE), has been genetically engineered to contain a single tryptophan (W344) that acts as the FRET donor in this study, whereas 2′(3′)-O-(N-methylanthraniloyl) (mant) fluorescent analogs of ATP and ADP bound to the active site are used as FRET acceptors. The single tryptophan substitution (F344W) is located on a loop between two α-helixes that traverse the myosin upper-50-kD subdomain at the front of the active site, whereas the mant moiety of the nucleotide analogs is known to sit in the nucleotide binding pocket (Fig. 1). Measurements made from superimposed crystal structures of Dictyostelium myosin II bound to mant nucleotides (4) and chicken smooth-muscle myosin bound to 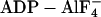 (2) show that the 344W residue is located ∼22 Å from the mant moiety, placing it within the useful range of measurement for this FRET donor-acceptor pair. Correlation of FRET efficiency with distance in each nucleotide state has allowed us to monitor a structural rearrangement of the active site that is consistent with the predictions of both Rayment et al. (14) and Holmes et al. (11).
(2) show that the 344W residue is located ∼22 Å from the mant moiety, placing it within the useful range of measurement for this FRET donor-acceptor pair. Correlation of FRET efficiency with distance in each nucleotide state has allowed us to monitor a structural rearrangement of the active site that is consistent with the predictions of both Rayment et al. (14) and Holmes et al. (11).
MATERIALS AND METHODS
Chemicals
Mant-labeled ATP and ADP were purchased from Molecular Probes (Eugene, OR). ATP and ADP were purchased from Sigma (St. Louis, MO). All other reagents were of the highest quality available.
Solutions
The standard buffer for fluorescence procedures was a 3-[N-morpholino] propanesulfonic acid (MOPS) buffer at pH 7.4 (20 mM MOPS, 20 mM KCl, 1 mM EGTA, 1 mM NaN3, 1 mM dithiothreotol, and 2 mM MgCl2) at room temperature. For protein purification procedures an imidazole buffer at pH 7.4 (10 mM imidazole-HCl, 90 mM NaCl, 1 mM NaN3, and 1 mM dithiothreotol) was used at 4°C.
cDNA construction
Site-directed mutagenesis was performed using a Quik-Change mutagenesis kit (Stratagene, LaJolla, CA) to create a mutant cDNA coding for a chicken smooth-muscle myosin heavy chain containing a single nonendogenous tryptophan at position 344 (highlighted as a space-filling model; Fig. 1). The cDNA used as a template (null-MDE) was a clone coding for the heavy chain containing the motor domain and light-chain binding regions (residues 1–819 kindly provided by Kathleen M. Trybus, University of Vermont) in which the seven endogenous tryptophans of the clone were mutated to phenylalanine (or methionine in the case of Trp-546) as previously described (19). The FLAG epitope (DYKDDDK) was attached to the C-terminus of the cDNA for purification purposes.
Protein expression and purification
The F344W-MDE and tryptophan null-MDE constructs were expressed in baculovirus by coinfecting Sf-9 cells with recombinant baculoviruses containing the mutant heavy chain and the essential light chain. Cells were incubated with baculovirus for 3 days, lysed, fractionated between 30% and 70% saturated ammonium sulfate and dialyzed overnight in imidazole buffer at 4°C as previously described (19). The resulting lysate was purified on an anti-FLAG antibody column, dialyzed overnight into MOPS buffer, and assayed for purity by SDS page on a Coomassie-stained gel (20). Protein concentrations were determined by the method of Edelhoch (21). Fluorescence experiments were performed on 10 such preparations of F344W-MDE. Actin used in actin-activated ATPase assays was purified from acetone powder as previously described (22). The purified actin concentration was determined spectroscopically using an extinction coefficient of 0.63 mg ml−1 cm−1 at 290 nm.
Enzymatic assays
Actin-activated ATPase assays were performed as previously described (23) at 37°C in low ionic strength MOPS buffer (10 mM MOPS, 1 mM EGTA, and 7 mM MgCl2) at pH 7.0 using a range of actin concentrations between 0 and 136 μM. ATPase rates were plotted as a function of actin concentration and fit to the Michaelis-Menten equation using a nonlinear least-squares routine in SigmaPlot 8.0 (SPSS, Chicago, IL) to determine values of Vmax and Km.
Steady-state fluorescence measurements
Steady-state fluorescence measurements of tryptophan fluorescence were obtained on a Quantamaster fluorimeter (Photon Technology International, South Brunswick, NJ) equipped with a 50-W xenon arc lamp excitation source, excitation/emission monochrometers, and a WG-320 cutoff emission filter. Protein samples were excited at 295 nm and tryptophan emission spectra were collected from 305 to 400 nm. Slit widths of 1 nm (excitation) and 5 nm (emission) were used. All spectra were corrected for Raman scatter and background fluorescence by subtraction of an appropriate buffer blank. Spectra were taken at a protein concentration of 1 μM in the standard MOPS buffer (defined above) at room temperature. Spectra of protein plus ATP or ADP were obtained at a saturating nucleotide concentration of 2 mM. Mant ATP and mant ADP were used at nonsaturating concentrations of 15 μM and 10 μM, respectively, to avoid inner-filter effects associated with the mant moiety. F344W-MDE plus mant nucleotide spectra were corrected for nonsaturating concentrations of mant nucleotides using
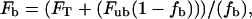 |
(1) |
where FT = total fluorescence, Fb = fluorescence of 344W in the nucleotide-bound state, fb = fraction of F344W-MDE bound to nucleotide, and Fub = fluorescence of 344W in the absence of nucleotide. Fb was determined by solving 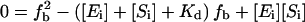 for fb, where [Ei] = initial protein concentration [Si] = initial nucleotide concentration, and Kd = the dissociation constant for nucleotide binding to F344W-MDE. Kd values were determined as discussed below. Quantum yields for tryptophan were calculated by comparison to an l-tryptophan standard (ф = 0.14) as previously described (24).
for fb, where [Ei] = initial protein concentration [Si] = initial nucleotide concentration, and Kd = the dissociation constant for nucleotide binding to F344W-MDE. Kd values were determined as discussed below. Quantum yields for tryptophan were calculated by comparison to an l-tryptophan standard (ф = 0.14) as previously described (24).
Fluorescence lifetime measurements
Lifetime measurements were made using a Photon Technology International (London, Ontario, Canada) Laserstrobe instrument. The excitation source for lifetime experiments was a nitrogen-pumped dye laser. Exciting radiation at 590 nm from the laser was modified to the final 295 nm using a frequency doubler, and fluorescence was collected by a time-delay gated photomultiplier tube. The instrument response function at 295 nm was determined using a dilute solution of nonfat dry milk as a scatterer. The F344W-MDE decays were deconvoluted from the instrument response function and fit to a two-exponential decay using FeliX 32 software provided by Photon Technology International.
Acrylamide quenching experiments
To determine the degree of exposure of the 344W residue to solvent, acrylamide quenching experiments were performed in various nucleotide conditions, including the presence of 2 mM ATP or 2 mM ADP, or in the apo state. The fluorescence values from the 333 nm emission peak in the absence of quencher (F0) were divided by the fluorescence in the presence of quencher (F), plotted against quencher concentration (Q), and fit to the Stern-Volmer equation using linear regression analysis in Kaleidagraph (Synergy Software, Reading, PA):
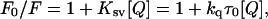 |
(2) |
where kq is the bimolecular quenching constant, τ0 is the 344W tryptophan lifetime in the absence of quencher, and Ksv is the Stern-Volmer constant (25). Thus, kq was calculated by dividing Ksv by τ0, determined by fluorescence lifetime measurements.
Fluorescence resonance energy transfer
The distance between the 344W donor and mant acceptor probes was measured using FRET according to the Förster energy transfer theory (26). We measured the efficiency (E) of FRET by observing the decrease in fluorescence of the 344W donor:
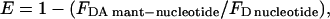 |
(3) |
where FDA mant-nucleotide is the tryptophan fluorescence of the F344 donor at 333nm in the presence of mant ATP or mant ADP nucleotide and FD nucleotide is the tryptophan fluorescence of the F344 donor at 333 nm in the presence of unlabeled ATP or ADP. All fluorescence values were corrected for the fraction of protein bound to nucleotide as described above. Fluorescence from the endogenous tyrosines in the 344W construct was corrected for by subtraction of values in the corresponding nucleotide states obtained for the null-MDE control containing no tryptophan residues. The distance (r) between the donor and acceptor probes was determined by Eq. 4:
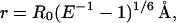 |
(4) |
where R0 is the Förster distance at which E = 50%.
R0 was calculated using Eq. 5:
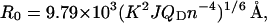 |
(5) |
where n is the refractive index of the buffer (assumed to be 1.4), QD is the quantum yield of 344W in the presence of ADP, mant ADP, ATP, and mant ATP (0.11, 0.10, 0.11, and 0.08, respectively), K2 is an orientation factor for which we assumed a value of 2/3 consistent with free rotation of the donor and acceptor probes, and J is the spectral overlap between the donor emission and acceptor excitation given by Eq. 6:
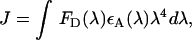 |
(6) |
where λ is the wavelength, FD is the normalized intensity of the unquenched donor, and εA is the extinction coefficient of the acceptor. J was determined to be 6.06 × 10−15 M−1 cm3 by numerical integration of our experimentally determined FD and εA values at 1-nm intervals for F344W-MDE in the presence of both mant ATP and mant ADP.
Equilibrium nucleotide binding affinity measurements
To determine F344W-MDE-mant ADP affinity by steady-state fluorescence, the protein was titrated with increasing concentrations of nucleotide in a series and the decrease in tryptophan fluorescence emission at 333 nm was monitored in the fluorimeter after excitation at 295 nm. These fluorescence values were corrected for inner-filter and dilution effects, normalized to the fluorescence value obtained for nucleotide-free protein and subtracted from 1. Plotting (1 − normalized fluorescence) versus [mant nucleotide] produces a rectangular hyperbola which approaches a maximum binding value (Bmax) asymptotically:
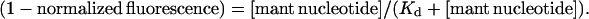 |
(7) |
The resulting plots were used to determine dissociation constant (Kd) values (equal to the nucleotide concentration at which 0.5·Bmax occurs) by fitting Eq. 7 to the data using SigmaPlot 8.0 (SPSS Corp., Chicago, IL). The affinity of the F344W-MDE protein for mant ATP was determined similarly by exciting at 295 nm and after both the tryptophan emission at 333 nm and the mant ATP fluorescence emission at 440 nm (in separate experiments). Unlike the case for mant ADP, these titrations could not be performed in a continuous series due to ATP hydrolysis during the experiments. Therefore, each specific concentration of mant ATP was added to protein and fluorescence at 333 nm was monitored in the fluorimeter as a function of time.
Transient kinetic analysis of mant-ADP binding
The affinity of F344W-MDE for mant ADP was also determined by making stopped-flow measurements of the binding and release rates of mant ADP, where Kd = koff/kon. To measure the apparent second-order binding constant (kon) for mant ADP, 2 μM F344W-MDE in one syringe was rapidly mixed with various concentrations of mant ADP (0.3–5 μM) in the second syringe. To determine the rate of mant ADP release (koff) from F344W-MDE, chase experiments were performed in which a complex of mant ADP (10 μM) bound to 2 μM F344W-MDE in one syringe was rapidly mixed with an excess of ATP (500 μM) in the second syringe. The rate of mant ADP binding or release was determined by following the change in mant ADP fluorescence above 420 nm with a 420 cutoff filter after excitation at 295 nm (5-nm bandpass). All transient kinetic experiments were done using a BioLogic μSFM-20 stopped-flow instrument (Claix, France) with a dead time of 2.1 ms under the flow-rate conditions used. The resulting data were fit using nonlinear regression analysis in Kaleidagraph (Synergy Software, Reading, Pa.). Experiments were performed in the MOPS buffer as described above for steady-state fluorescence experiments at 25°C.
Protein structural comparisons
The three-dimensional structures of myosin isoforms were superimposed and visualized in Swiss-PdbViewer (GlaxoSmithKline, Middlesex, UK).
RESULTS
Actin-activated ATPase assays
ATPase assays performed on the F344W-MDE protein (Table 1) demonstrated that the conservative F344W mutation did not adversely affect the ability of the mutant protein to interact with actin or hydrolyze nucleotide. Compared to WT-MDE protein, F344W-MDE had a threefold lower Vmax value (0.23 s−1 vs. 0.76 s−1), but a >4 times higher basal rate of hydrolysis in the absence of actin (0.35 s−1 vs. 0.08 s−1). However, because the mutant protein also had a fourfold tighter Km value for actin (15 μM vs. 62 μM), the ratio of Vmax to Km of F344W-MDE was slightly higher than that of WT-MDE (1.5 × 104 M−1 s−1 vs. 1.2 × 104 M−1 s−1). Similar results, i.e., lower Vmax and lower Km values for the mutant protein versus the wild-type, have been obtained for several other single-tryptophan mutants of myosin created in our lab (19,27). Thus, the F344W-MDE protein behaves much like other single-tryptophan MDE mutant proteins and the wild-type chicken smooth-muscle MDE protein in terms of its ability to hydrolyze ATP in an efficient and actin-dependent manner.
TABLE 1.
Kinetic parameters of MDE constructs
| MDE construct | KM (μM)* | Vmax (s−1)* | Vmax/KM (μM−1 s−1) |
|---|---|---|---|
| WT-MDE | 62 | 0.74 | 1.2 × 104 |
| F-344W-MDE | 15 | 0.23 | 1.5 × 104 |
Km and Vmax averaged values were determined from two separate protein preparations assuming Michaelis-Menten kinetics by plotting actin-activated ATPase rates versus actin concentration for 23, 46, 92, 115, and 136 μM actin at 37°C.
Steady-state fluorescence in the absence of FRET
To characterize the tryptophan donor signal in the absence of acceptor, we examined the steady-state fluorescence emission spectra of the F344W-MDE protein in the absence of nucleotide and in the presence of unlabeled ADP and ATP. In the presence of saturating concentrations of ADP or ATP, the intrinsic fluorescence of F344W-MDE was quenched relative to the apo state by 81 ± 5% and 76 ± 3%, respectively (Fig. 2 and Table 2), showing that the 344W residue experiences a different environment in each of these conditions. Addition of saturating ADP to a solution of myosin is known to produce a strongly actin-bound myosin state (28). However, addition of saturating ATP leads to a more complex situation as hydrolysis of the triphosphate can occur and thus myosin will be found in a mixture of nucleotide states with the percentage of myosin in each state being determined by the rate constants for the protein in the ATPase cycle, as discussed below. No spectral shift of the F344W-MDE emission maxima of 333 nm was seen in the steady-state spectra in the presence of ATP or ADP; the implications of this are also discussed below.
FIGURE 2.
Normalized F344W-MDE fluorescence emission spectra in the presence of (A) ADP (+) and mant ADP (◊) or (B) ATP (+) and mant ATP (◊) relative to the apo state (○). Spectra are corrected for the fraction of F344W-MDE not bound to nucleotide. Quenching of 344W fluorescence is observed in the presence of each nucleotide: ADP, 81% apo; mant ADP, 76% apo; ATP, 76% apo; mant ATP, 58% apo. Quenching of fluorescence relative to the apo state in the presence of nucleotides lacking the mant moiety is due to changes in the 344W environment. Additional loss of intensity in the presence of mant nucleotides (e.g., mant ATP compared to ATP or mant ADP compared to ADP) is due directly to FRET between 344W and the mant moiety at the active site.
TABLE 2.
Determination of FRET efficiencies
| Nucleotide state | Kd (μM) | fb | D (% apo) | DA (% apo) uncorrected | DA (% apo) corrected | Efficiency (%) | Ro (Å) | r (Å) |
|---|---|---|---|---|---|---|---|---|
| (mant) ADP | 8.2 ± 0.02 | 0.53 ± 0.02 | 81 ± 5 | 82 ± 4 | 76 ± 4 | 6.4 ± 0.4 | 21.3 | 33.4 ± 2.4 |
| (mant) ATP | 0.84 ± 0.02 | 0.94 ± 0.01 | 76 ± 3 | 58 ± 4 | 58 ± 4 | 23.8 ± 2.3 | 20.5 | 24.9 ± 2.2 |
Kd; dissociation constant for mant nucleotide binding to F344W-MDE; fb; fraction of mant nucleotide bound to F344W at experimental concentrations; D, donor fluorescence in the presence of unlabeled nucleotide as a percentage of the apo signal; DA uncorrected; donor fluorescence in the presence of mant nucleotide as a percentage of the apo signal; DA corrected, donor fluorescence in the presence of mant nucleotide acceptor as a percentage of the apo signal corrected for inner-filter effects and fraction of bound nucleotide; Efficiency, efficiency of FRET as determined from D and DA; R0, the distance at which energy transfer is 50%; and r, the distance between 344W and the mant moiety in each nucleotide state.
Acrylamide quenching experiments
To determine the contribution of changes in solvent exposure to the steady-state spectra of 344W described above, we performed acrylamide quenching experiments in the apo state and in the presence of ADP and ATP (Fig. 3). From the resulting plots of F0/F versus [Q] Stern-Volmer constants of 1.75 ± 0.07 M−1, 1.65 ± 0.07 M−1, and 1.45 ± 0.21 M−1 were determined for the apo, ADP, and ATP conditions, respectively, by fitting to the Stern-Volmer equation. Fluorescent lifetime measurements were performed on the F344W-MDE in the apo state and in the presence of ADP and ATP, which yielded lifetime values of 3.54 ± 0.32 ns, 2.58 ± 0.43 ns, and 2.64 ± 0.14 ns, respectively (data not shown). Dividing each Stern-Volmer constant by the appropriate lifetime yields kq values of 5.1 × 108 ± 0.1 M−1 s−1, 5.4 × 108 ± 0.3 M−1 s−1, and 5.7 × 108 ± 0.1 M−1 s−1 for the apo, ADP, and ATP conditions, respectively. Such low bimolecular quenching constant values are indicative of a highly buried residue, suggesting that 344W remains relatively buried in each nucleotide state tested. The small variation between these constants shows that there was little change in the degree of accessibility of the 344W residue to solvent in the presence of ATP or ADP, or in the absence of nucleotide.
FIGURE 3.

Acrylamide quenching of F344W-MDE under apo (○), ADP (♦), and ATP (□) conditions. F0/F was plotted against [acrylamide] and fit to the Stern-Volmer equation (Eq. 2). The resulting Stern-Volmer constants were divided by the fluorescence lifetime under each experimental condition in the absence of acrylamide to determine the appropriate bimolecular quenching constants (kq).
Affinity determinations for mant nucleotides
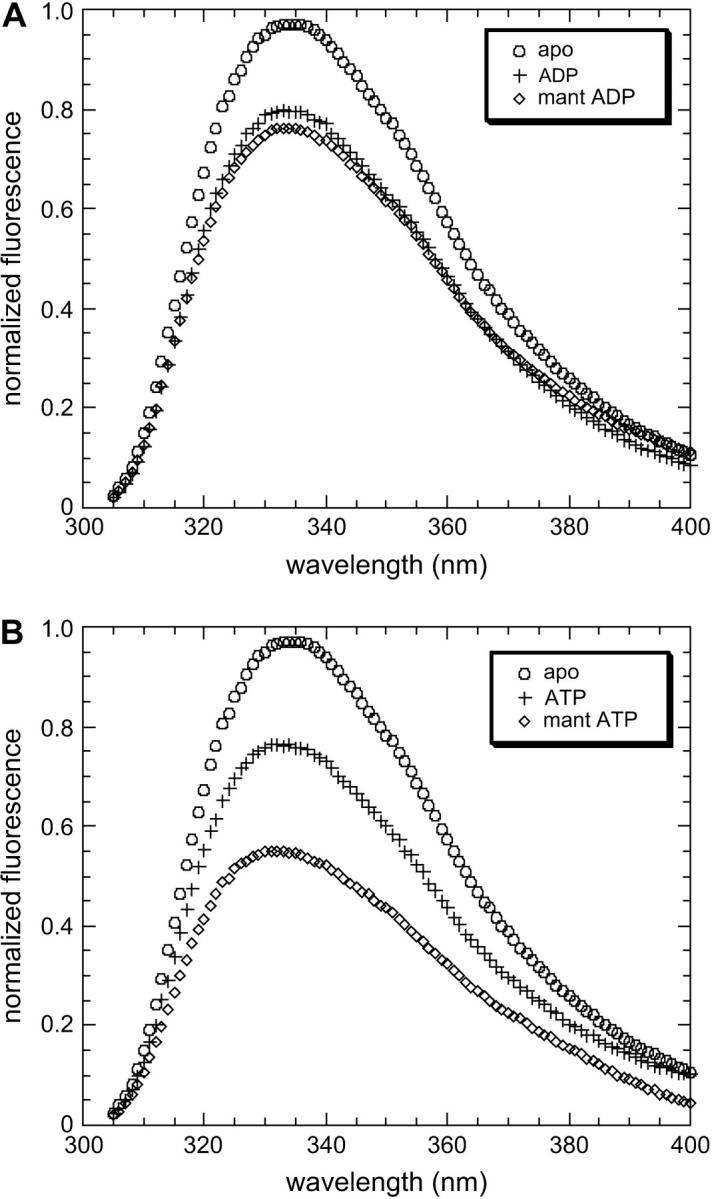
Because accurate determination of the affinity of F344W-MDE for mant nucleotides was so crucial to the accuracy of the corrected FRET results, the affinities of F344W-MDE for mant ADP and mant ATP were determined by multiple methods. Affinity determinations were performed for mant ATP by following the attenuation of the 344W fluorescent signal (Fig. 4) and also the enhancement of the mant signal (data not shown) with respect to nucleotide concentration. The Kd values of 0.84 ± 0.56 μM and 1.24 μM determined by following the 344W signal and the mant ATP signal, respectively, were in excellent agreement, suggesting tight binding of mant ATP to F344W-MDE. The affinity of F344W-MDE for mant ADP was also determined by following the attenuation of the 344W fluorescent signal (Fig. 4), yielding a Kd value of 10.5 μM ± 0.8 μM. However, a significant inner filter effect in the presence of high concentrations (>12 μM) of mant nucleotides prevented an accurate determination of the saturation level of mant ADP binding to F344W-MDE. Thus we also determined the affinity of 344W-MDE for mant ADP from stopped-flow measurements of the reverse and forward mant ADP binding rates (Fig. 5). We determined an apparent second-order rate constant for binding of mant ADP to F344W-MDE of 0.79 μM−1 s−1 and an F344W-MDE-mant ADP dissociation rate of 6.47 s−1 ± 0.10 s−1. The affinity of F344W-MDE for mant ADP determined by the ratio of these rates was 8.2 ± 0.02 μM, in very good agreement with the 10.5 ± 0.8 μM value determined by steady-state titration. F344W-MDE was determined to be 53 ± 2% saturated in the presence of 10 μM mant ADP using the Kd of 8.2 μM for mant ADP determined by the kinetic measurements. In the presence of 15 μM mant ATP F344W-MDE was determined to be nearly saturated (94 ± 1%), consistent with the steady-state titration data obtained at concentrations >6 μM. The protein was completely saturated with nucleotide in the presence of 2 mM ATP or ADP (data not shown).
FIGURE 4.

Determination of F344W-MDE affinity for mant ATP and mant ADP by steady-state titration. Association curves showing mant ADP (♦) and mant ATP (○) binding to F344W-MDE as a function of [mant nucleotide]. The determined Kd values (Eq. 7) indicate much tighter binding of mant ATP (Kd = 0.84 ± 0.02 μM) to F344W-MDE as compared to mant ADP (Kd = 10.5 ± 0.8 μM).
FIGURE 5.
Determination of the F344W-MDE-mant ADP binding and dissociation rates. (A) Linear fit to the plot of the observed rate of binding to F344W-MDE versus mant ADP concentration gives an apparent second-order binding rate constant of 0.79 μM−1 s−1. Attenuation of intrinsic fluorescence from 344W was followed as a function of time as 2 μM F344W-MDE was rapidly mixed with 0.3–5 μM mant ADP. The nonzero y-intercept is equal to the mant ADP release rate and is in good agreement with the value measured experimentally by the chase experiment. The inset shows an averaged trace composed of five stopped-flow records at 2.5 μM mant ADP. (B) Chase experiment to determine the rate of mant ADP release from F344W-MDE. Two micromoles F344W-MDE in the presence of 10 μM mant ADP was rapidly mixed with 500 μM unlabeled ATP, and the resulting attenuation of mant fluorescence was followed as a function of time (averaged data from four traces). A single exponential fit of the data gives an F344W-MDE-mant ADP dissociation rate of 6.47 ± 0.10 s−1. All stopped-flow experiments were performed at 25°C.
Steady-state FRET experiments
To determine the distance between 344W at the front of the active site and the mant moiety on the ribose ring of the nucleotide sitting at the back of the active site we examined tryptophan fluorescence in the presence of mant nucleotides and compared the fluorescence emission to that obtained in the presence of the unlabeled nucleotides (Fig. 2 and Table 2). Spectra obtained from additions of mant nucleotides to F344W-MDE showed quenches of intrinsic fluorescence in the presence of mant ADP and mant ATP of 76 ± 4% and 58 ± 4%, respectively, after correction for inner-filter effects and fraction of protein bound to nucleotide (Fig. 2). This additional decrease in fluorescence intensity relative to the nonmant nucleotides is directly due to resonance energy transfer from 344W to the mant fluorophore on the ribose ring of the bound nucleotide. FRET efficiencies of 6.4 ± 0.4% for mant ADP and 23.8 ± 2.3% for mant ATP were obtained as described in Eq. 3. R0 determinations showed similar values of 21.3 Å for the 344W-mant ADP donor-acceptor pair and 20.5 Å for the 344W-mant ATP donor-acceptor pair. Distances were calculated to be 33.4 ± 2.4 Å between the 344W and mant ADP and 24.9 ± 2.2 Å between 344W and mant ATP using Eq. 4, indicating an 8.5-Å movement between 344W and the mant moiety in the strongly and weakly actin-bound states of the smooth-muscle myosin ATPase cycle.
In addition to the strongly fluorescent 344W residue, the F344W-MDE contains 28 endogenous weakly fluorescent tyrosines. Although our experiments were done at an excitation wavelength of 295 nm, which should produce little or no tyrosine excitation, we controlled for the contribution of tyrosine fluorescence by subtracting emission spectra obtained under identical experimental conditions of a “null-MDE” mutant from our F344W-MDE emission spectra. The null-MDE is identical to the F344W-MDE except for the presence of a phenylalanine at position 344 and thus contains no tryptophan residues. Our subtractions of null-MDE spectra from F344W-MDE spectra confirm that the tyrosines in our MDE construct make no contribution to our FRET results (data not shown).
DISCUSSION
Interpretation of our FRET results
FRET measurements between 344W and mant nucleotide have allowed us to compare the distance between 344W and the mant moiety in both weakly (presence of ATP) and strongly (presence of ADP) actin-bound states of the ATPase cycle. Because mant nucleotide was the FRET acceptor in our experiments it was not possible to measure this distance in a nucleotide-free state. However, comparisons of the FRET signal in the presence of ATP and ADP show a unique position of the upper-50-kD subdomain of smooth-muscle myosin, and provide the first experimental evidence in solution of a substantial structural change around the myosin active site. Our results are consistent with both the structural rearrangement of the active site predicted by Rayment et al. (14) and the pivoting of the upper-50-kD subdomain used by Holmes et al. (11) to fit myosin ADP structures to cryo-EM reconstructions.
Although FRET results in general should not be misinterpreted as giving accurate determinations of distance, our FRET-measured distance change of 8.5 Å is of sufficient magnitude and certainty to show a definite distance change between the 344W residue and mant moiety in the presence of ADP as compared to the presence of ATP. The most straightforward explanation of this opening is movement of the upper-50-kD subdomain. However, it is also possible that our FRET results reflect a more local rearrangement of the short loop that contains the 344W residue, and/or that the mant moiety is repositioned during the ATPase cycle, contributing to the observed distance change. Even in the case in which a combination of nucleotide repositioning and local 344 loop movement causes the opening between the 344W residue and mant moiety in the ADP state, our results still demonstrate a rearrangement of the core active site. Thus, our results provide the first evidence in solution consistent with a movement of upper-50-kD subdomain that opens the myosin active site in a strongly actin-bound state of the ATPase cycle. Such an opening may be necessary to coordinate nucleotide hydrolysis to actin binding. We therefore propose a model of myosin domain coordination in which movement of the 50 kD cleft during actin binding is communicated to the active site through a pivoting of the upper-50-kD subdomain as the 50-kD cleft closes on actin, effectively opening the active site and increasing the rate of product release. Changes in the interactions and positions of switch I and switch II upon product release would then be communicated to the converter domain through the switch II helix, triggering the powerstroke only after myosin has entered the strongly bound state. As the power stroke is almost certainly timed to occur when myosin is strongly bound to actin, the necessity of such a mechanism to link product release to actin binding is understandable.
Interpretation of our steady-state results using unlabeled nucleotides
Our steady-state results using unlabeled ATP and ADP suggest that structural changes occur during the ATPase cycle that place the 344W residue in a different environment in the apo, ADP, and ATP states. As no spectral shift was seen in the presence of either nucleotide, it is likely that the attenuation of fluorescence was not the result of a large change of exposure of 344W to solvent, but instead probably reflects closer proximity of 344W to a charged protein side chain capable of altering the fluorescence emitted by 344W. Research on mechanisms leading to fluorescence quenching of tryptophans in proteins suggests that charged residues can influence quantum yield by stabilizing a charge-transfer state in which the peptide bonds accept the excited electron from tryptophan (29). Inspection of the chicken smooth-muscle myosin amino acid structure shows several such residues, including a string of three glutamic acid residues located at positions 346–348, which could provide such a function. This conclusion is supported by our quenching results, which showed nearly identical bimolecular quenching constants for the apo, ADP, and ATP states, respectively. Regardless of the underlying basis for the differential quenching observed in the ADP and ATP states, we have obtained the appropriate donor-only signals in each state to use in determinations of FRET efficiency in the presence of mant nucleotide.
Technical issues and caveats
Because the mant moiety used as our FRET acceptor absorbs strongly at both the excitation wavelength used in our experiments (295 nm) and at the emission maxima of the 344W tryptophan (333 nm), accurately correcting for the substantial inner-filter effects obtained at high mant nucleotide concentrations represented a difficulty. We avoided this inaccuracy by performing our fluorescence experiments at nonsaturating mant nucleotide concentrations of 10 μM mant ADP and 15 μM mant ATP. Therefore our determinations of FRET efficiency were dependent on knowing the fraction of protein bound to mant nucleotide, which we calculated based on the affinities of mant nucleotides for F344W-MDE. The excellent agreement between the two very different methods we used to determine the affinity of F344W-MDE for mant ADP affinity gives us confidence in our correction for the fraction of mant ADP bound to F344W-MDE in the FRET experiments. In the case of mant ATP we also had excellent agreement between two different measurements of Kd. Due to the hydrolysis of mant ATP during each titration it was not possible to titrate with very low concentrations of mant ATP. However, the concentration of mant ATP used in the FRET experiments (15 μM) relative to the saturation point observed in our steady-state titrations (>6 μM) strongly suggests that F344W-MDE was indeed almost completely saturated.
The Kd values obtained for F344W-MDE (mant ATP, 0.8 μM; mant ADP, 8.2 μM) are in close agreement with values we obtained for the null-MDE (mant ATP, 0.5 μM; mant ADP, 8 μM) and Kd values determined for W29-MDE, another single tryptophan MDE mutant protein constructed in our lab (mant ATP, 1 μM; mant ADP, 7 μM; data not shown). Working with smooth-muscle wild-type S1 myosin II, Cremo and Geeves (28) obtained a Kd of 0.075 μM for mant ADP and 16-fold higher Kd for ADP of 1.2 μM. However, Kd values for various myosin II S1 proteins for ADP range widely, from 0.33 μM for cardiac S1 (30) to 37 μM for Dictyostelium myosin II (31), and thus it is not surprising that the single tryptophan-MDE mutant smooth-muscle myosin varies from the wild-type S1 in its nucleotide affinities.
There are a number of caveats that must be addressed regarding interpretation of our FRET data. When F344W-MDE is combined with ATP or mant ATP, we would expect myosin to be found in a mixture of intermediate states, with the weakly actin-bound M·ATP and M·ADP Pi species predominating. Rate constants previously determined for the elemental steps of ATP hydrolysis by smooth-muscle myosin II suggest that the M·ADP Pi species should be the most populated state, as the forward hydrolysis step is favored over the reverse hydrolysis step and phosphate release is rate-limiting (32). Our results in the presence of mant ATP therefore most likely represent a mixture of weakly actin-bound states of the myosin ATPase cycle.
Because our FRET study was done on a population of molecules in solution, the FRET distances reflect average distances between 344W and the mant moiety for the population. As it is a near certainty that myosin interconverts between a wide range of conformations in each nucleotide state (i.e., the myosin energy landscape is populated by many variants in each nucleotide state of the ATPase cycle), our FRET study cannot distinguish these conformations and dynamic motions within them. In fact, conformations that are relatively rare and thus contribute little to FRET may be highly important in terms of experiencing interactions that drive myosin through the ATPase cycle. However, this caveat is true of any FRET solution study and does not decrease the value of our results.
Finally, in calculating R0 between the 344W and mant nucleotides we made the assumption that the orientation factor, K2, is equal to 2/3. This implies that the 344W-mant nucleotide FRET donor-acceptor pair are free to rotate and thus rotational diffusion between the probes will occur after excitation but before energy transfer takes place. Although the possible range of K2 values is 0–4, when the 2/3 assumption is made the FRET distance cannot be in error by >35% (33). For example, if K2 was in reality 0.1 in the presence of both mant ADP and mant ATP, the distance change between the weakly and strongly actin-bound states of the ATPase cycle decreases to only 6.2 Å, still a significant difference. An increase in K2 would only serve to increase the observed difference in distance between these states. Thus it is clear that variation in K2 from the assumed 2/3 value would not significantly affect our basic conclusion that there is a large-scale closure between the 344W residue and the mant moiety during the transition from the strongly (ADP) to weakly (ATP) actin-bound states of the myosin ATPase cycle.
Comparison to available crystal structure data
Superimposition of mant nucleotide coordinates from Dictyostelium myosin II (4) on the chicken smooth-muscle myosin MDE structure (2) showed a distance between the mant moiety and 344W of 22.4 Å, in very good agreement with our FRET-determined distance. Distance measurements from both the P-loop and the ribose ring of the bound nucleotide to F344 or its equivalent show that little variation in these distances occurs among nucleotide-bound myosin II structures, most likely because these structures capture weak actin-binding states with switch I closed and nucleotide tightly bound.
Recently Coureux et al. (9,34) obtained chicken myosin V structures in the absence of nucleotide or soaked with ADP that show switch I in an open position, as well as an ADP.BeFx structure that resembles previous myosin II structures with switch I closed. Another recent apo structure for Dictyostelium myosin II (8), is in close agreement with the myosin V apo structure. By comparison with the myosin II nucleotide-bound structures the P-loop has moved closer to the 344 position in the myosin V ADP and apo structures (Fig. 6). Thus, the conformational state observed for smooth-muscle myosin in the presence of ADP in this study, in which 344W has moved further away from the nucleotide, is likely to be unique compared to the ADP or apo structures seen in myosin V. Although the myosin V-ADP structure does capture switch I in an open position, the authors state that this structure likely corresponds to a weakly ADP-bound state of myosin V (34). In contrast, the ADP state captured in our study is likely a tightly ADP-bound state. Rosenfeld et al. (35) demonstrated that, unlike skeletal and Dictyostelium myosin II, smooth-muscle myosin is capable of adopting a unique strong actin-binding, tightly ADP-bound state, i.e., actin and ADP binding can be loosely coupled in smooth-muscle myosin. They argued that the tight affinity for ADP in this state may be mediated by loop 1, which would explain the fact that the active site in our smooth-muscle myosin construct is open and yet binds ADP relatively tightly. A unique strongly bound ADP state has also been observed in smooth-muscle myosin by Whittaker et al. (36), who used cryo-EM and helical image reconstruction to demonstrate that ADP release generates an additional tilt of 23° of the regulatory domain in chicken gizzard smooth-muscle myosin not seen in skeletal and Dictyostelium myosin II. Therefore, our smooth-muscle ADP FRET result may capture a myosin state that does not exist in skeletal and Dictyostelium myosin II isoforms.
FIGURE 6.

Superimposition of Dictyostelium myosin II mant ADP (1LVK.pdb (4)) with myosin V apo (1OE9.pdb (9)) and ADP structures (1W7I.pdb (34)). The P-loop and 344W equivalents are shown in red (myosin II mant ADP structure), green (myosin V ADP structure), and blue (myosin V apo structure). The distance between the 344W position and the P-loop decreases in the myosin V apo and ADP structures relative to that in the myosin II mant ADP structure due to movement of the P-loop.
In addition, whereas the nucleotide tends to shift along with the P-loop in the myosin V structures, the position of the mant attachment, the 3′ carbon of the ribose ring, shows little change relative to 344W in any of the structures. Thus, the evidence from crystal structures suggests that movement of the mant moiety is not the source of the distance change in our FRET study. Therefore, the simplest interpretation of our FRET results is that we are observing movement of the upper-50-kD domain. The 8-Å movement of 344W away from the nucleotide we observe in the presence of ADP is consistent with the model of Holmes et al. (11), in which rigid rotation of the upper-50-kD subdomain upon actin binding serves to close the actin-binding cleft and open switch I at the active site. The presence of ADP may stabilize the active site in a more open conformation and/or the actin-binding cleft in a more closed conformation in the absence of actin. Another intriguing possibility is that a more open active site in the ADP state is physiologically relevant in terms of product release.
Future work
The ability of the 344W residue to indicate movement of the upper-50-kD subdomain, which correlates to changes between weakly and strongly actin-bound states, provides a useful fluorescent signal for use in future kinetics studies to correlate structural rearrangements of the myosin active site to the biochemical events of the ATPase cycle. As well, such studies can provide a means to correlate active-site movement with motions of other myosin domains that must be coordinated to produce directed force and motion during muscle contraction and other forms of cell motility.
Acknowledgments
We thank Kathy Trybus (University of Vermont) for her kind donation of the wild-type MDE cDNA, and Steven Rosenfeld (University of Alabama at Birmingham) for his generous gift of mant nucleotides. We also thank Christopher Yengo, Art Rovner, Patty Fagnant, Marilyn van Duffelen, Nicholas James, Scott Berbick, and Marque Moffett for their technical and intellectual contributions to this project.
This work was supported by a grant to C.L.B. from the National Institutes of Health, HL 63798.
References
- 1.Lymn, R. W., and E. W. Taylor. 1971. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry. 10:4617–4624. [DOI] [PubMed] [Google Scholar]
- 2.Dominguez, R., Y. Freyzon, K. M. Trybus, and C. Cohen. 1998. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell. 94:559–571. [DOI] [PubMed] [Google Scholar]
- 3.Houdusse, A., V. N. Kalabokis, D. Himmel, A. G. Szent-Gyorgyi, and C. Cohen. 1999. Atomic structure of scallop myosin subfragment S1 complexed with MgADP: a novel conformation of the myosin head. Cell. 97:459–470. [DOI] [PubMed] [Google Scholar]
- 4.Bauer, C. B., P. A. Kuhlman, C. R. Bagshaw, and I. Rayment. 1997. X-ray crystal structure and solution fluorescence characterization of Mg.2′(3′)-O-(N-methylanthraniloyl) nucleotides bound to the Dictyostelium discoideum myosin motor domain. J. Mol. Biol. 274:394–407. [DOI] [PubMed] [Google Scholar]
- 5.Fisher, A. J., C. A. Smith, J. B. Thoden, R. Smith, K. Sutoh, H. M. Holden, and I. Rayment. 1995. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP.BeFx and MgADP.AlF4. Biochemistry. 34:8960–8972. [DOI] [PubMed] [Google Scholar]
- 6.Gulick, A. M., C. B. Bauer, J. B. Thoden, and I. Rayment. 1997. X-ray structures of the MgADP, MgATPgammaS, and MgAMPPNP complexes of the Dictyostelium discoideum myosin motor domain. Biochemistry. 36:11619–11628. [DOI] [PubMed] [Google Scholar]
- 7.Rayment, I., W. R. Rypniewski, K. Schmidt-Base, R. Smith, D. R. Tomchick, M. M. Benning, D. A. Winkelmann, G. Wesenberg, and H. M. Holden. 1993. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 261:50–58. [DOI] [PubMed] [Google Scholar]
- 8.Reubold, T. F., S. Eschenburg, A. Becker, F. J. Kull, and D. J. Manstein. 2003. A structural model for actin-induced nucleotide release in myosin. Nat. Struct. Biol. 10:826–830. [DOI] [PubMed] [Google Scholar]
- 9.Coureux, P. D., A. L. Wells, J. Menetrey, C. M. Yengo, C. A. Morris, H. L. Sweeney, and A. Houdusse. 2003. A structural state of the myosin V motor without bound nucleotide. Nature. 425:419–423. [DOI] [PubMed] [Google Scholar]
- 10.Volkmann, N., G. Ouyang, K. M. Trybus, D. J. DeRosier, S. Lowey, and D. Hanein. 2003. Myosin isoforms show unique conformations in the actin-bound state. Proc. Natl. Acad. Sci. USA. 100:3227–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmes, K. C., I. Angert, F. J. Kull, W. Jahn, and R. R. Schroder. 2003. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 425:423–427. [DOI] [PubMed] [Google Scholar]
- 12.Yengo, C. M., E. M. De La Cruz, L. R. Chrin, D. P. Gaffney 2nd, and C. L. Berger. 2002. Actin-induced closure of the actin-binding cleft of smooth muscle myosin. J. Biol. Chem. 277:24114–24119. [DOI] [PubMed] [Google Scholar]
- 13.Conibear, P. B., C. R. Bagshaw, P. G. Fajer, M. Kovacs, and A. Malnasi-Csizmadia. 2003. Myosin cleft movement and its coupling to actomyosin dissociation. Nat. Struct. Biol. 10:831–835. [DOI] [PubMed] [Google Scholar]
- 14.Rayment, I., H. M. Holden, M. Whittaker, C. B. Yohn, M. Lorenz, K. C. Holmes, and R. A. Milligan. 1993. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 261:58–65. [DOI] [PubMed] [Google Scholar]
- 15.Vale, R. D. 1996. Switches, latches, and amplifiers: common themes of G proteins and molecular motors. J. Cell Biol. 135:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naber, N., T. J. Minehardt, S. Rice, X. Chen, J. Grammer, M. Matuska, R. D. Vale, P. A. Kollman, R. Car, R. G. Yount, R. Cooke, and E. Pate. 2003. Closing of the nucleotide pocket of kinesin-family motors upon binding to microtubules. Science. 300:798–801. [DOI] [PubMed] [Google Scholar]
- 17.Nassar, N., G. Horn, C. Herrmann, A. Scherer, F. McCormick, and A. Wittinghofer. 1995. The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature. 375:554–560. [DOI] [PubMed] [Google Scholar]
- 18.Kawashima, T., C. Berthet-Colominas, M. Wulff, S. Cusack, and R. Leberman. 1996. The structure of the Escherichia coli EF-Tu.EF-Ts complex at 2.5 A resolution. Nature. 379:511–518. [DOI] [PubMed] [Google Scholar]
- 19.Yengo, C. M., P. M. Fagnant, L. Chrin, A. S. Rovner, and C. L. Berger. 1998. Smooth muscle myosin mutants containing a single tryptophan reveal molecular interactions at the actin-binding interface. Proc. Natl. Acad. Sci. USA. 95:12944–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- 21.Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 6:1948–1954. [DOI] [PubMed] [Google Scholar]
- 22.Pardee, J. D., and J. A. Spudich. 1982. Purification of muscle actin. Methods Enzymol. 85 Pt. B:164–181. [DOI] [PubMed] [Google Scholar]
- 23.White, H. D. 1982. Special instrumentation and techniques for kinetic studies of contractile systems. Methods Enzymol. 85 Pt. B:698–708. [DOI] [PubMed] [Google Scholar]
- 24.Valeur, B., and G. Weber. 1977. Resolution of the fluorescence excitation spectrum of indole into the 1La and 1Lb excitation bands. Photochem. Photobiol. 25:441–444. [DOI] [PubMed] [Google Scholar]
- 25.Eftink, M. R., and C. A. Ghiron. 1981. Fluorescence quenching studies with proteins. Anal. Biochem. 114:199–227. [DOI] [PubMed] [Google Scholar]
- 26.Förster, T. 1965. Delocalized excitation and excitation transfer. In Modern Quantum Chemistry. Vol. 3, Action of Light and Organic Crystals. O. Sinanoglu, editor. Academic Press, New York.
- 27.Yengo, C. M., L. R. Chrin, A. S. Rovner, and C. L. Berger. 2000. Tryptophan 512 is sensitive to conformational changes in the rigid relay loop of smooth muscle myosin during the MgATPase cycle. J. Biol. Chem. 275:25481–25487. [DOI] [PubMed] [Google Scholar]
- 28.Cremo, C. R., and M. A. Geeves. 1998. Interaction of actin and ADP with the head domain of smooth muscle myosin: implications for strain-dependent ADP release in smooth muscle. Biochemistry. 37:1969–1978. [DOI] [PubMed] [Google Scholar]
- 29.Callis, P. R., and T. Liu. 2004. Quantitative prediction of fluorescence quantum yields for tryptophan in proteins. J. Phys. Chem. B. 108:4248–4259. [Google Scholar]
- 30.Siemankowski, R. F., and H. D. White. 1984. Kinetics of the interaction between actin, ADP, and cardiac myosin-S1. J. Biol. Chem. 259:5045–5053. [PubMed] [Google Scholar]
- 31.Ritchie, M. D., M. A. Geeves, S. K. Woodward, and D. J. Manstein. 1993. Kinetic characterization of a cytoplasmic myosin motor domain expressed in Dictyostelium discoideum. Proc. Natl. Acad. Sci. USA. 90:8619–8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marston, S. B., and E. W. Taylor. 1980. Comparison of the myosin and actomyosin ATPase mechanisms of the four types of vertebrate muscles. J. Mol. Biol. 139:573–600. [DOI] [PubMed] [Google Scholar]
- 33.dos Remedios, C. G., and P. D. Moens. 1995. Fluorescence resonance energy transfer spectroscopy is a reliable “ruler” for measuring structural changes in proteins. Dispelling the problem of the unknown orientation factor. J. Struct. Biol. 115:175–185. [DOI] [PubMed] [Google Scholar]
- 34.Coureux, P. D., H. L. Sweeney, and A. Houdusse. 2004. Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 23:4527–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenfeld, S. S., J. Xing, M. Whitaker, H. C. Cheung, F. Brown, A. Wells, R. A. Milligan, and H. L. Sweeney. 2000. Kinetic and spectroscopic evidence for three actomyosin:ADP states in smooth muscle. J. Biol. Chem. 275:25418–25426. [DOI] [PubMed] [Google Scholar]
- 36.Whittaker, M., E. M. Wilson-Kubalek, J. E. Smith, L. Faust, R. A. Milligan, and H. L. Sweeney. 1995. A 35-A movement of smooth muscle myosin on ADP release. Nature. 378:748–751. [DOI] [PubMed] [Google Scholar]