

Abstract
Nuclear receptor (NR) ligands occupy a pocket that lies within the core of the NR ligand-binding domain (LBD), and most NR LBDs lack obvious entry/exit routes upon the protein surface. Thus, significant NR conformational rearrangements must accompany ligand binding and release. The precise nature of these processes, however, remains poorly understood. Here, we utilize locally enhanced sampling (LES) molecular dynamics computer simulations to predict molecular motions of x-ray structures of thyroid hormone receptor (TR) LBDs and determine events that permit ligand escape. We find that the natural ligand 3,5,3′-triiodo-L-thyronine (T3) dissociates from the TRα1 LBD along three competing pathways generated through i), opening of helix (H) 12; ii), separation of H8 and H11 and the Ω-loop between H2 and H3; and iii), opening of H2 and H3, and the intervening β-strand. Similar pathways are involved in dissociation of T3 and the TRβ-selective ligand GC24 from TRβ; the TR agonist IH5 from the α- and β-TR forms; and Triac from two natural human TRβ mutants, A317T and A234T, but are detected with different frequencies in simulations performed with the different structures. Path I was previously suggested to represent a major pathway for NR ligand dissociation. We propose here that Paths II and III are also likely ligand escape routes for TRs and other NRs. We also propose that different escape paths are preferred in different situations, implying that it will be possible to design NR ligands that only associate stably with their cognate receptors in specific cellular contexts.
INTRODUCTION
The nuclear-receptor (NR) superfamily of transcription factors includes receptors for thyroid hormone (TH), retinoids, steroids, vitamin D, xenobiotics, fatty acids, bile acids and cholesterol derivatives, and orphan receptors for which ligands have not been identified (1–3). NRs play widespread roles in development, homeostasis, and disease and, consequently, are major targets for pharmaceutical development. NRs are composed of three domains, named the N-terminal domain, DNA-binding domain (DBD), and a discrete C-terminal ligand-binding domain (LBD) (1). The LBD consists of ∼250 residues and is the most highly conserved domain in NRs. Apart from ligand binding, it contains dimerization surfaces and cofactor binding sites. The DBD is formed by ∼70 residues, which fold into two zinc-finger motifs, and is responsible for recognizing specific DNA response elements. The N-terminal domain varies markedly between receptors. It may contain from 24 residues (vitamin D receptors) up to more than 600 residues (mineralocorticoid receptors). The N-terminal domain contains ligand-dependent and ligand-independent transactivation functions (1). To some extent, the three domains are modular to the point that the LBD of one receptor can be linked to the DBD of another receptor in such a way that the hybrid receptor maintains activity, responds to the hormone of one receptor, and regulates the transcription of the gene of the second receptor (4). NR ligands bind to the LBD, thereby influencing NR subcellular localization, coregulator recruitment, oligomerization, and activities of the receptor N-terminal and DBDs (5). X-ray structures of many NR-LBDs reveal that the ligand is completely buried in the hydrophobic core of the domain. However, most NR-LBD structures lack obvious entry/exit routes for the ligand (1,2,6–8). Thus, significant conformational rearrangements must accompany ligand entry and exit from the enclosed pocket. The portion of the estrogen receptor (ER) and peroxisome proliferator activated receptor (PPAR) LBDs that envelopes the ligand becomes highly disordered in the absence of ligand and may even adopt a highly mobile molten globular state (9–12). Thus, ligand entry and exit probably involve rearrangements within this part of the LBD. The precise nature of these rearrangements is not understood.
Analysis of static x-ray crystal structures of NR LBDs suggests one possible entry/exit route. Comparisons of NR LBDs in complex with agonists or antagonists, and a few available unliganded LBDs, reveal that ligand induces tight packing of the LBD C-terminal helix (H) 12 against the body of the receptor (13,14). This event occludes part of the specific corepressor-binding site that forms on the unliganded NR surface and induces formation of coactivator binding site on the liganded NR surface, thereby influencing gene expression (15). It is proposed that repositioning of H12 also regulates access of ligand to the pocket. Ligand would enter the pocket under H12, which then folds over and traps the ligand until H12 becomes displaced and ligand is released (16–18). Accordingly, many studies document a functional connection between the NR H12 agonist position and stable ligand binding. Factors that stabilize H12 usually reduce rates of ligand dissociation, and factors that destabilize H12 increase rates of ligand association (9,11,12,19,20).
Analysis of NR structures suggests that there may be other possibilities. Determination of temperature factors (B-factors) within TR x-ray crystal structures, which provides information about the distribution of electron density and serves as an indirect indication of protein mobility, reveals that the H1–H3 loop region and the associated β-sheets are highly flexible (6,21–25). Further, the surfaces of the liganded retinoic acid receptor (RAR) and both unliganded and liganded PPAR LBDs exhibit visible cavities within the H1–H3 loop/β-sheet regions (16,26). Thus, the H1–H3 loop/β-sheets could harbor a site of ligand entry or release (6).
At best, however, x-ray crystal structures represent snapshots of particular protein conformations and provide only limited information about mobility. Further analysis of single x-ray crystal structures is unlikely to help us distinguish 1) whether H12 or the H1–H3 loop region represents a plausible entry/exit route for various NRs, 2) whether other entry/exit routes exist, and 3) the dynamic structural alterations involved in ligand entry and exit.
Molecular dynamics (MD) simulations methodologies have been used for the study of the molecular mobility of proteins in several contexts with great success and can be modified to examine interactions of proteins with their cognate ligands (17,28–31). Two groups have applied different MD techniques to examine how retinoic acid (RA) might dissociate from the RAR LBD. Karplus and colleagues used a technique called locally enhanced sampling (LES) MD, which detects random fluctuations in the protein structure that permit buried ligands to escape, and concluded, essentially, that RA dissociates through a single exit channel formed by opening of H12 (17). Schulten and colleagues employed a technique called steered MD, in which ligand is forced out of the protein along particular paths proposed by the investigator, and concluded that ligand could either escape under H12 or through expansion of the small surface aperture near the H1–H3 loop (31). Thus, MD simulations confirm that the RAR H12 region contains one likely site RA entry/exit route, but studies of Schulten et al. suggest that RA can dissociate through at least one alternate pathway.
The application of MD methodologies to the problem of ligand dissociation from NRs is subject to some limitations. First of all, every simulation requires an adequate initial structure, which usually can be obtained from crystallographic models. The quality of these structures depends on their resolution and completeness and may possibly be affected by crystal packing effects on the observed structures. Furthermore, ligand dissociation dynamics is extremely slow in comparison to the timescales accessible to current simulation techniques and computer resources: The mean residence time (half-life) of the ligand inside the LBD is of several minutes (32), which is several orders of magnitude longer than the longest simulations ever reported to date. This does not mean necessarily that the actual event of ligand dissociation takes so long, but it is clear that conformational sampling cannot be done effectively in a conventional MD simulation. Other well-known shortcomings also affect the simulations of proteins: the use of classical effective potential energy functions, which ignores quantum effects (e.g., proton transfer), the lack of knowledge about the protonation state of acid and basic residues, which is particularly critical for histidine residues, and the need for a realistic modeling of the solvent, to name a few. Despite that, MD simulations can provide valuable insights into the complex dynamical behavior of the NRs.
The thyroid hormone receptor (TR) has received considerable attention in terms of LBD structure and pharmaceutical development (6,21–25,27,33,35). There are two TR isoforms, named TRα and TRβ, which are products of different genes. The LBDs of these two isoforms share 86% similarity in their amino acid sequences and the DBDs are 88% similar, but there is no similarity between their N-terminal domains. The LBDs of both isoforms contain ∼260 residues, which correspond to ∼60% of these receptors' full length. Their structures are very similar and, in particular, only a single amino acid residue differs in their ligand-binding pocket (22). Both isoforms bind to the same natural ligands T3 and T4 (Fig. 1, a and e) but are found in different concentrations in different tissues. TRβ is found particularly in the liver, whereas TRα is found predominantly in the heart. The possibility of modulating the receptor activities separately is of great pharmaceutical value. For instance, TH analogs that bind selectively to TRβ, such as IH5, GC-24, or GC-1 (Fig. 1, c, d, and f), reduce body fat content and circulating levels of cholesterol, triglycerides, and lipoprotein Lp(a) without eliciting other unwanted effects of THs (34,36–40). TR antagonists could act as rapid treatments for elevated TH (hyperthyroidism) and for cardiac arrhythmias (14,33,41). Improved understanding of the steps involved in formation of stable TR-ligand complexes and the subsequent dissociation of ligand should be helpful for developing these classes of compounds and other NR ligands. The LBDs of both TR isoforms have been crystallized in complex with the major form of TH (triiodothyronine, T3) and several alternate agonists (6,21–23,27,33–35). Also available are structures of TRβ mutants that arise in an inherited human disease, TH resistance syndrome (RTH), and exhibit enhanced rates of T3 dissociation in vitro (24,25).
FIGURE 1.

Structures of TR ligands. Simulations were done with (a) T3, (b) Triac, (c) IH5, and (d) GC24. The ligand T4 (e) is an important natural ligand, and the ligand GC1 (f) is a synthetic TRβ-selective agonist.
In this work, we apply a slight variant of the LES MD simulation technique to several representative TR structures to assess likely pathways of ligand dissociation. We detect three competing ligand escape paths from TRs: one near H12, one that involves rearrangements in the H1–H3 loop/β-sheets, and a novel escape route that involves formation of an aperture between H8 and H11. We propose that these pathways are general for the NR family but that different pathways may predominate in different contexts. Overall, we report here results of nearly 70 LES MD simulations and discuss the implications of our findings within the context of recent experimental works on the mechanisms of ligand binding and dissociation from NRs.
METHODOLOGY
X-ray crystal structures
Coordinates for LBD structures used in the simulations were obtained from Prof. Robert Fletterick's laboratory home page for TRα1 isoform refined to 2.0 Å resolution (6); the Protein Data Bank for structures of TRα1 bound to IH5, TRβ bound to IH5 and GC24, and the Triac-bound mutants TRβA234T and TRβA317T (PDB ids and resolution: TRα1-IH5: 1NAV, 2.5 Å; TRβ-IH5: 1NAX, 2.7 Å; TRβ-GC24: 1Q4X, 2.8 Å; TRβ-A234T-Triac: 1NQ0, 2.4 Å; TRβ-A317T-Triac: 1NQ2, 2.4 Å) (24,25,27); and our unpublished data for TRβ bound to T3 at 2.6 Å resolution. Some missing residues were modeled in all structures, particularly the Ω-loop in TRβ structures. Structures were locally minimized with the LBFGS (42) optimization algorithm to relieve bad contacts in the computer package TINKER (43). Fig. 4 was built with Molscript (44) and Raster3D (45). Figs. 5 and 6 were built with the Visual Molecular Dynamics package (VMD) (46). The probe radius used to build the surfaces on Fig. 5 was 1.4 Å, which is the default value in VMD.
FIGURE 4.
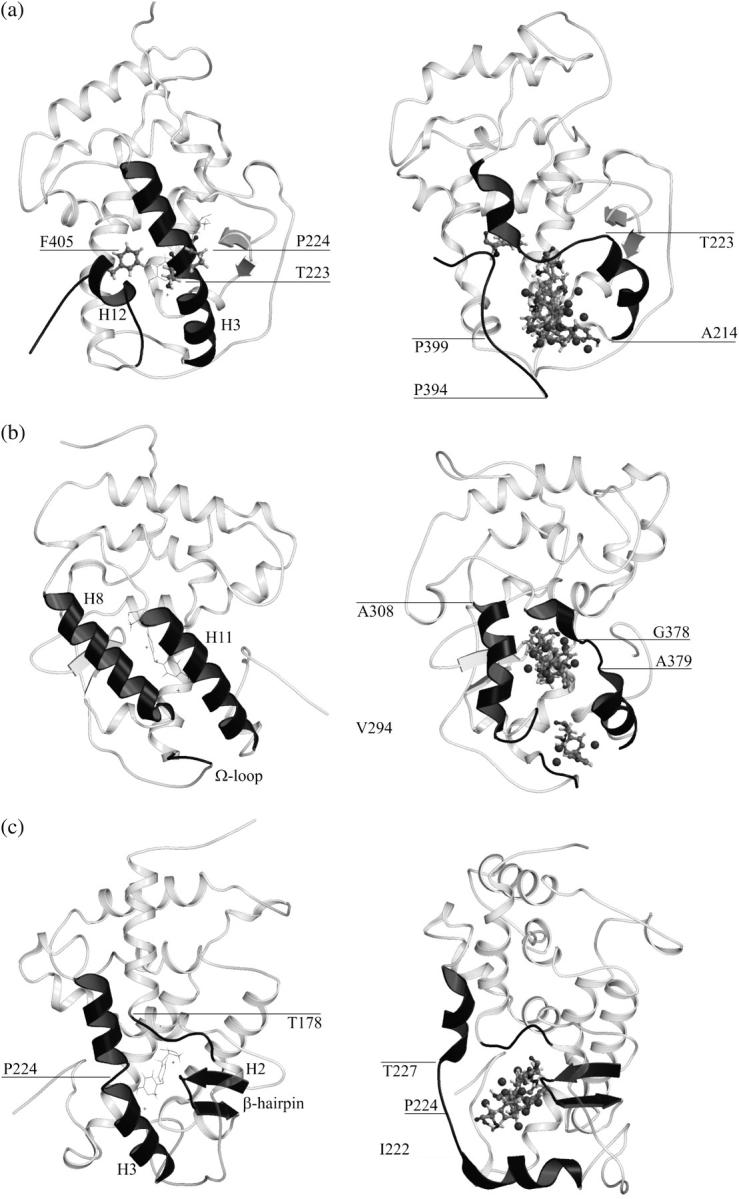
Three ligand escape paths. Snapshots of the simulations showing global views of the paths found for T3 escape from TRα1 are on the right. Corresponding views of the crystallographic structure are on the left. (a) Path I, (b) Path II, and (c) Path III. T3 molecules are shown as lines on the left and as ball and sticks on the right. Some T3 replicas were omitted for clarity.
FIGURE 5.
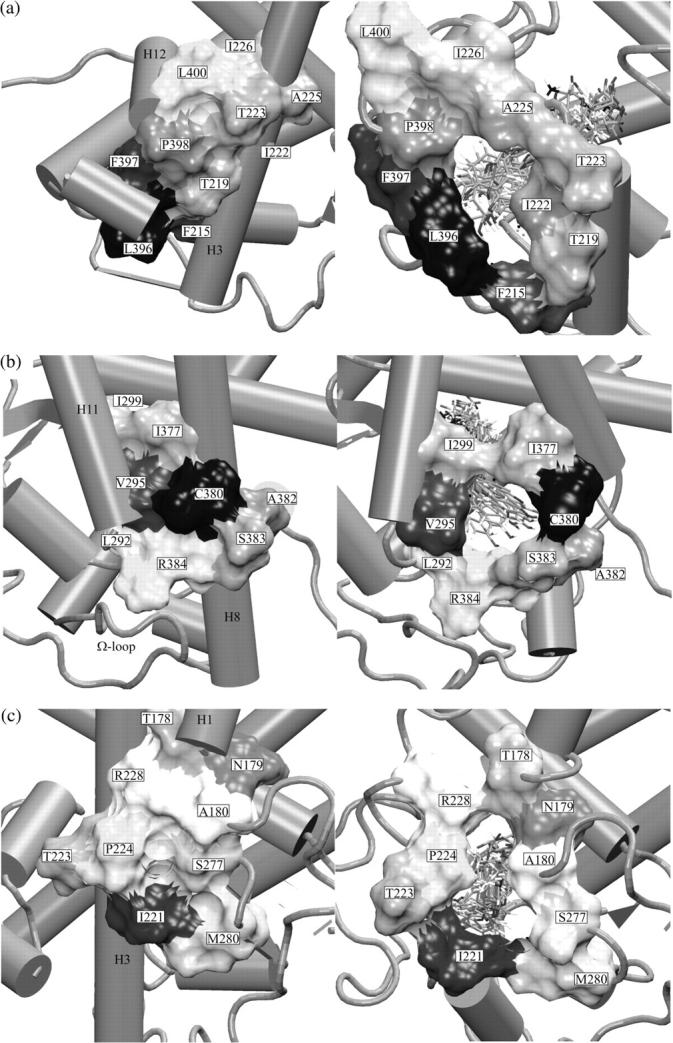
Formation of ligand escape cavities. The relative position of amino acids residues are presented in the crystallographic structure (left) and in a snapshot of the initial stages of the simulation (right). The images are from (a) Path I, (b) Path II, and (c) Path III. Different ligand (T3) copies are shown by sticks.
FIGURE 6.

Ligand-bound structures of the NR LBDs suggest the generality of ligand escape pathways in the NR superfamily. The structures are (a) ER-estradiol complex; (b) ER bound to the selective antagonist, 4-hydrotamoxifen; and (c) the GR-antagonist (R486) complex. (d) GR receptor bound to dexamethasone. (e) The AR bound to dihydrotestosterone. (f) Another dexamethasone-GR structural model. (g) The PXR bound to hyperforin. (h) The FXR complexed with the high affinity ligand fexaramine. (i) The PPARγ-rosiglitazone complex. Structures a–c suggest the displacement of the H12 apart from H3 as in Path I. Structures d–f lack the Ω-loop, and the ligand is solvent accessible in the region between H8 and H11, similar to Path II. Structures g–i ligands have extensions that abut the mobile region comprised by the H1–H3, suggesting a dissociation pathway similar to Path III.
MD simulations
All simulations were performed with the TINKER package (43). Before any data analysis, we performed 100 ps simulations on minimized structures with thermalization at every 1 ps at room temperature (300 K) for all structures with one ligand. These thermalized structures were used as starting configurations for the control simulation and for simulations presented here with multiple copies (LES). The control simulations, aimed at testing the overall validity of our model (Fig. 2), were conducted in the NVE ensemble using conventional MD techniques for one ligand copy and lasted 1 ns.
FIGURE 2.

Accessing the validity of the simulations. (a) Convergence of the cumulative mean-square displacement per atom in the control simulation (see Methodology). The small slope of the line that best fits the curve in the last 200 ps (0.27 Å ns−1) shows that the observed mobility is due to fluctuations around an average structure. (b) MD simulations reproduce relatively well the molecular mobility in TRs. Comparison of B-factors obtained from the TRα1 crystal structure with those observed in the 1 ns control MD simulation performed with the TRα1 crystal structure.
All LES simulations were performed in the NVE ensemble with the velocity Verlet algorithm to integrate the equations of motion (47). Short range van der Waals interactions were cut off at 9 Å. No cutoff was used for electrostatic interactions (all interactions were directly computed). The systems comprised the ligands and the protein in vacuum, so no periodic boundary conditions were applied.
Measures of protein mobility in the control simulation
Time-averaged cumulative root mean-square deviation
We use the cumulative, time-averaged, root mean-square deviation (RMSD) to probe the convergence of the fluctuations experienced by the protein in the control simulations. The RMSD is computed by summing the deviation of all atoms from the initial structure from the start of the simulation (t = 0) up to time t and dividing by t:  If the observed mobility results from fluctuations around an average structure, 〈RMSD〉 must be constant for large t, otherwise it is expected to increase steadily.
If the observed mobility results from fluctuations around an average structure, 〈RMSD〉 must be constant for large t, otherwise it is expected to increase steadily.
Calculation of temperature factors
The Debye-Waller factor (Temperature factor, B-factor) corresponds to the theoretical temperature factor given by the mean-square deviation of atomic positions, namely, 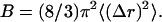 We have computed B from MD control simulations and compared it to the experimental temperature factors according to previously published protocols (31).
We have computed B from MD control simulations and compared it to the experimental temperature factors according to previously published protocols (31).
LES simulations
LES simulations were proposed by Elber and Karplus and applied originally to the problem of carbon monoxide escaping through myoglobin (28). The method is designed to the study of the molecular motions of a small part (e.g., a ligand) of a given large molecular system (the ligand-protein complex). To obtain a broader sampling of the structure and dynamics of the system, the subset of interest is replicated and the simulation is performed for the replicated set in the presence of a unique copy of the rest of the system. This method has been extensively used to study ligand diffusion through globins, and several results were confirmed experimentally (48–50). In a ligand-protein complex, for example, the ligand may be replicated several times. The ligands do not interact with each other, and the protein-ligand interactions are scaled according to the number of ligands present. Since there are several copies of the ligand, the probability of observing a rare event (e.g., ligand dissociation) is increased. Furthermore, since the ligand-protein interactions are scaled by the number of copies, the energy barriers that must be overcome to observe ligand dissociation are also decreased, and therefore the ligand dissociation rates are increased exponentially (51). There have been important theoretical developments of the LES technique: Straub and Karplus have shown that energy equipartition is not satisfied by the conventional implementation of LES and that the kinetic energy of the ligands increases as the number of ligands increase (52). Ulitksy and Elber have shown that time-dependent and several thermodynamic properties are not well represented by LES. They have proposed a correction to LES (cLES) that increases the accuracy of the temperature distribution and yields the correct time-dependent properties of the enhanced sampled subset of the system (51,53). They have shown, for instance, that the diffusion rates are well reproduced by cLES, as opposed to the conventional LES technique, which yields much faster diffusion.
The NR-LBD-ligand complex possesses some very particular properties that dictate the choice of a suitable form of the LES implementation: First, in this type of problem one wishes primarily to search for likely pathways for ligand escape from the LDB pocket. Second, ligand dissociations from the protein are regarded as rare events, given that the (inverse) escape rate is experimentally determined to be several minutes (23). Therefore, to observe dissociation in a simulation, the technique must provide three main features: i), the enhanced sampling provided by the number of ligand copies, ii), lowering of the energy barriers provided by the reduced potentials, and iii), faster diffusion rates. The necessity of large diffusion rates prevents us from using the corrected cLES method of Ulitsky and Elber (notice that cLES was not used either in the study of RA dissociation from its NR (17)). Furthermore, the ligands are relatively large (29–51 atoms in our case) and have several intramolecular degrees of freedom. Thus, the shortcomings associated with high subsystem temperatures should be avoided for the ligands internal motions as well. Following these guidelines, we have implemented a variant of the LES method in the TINKER (43) package in which the ligand-protein (and protein-ligand) interaction potentials are divided by the number of copies, whereas the interaction between ligands is null. The masses of the ligands are not scaled, and the intramolecular potentials of the ligands and of the protein amino acids are treated as full potentials. In this way, the dynamics of the system is fully Newtonian and the average kinetic energy per atom is 3kBT/2, independent of the number of the copies used (energy equipartition is satisfied). Unlike cLES, the drawback of our implementation is that the dynamics of the ligand copies will not be mapped onto the dynamics of a single-ligand protein system. Therefore, although energy equipartition is satisfied, the time-dependent properties will not correspond to the time-dependent properties of a conventional ligand-protein system. This implementation is related to the thermalization scheme proposed by Straub and Karplus to overcome the energy equipartition problem for nonNewtonian systems (17,27,28,52). From our implementation we would not expect to observe the time dependence of ligand escape, but we certainly hope to provide valuable structural insights into the mechanisms of ligand dissociation from the TRs.
Initial atom velocities are randomly attributed according to the Boltzmann distribution for the desired temperature of 300 K. The kinetic energy (and, hence, total energy) of the system increases with the number of ligand copies.
Individual hormone and protein atoms are explicitly treated; i.e., all atoms were included in the MD simulations. The potential energy parameters for the protein atoms were obtained from the OPLS-AA force field for protein simulation (54). Simulations were performed with the minimum number of ligands such that dissociation was observed in the timescale of our simulations (∼150 ps), namely, 10–50 copies of the T3 and 50 copies of other ligands. We also performed LES simulations of dissociation of 9-cis-retinoic acid dissociation from the RAR (not shown; part of these results appeared in Sonoda et al. (55)). Our simulations use OPLS-AA force field parameters, whereas previous studies used CHARMM parameters (17). Nevertheless, we observed RA dissociation through Path I in similar timescales, suggesting that simulated ligand dissociation pathways are independent of details of force field parameters and that our implementation of LES (55,56) can be directly compared to those of others (17).
Ligands
Our simulations treat the ligands as fully flexible molecules. The van der Waals and torsional parameters for the T3, IH5, GC24, and Triac molecules were obtained by group analogy in the OPLS-AA set (57), except for the iodine atoms for which the van der Waals interaction parameters are those reported in Blaney et al. (58). Partial atomic charges for T3 and Triac were computed from the optimized structure at the MP2/Lanl2DZ level of theory. This basis set was chosen because of the iodine atoms in these ligands. Charges for IH5 and GC24 were calculated from the optimized structures at the Hartree-Fock 6-31G(d,p) level of theory. All ab initio calculations were performed with Gaussian98 (59). The charges for T3 and Triac were computed with the Mulliken protocol because ESP fitting protocols such as Merz-Kollman or ChelpG are not parameterized for iodine. Although the use of ESP charges is recommended for the OPLS force field, the use of Mulliken charges in this case is not critical due to the approximate character of the interaction between the ligands and the protein. The charges for IH5 and GC24 where computed by the Merz-Kollman scheme. All the parameters are supplied as Supplementary Material.
RESULTS AND DISCUSSION
MD simulations reproduce observed molecular mobility in TRα
First, to address whether the mobility of the protein structure observed in our control simulation is a consequence of fluctuations around an average structure (and not a constant drift from the initial structure), we have computed the cumulative time-averaged root mean-square displacement. Such a measure of the mobility must converge if the protein motions are characterized by fluctuations around an average structure. As shown in Fig. 2 a, our 1 ns control simulation exhibits a 〈RMSD〉(t) drift of only 0.27 Å ns−1 for the last 200 ps, revealing that, indeed, there is no significant drift of the positions from the initial structure and, therefore, the mobility observed is reasonably well converged.
Since the temperature factors (B-factors) observed in actual crystal structures represent one indication of protein mobility, it is possible to obtain a crude indication of whether MD simulations provide realistic information about actual protein mobility by comparing motions observed in MD simulations with B-factors derived from the crystal structure. The results of such a comparison, performed with the rat TRα1 LBD in complex with T3 resolved to 2.0 Å (6), are shown in Fig. 2 b. Overall, the same areas of TRα that exhibit high mobility in the simulation also exhibit high B-factors in the crystal, indicating that our MD simulations provide useful information about the TR-LBD mobility. Some discrepancies are clearly present. Surface amino acids Pro193-Ser199 (H2) and Ile360-Trp364 (top of H11) display higher mobility in the simulation than in the crystal. This is likely due to the fact that both regions of the TR-LBD engage in contacts with neighboring LBDs in the crystal (6), as opposed to our TR monomer structure simulations in which these contacts are obviously absent. This simulation also confirms that the overall structure of the protein is conserved even in vacuum in the timescale of the simulations presented here. As already mentioned, conventional MD simulations do not detect dissociation of buried ligands from proteins; these events occur on timescales that are beyond the reach of current computing techniques. Accordingly, we did not detect dissociation of bound T3 from the TRα-LBD during the 1 ns control simulation.
To test our LES simulations for unphysical results (for instance, unphysically close contacts), we have computed the intermolecular interaction energy of the system while varying the number of copies. We have not found any unrealistic intermolecular interaction energies for any number of ligand copies (Supplementary Material, Fig. S1).
Three pathways for T3 dissociation from TRα1
We performed several sets of LES simulations with the TRα1 crystal structure, described above, in the presence of 10–50 T3 copies. Ligand dissociation was detected frequently and involved rearrangements of the region of the LBD that envelops T3. More surprisingly, T3 dissociated from the LBD via three distinct routes rather than a single preferred pathway. A schematic of Paths I–III is shown in Fig. 3. Ligands dissociate either i), through a space between H3 and H11 that lies under H12 and is vacated by repositioning of H12 as predicted by models described in the Introduction; ii), between H8 and H11 and the Ω-loop in a pathway that has never previously been observed in MD simulations or predicted from analysis of static protein structures; or iii), through an aperture between H3, the β-hairpin formed by β-sheets S3 and S4, and the loop between H1 and H2 and analogous to previously proposed entry or escape routes within this region of TR, RAR, and PPAR LBDs (6,16,26). Path I was observed most commonly in our simulations (14 times in 33 simulations performed with T3), Path II relatively infrequently (6 times in 33 simulations), and Path III almost as frequently as Path I (10 times in 33 simulations) (Table 1).
FIGURE 3.

Schematic representation of the three paths. (a) In Path I, H3 breaks down in two helices and H12 swings apart from it, forming the escape cavity. (b) The joint bending of H8 and the Ω-loop away from H11 allows ligand escape in Path II. (c) In Path III, the breakdown of H3 allows for the formation of a cavity between it and the β-hairpin through which ligands escape. Arrows indicate protein movements that from the bound sturcture (dotted) allow the ligands to escape.
TABLE 1.
Escape paths found in separate MD simulations with TRα-T3
| Number of copies | Escape paths found | |||||
|---|---|---|---|---|---|---|
| 13 | II | —* | I | |||
| 14 | III | II | III | |||
| 15 | I | I | I | |||
| 16 | I | I | —* | |||
| 17 | I | III | I | |||
| 18 | II | III | III | |||
| 19 | —* | II | I | |||
| 20 | III | II | I | |||
| 25 | III | I | III | |||
| 50 | I | I | III | I | II† | III |
Three simulations were performed with 13–25 copies, and six simulations were performed with 50 copies, as shown.
No escape found during the entire course of the simulations (∼150 ps).
In this simulation, three copies left the binding pocket through Path I, whereas the other 47 left through Path II.
Ligand escape cavities
The precise events that result in formation of ligand escape cavities and the nature of these cavities as observed in our simulations are described in detail in this section. As discussed previously, the reliability of the simulations decreases, while the positions of the ligands diverge. Therefore, our analysis considers two phases of the simulations separately. The very first stages of the simulations, when the ligands approximately share the same positions, are employed to identify the protein-protein contacts that are broken to form an aperture in the protein surface which permits ligand escape. We propose that the residues involved in these contacts should be explored in detail by mutational analysis or by means of other simulation techniques. Second, we describe the overall aspects of the pathways encountered, which include protein motions that take place after the dissociation of several ligands. Interpretation of these motions requires care, since the fluctuations may be a consequence of the systems approximations. These fluctuations provide insights into the stability of different parts of the protein but may not be interpreted as movements that one would actually expect in a real system.
Dissociation along Path I
Path I involves displacement of H12 (Figs. 3 a and 4 a). The sole difference between events observed here and in previous simulations performed with RARs is that TRα H3 breaks into two segments during the simulation, whereas RAR H3 tends to stiffen and forms an extended α-helix ((17), data not shown, see Discussion). Outward motion of the N-terminal H3 segment helps to create the escape route.
The rearrangements that lead to ligand escape involve the regions of the TR that pack against the first thyronine ring of the ligand (6). First, H12 and the associated H11–H12 loop dislocate from H3. This is followed by simultaneous rearrangements in H12 and H3: i), H12 undergoes denaturation and unwinding between amino acids Leu400–Phe405. Phe405, which lies on the inner face of H12 and contacts ligand directly in the TR structure, faces inward toward the pocket throughout the simulation; and ii), H3 breaks into two helices, which are preserved throughout the remainder of the simulation, separated at amino acid residues Thr223 and Pro224. The lower portion of H3 moves outward, opening up the interior of the LBD. Denaturation of H12 and breakdown of H3 are followed by further dislocation of the H11–H12 loop (Thr394–Pro399) from the lower part of H3 (Ala214–Thr223) as H3 moves outward. T3 finally passes through the cavity formed between the H11–H12 loop and the lower part of H3.
The Path I escape cavity is shown in detail in Fig. 5 a. Essentially, the cavity is formed by disruption of hydrophobic interactions between Thr223 on H3 and Leu400 on H12 and between Thr219 (from H3) and Pro398 and Leu396 (both in the H11–H12 loop) as H3 pulls away from the H11–H12 loop and H12.
Dissociation along Path II
The main features of Path II are separation of H8 and H11 and concurrent dislocation of the associated Ω-loop that links H2 to H3 (residues Pro200–Asp211) (Figs. 3 b and 4 b). Ω-loops are frequently found in globular proteins and have been reported in a number of instances to work as “lids” to control enzyme substrate binding and release (60).
The regions of TR that rearrange in ligand escape (H11 and H8) participate in hydrophobic interactions with the phenyl rings of the ligand, and His381 on H11 also forms a hydrogen bond with the first thyronine ring. The dynamics of the pathway evolve as follows: H11 undergoes local denaturation at residues Gly378 and Ala379. The integrity of H8 is preserved, but H8 residues Val295–Ala308 swing apart from H11 as the ligand moves between the helices, disrupting their native contacts. The Ω-loop bends together with H8 as it pulls apart from H11, as seen in Figs. 3 b and 4 b. Val295 on H8 hydrogen bonds with the side chain of Asp208 at the start of the simulation, and this contact is preserved throughout the rest of the simulation, leading to joint displacement of H8 and the Ω-loop.
The escape cavity is shown in detail in Fig. 5 b. Cys380 (H11), in close contact with Val295 and Arg284 (H8) at the start of the simulation, pulls apart from both of these amino acids thereby permitting ligand escape. The cavity border comprises mostly hydrophobic residues: Cys380, Ser383, Arg384, Ile377 (H11), and Val295, Ile299 (H8).
Dissociation along Path III
Path III involves a region of TRα that was previously proposed to represent a ligand entry route (6) and strongly resembles the mechanism proposed for the dissociation of RA from RARγ on the basis of steered MD simulations by Schulten et al. (31). Here, ligands leave on the opposite side of the receptor from H12 via an aperture formed between H3, the β-hairpin formed by β-sheets S3 and S4, and the loop between H1 and H2 (Figs. 3 c and 4 c).
The specific residues directly involved in Path III are within the H1–H2 loop (Thr178–Gly182), the lower (N-terminal) portion of H3 (Lys220–Val229), and the β-hairpin (Thr275–Met280). Several amino acids that directly contact T3 (Ile221, Ile222, Ala225, Arg228, and Leu276) also participate. The dynamics of the pathway evolve as follows: H3 breaks into two helices—residues Leu212–Pro224 and Ile226–Lys234, as observed for Path I. The lower helical portion of H3 (amino acids 212–224) dislocates from the associated β-hairpin and moves outward. The internal structure of the β-hairpin is conserved while it becomes displaced and moves away from H3 (Figs. 3 c and 4 c).
The escape cavity is shown in Fig. 5 c. The region that unfolds is hydrophilic. Before unfolding, this region of the TRα is stabilized by hydrogen bonds between different amino acid residues and between amino acid residues and the carboxyl group of the ligand (6,22). In particular, the hydrophilic interaction between Arg228 (in the pocket at the top of H3 and in contact with the carboxyl group of the ligand at the start of the simulation) and the side chain of Ser277 (in the pocket in the β-sheets) is broken during ligand escape, and these residues eventually form opposite ends of the escape cavity at the end of the simulation. Interactions between Ser277 and Pro224 (H3) and Arg228 and Ala180 (H1–H2 loop) are also lost. The resulting loss of contacts between Pro224 and surrounding residues probably increases the helix breaking activity of the Pro residue and facilitates H3 disruption, an essential feature of this pathway. Each of the aforementioned amino acid residues and residues Ile221, Asn179, and Thr178 form the border of the cavity that permits ligand escape. Unlike escape cavities involved in Paths I and II, the Path III escape cavity is hydrophilic and will be more favored in aqueous solution.
Different escape pathways predominate in different TR structures
Next, we investigate whether Paths I–III are general for both α- and β-TRs and alternate TR ligands by performing LES simulations with structures of TRβ in complex with T3 (I. Polikarpov, unpublished) and the highly TRβ-selective TR agonist GC-24 (27), and with structures of both TR isoforms in complex with the high affinity TRβ-selective agonist IH5 (Pdb IDs 1NAV; 1NAX). In addition, we performed simulations with structures of two human TRβ mutants, A234T and A317T, that arise in the human syndrome of resistance to thyroid hormone (RTH) and exhibit increased rates of ligand dissociation in vitro (24,25). Simulations were performed six times with 50 copies of each ligand and utilized x-ray crystal structures that were refined to high (2.4–2.8 Å) resolution (see Methodology).
Overall, we detect the same dissociation pathways that were identified for TRα (Table 2). Paths I and III appeared frequently. Path II (which appeared at low frequency with TRα) was only detected in simulations with TRβ mutants and once in a simulation that utilized a TRβ/T3 model (not shown), although this structure was solved at relatively low resolution (3.7 Å; PDB id. 1BSX (21)) and may not yield reliable results. Thus, Paths I and III, at least, are common to both TRs.
TABLE 2.
Escape pathways found for selective ligands, different isoforms, and mutants in auxiliary simulations
| System | Escape paths found | |||||
|---|---|---|---|---|---|---|
| TRβ with T3 | I | I | I | I | I | I |
| TRβ with GC24 | III | III | III | III | III | III |
| TRβ with IH5 | I | I | I | I | III | I |
| TRα1 with IH5 | III | III | I* | I | III | I |
| TRβ A317T with Triac | II | I | III | II | I | II |
| TRβ A234T with Triac | II | II | II | II | II | II |
All simulations here were performed with 50 copies of the ligand.
One of the 50 copies left the binding cavity through Path III.
More interestingly, individual escape paths were detected at different frequencies in different contexts (Table 2). LES simulations performed with the TRα-T3 structural model detect Path I and Path III in relatively even proportions and Path II at low frequency (Table 1), but LES simulations performed with structural models of TRβ-T3 only detected Path I (six of six simulations, Table 2). Similarly, simulations performed with the TRα-IH5 structural model detected Paths I and III with equivalent frequencies (each in three of six simulations), whereas Path I predominated with the TRβ-IH5 structural model (five of six simulations). Nevertheless, Path III predominated in simulations that utilized the TRβ-GC24 model (six of six simulations).
The TRβ RTH mutants also exhibited unique preferences for particular dissociation paths; simulations performed with TRβA234T revealed a strong preference for Path II (six of six simulations) and a mixture of paths for TRβA317T mutant (Path I: two simulations; Path II: three simulations; Path III: one simulation). Although these preferences were detected in a small number of simulations, we suggest that they are consistent with observed features of TRα and TRβ crystal structures and reflect meaningful differences in the choice of dissociation paths (see Concluding Remarks).
CONCLUDING REMARKS
In this study, we used x-ray structures of liganded TR-LBDs in LES simulations to identify ligand escape pathways from the enclosed TR pocket. As expected, T3 dissociates from TRα1 through a cavity formed by dislocation of H12 from H3 and H11 (Path I). Nevertheless, T3 can also dissociate through a completely unsuspected escape cavity formed by joint displacement of H8, H11, and the Ω-loop (Path II) or through a hydrophilic cavity formed by rearrangements within the H1–H3 loop and associated β-sheets (Path III). Paths I and III were also detected in auxiliary LES simulations that explored dissociation of T3 and GC24 from TRβ and IH5 from both TRs, albeit at different frequencies, and Path II was detected in simulations that utilized TRβ RTH mutants. Thus, we propose that TR ligands dissociate from the LBD along three competing pathways, rather than via a single entry/exit route.
Functional evidence supports the idea that TR ligands dissociate from the LBD in more than one way. Suboptimal packing of TR H12 over the thyroxine (T4) 5′ iodine group correlates with very high dissociation rates of T4 relative to T3, which lacks the 5′ iodine (23). Thus, destabilization of H12 (Path I) promotes increased rates of ligand release. Nevertheless, x-ray crystal structures of the TRβ RTH mutants used here reveal that increased rates of T3 dissociation are accompanied by specific increases in disorder within the H1–H3 loop (24,25). Thus, destabilization of the Ω-loop/H1-H3 loop region (involved in Paths II and III) also promotes ligand escape, as predicted in our simulations.
As described in the Introduction, NR H12 position regulates ligand association and dissociation rates, and H12 covers a possible ligand entry/exit route. This is illustrated in x-ray structures of ERα and glucocorticoid receptor (GR) in complex with tamoxifen and RU486 (Fig. 6, a–c) (61–63). Nevertheless, our analysis of static x-ray structures of a number of NR-LBDs reveals features that may be consistent with ligand escape along Paths II and III. Liganded GR and androgen receptor (AR) LBDs lack the Ω-loop and only contain a short H8 (Fig. 6, d–f) (63–66). Both of these features could favor ligand escape along Path II. Further, structures of the pregnane X receptor (PXR) bound to hyperforin (67), farnesoid X receptor (FXR) bound to fexaramine (65), and PPARγ bound to rosiglitazone (26), reveal cavities between the bottom of H3 and associated β-sheets in the case of PXR and in the H1–H3 region in the case of FXR and PPAR. This, coupled with the facts that human RTH mutations which destabilize the TR H1–H3 region result in increased rates of ligand dissociation (24,25), that steered MD simulations, indicate that RA escape from the RAR LBD H1–H3 region is energetically favorable (31), and that Path III uniquely involves expansion of a hydrophilic cavity that will be favored in living cells (Fig. 5 c), suggests that Path III is a common mode of ligand escape from many NRs.
Could the dissociation pathways described here function in reverse as ligand association pathways? Our studies do not address this question, but we note that NR H12 is implicated in regulation of the rates of ligand dissociation and association (19,20,67), whereas our preliminary results suggest that RTH mutations that enhance ligand exit (possibly through Paths II and III (Table 2)) enhance T3 dissociation rates but do not affect ligand association rates (Cunha Lima, unpublished). Perhaps Path I represents a viable ligand entry route, but Paths II and III do not.
We observe differences in relative frequencies of Paths I, II, and III in simulations performed with different structures. Paths I and III are detected at approximately equivalent frequencies in simulations performed with TRα, whereas previous LES simulations and our unpublished simulations performed with RARs only detect Path I (17,55). The likely explanation for this difference centers upon the tendency of TR H3 to break into short α-helices around a unique Pro residue not conserved in RARs (Fig. 7) or other NRs (not shown). Disruption of TR H3 promotes ligand escape via Path III through spaces vacated by the N-terminal portion of H3. RARs contain a Lys residue at the equivalent position, which lacks the propensity of Pro to disrupt α-helices, and RAR H3 stiffens during simulations to form an extended α-helix that “pinches” the pocket, precluding ligand escape through the bottom of H3 and forcing RA out under H12 (17).
FIGURE 7.

H3 of RA and TRs. The structurally important Pro residue, which could be a key amino acid required to promote dissociation through Path III, is highlighted in TR sequences.
It is not clear whether ligand escape from RARs is truly restricted to Path I or whether RA can escape from the H1–H3 loop via rearrangements that are too subtle to be detected in LES. We favor the latter possibility, since there are strong similarities between hydrophilic escape cavities in the TR H1–H3 region and detected in the RAR H1–H3 region in steered MD (31).
Our simulations also detected preferences for particular escape paths among TR isoforms, TRs in complex with different ligands and between wild-type TRβ and TRβ RTH mutants. Each of these preferences is consistent with observed features of TR structures: i), T3 and IH5 dissociate from TRβ along Path I but from TRα along Paths I and III. This is consistent with the fact that the TRβ H11–H12 region (involved in Path I) is often less stable than the equivalent region of TRα, as judged by B-factors (6,21–23). and ii), T3 dissociates from TRβ along Path I, whereas GC24 dissociates from TRβ along Path III. GC24 contains a bulky 3′ benzyl extension accommodated by rearrangements in H3 and H11 that extend the functional ligand-binding pocket (27). The GC24 extension contacts regions of TR H3 and H11 that are exposed in Path I, and we suggest that GC24 blocks Path I and favors Path III. Finally, as alluded to above, we suggest that ligand dissociates from wild-type TRβ along Path I but from TRβA234T along Path II and from TRβA317T along a mix of routes because RTH mutations destabilize the H1–H3 loop involved in ligand exit through Paths II and III (24,25).
The notion that TR and other NRs harbor multiple ligand dissociation pathways, and that different pathways may predominate in different contexts, has interesting implications for NR function and pharmaceutical design; TR ligands that are tailored to escape from the LBD along particular pathways will exhibit context selective binding. The strong TRβ selectivity of GC24 may be related to its preference for dissociation along Path III. Moreover, our studies assumed isolated LBDs, although NRs form large complexes with other coregulatory molecules that may differentially stabilize various portions of the LBD and influence the choice of entry/exit route. For example, coactivator binding stabilizes H12 over the putative Path I entry/escape route. Thus, it is possible that ligands that are committed to Path I would dissociate very slowly from TR/coactivator complexes and promote stable transcriptionally active TR complexes.
At the least, our simulations identify flexible regions of TR that could harbor compounds with bulky extensions such as the GC24 3′ benzyl group, accommodated within the region of TRβ that rearranges in Path I (14,27). Perhaps other regions of flexibility identified in our simulations could also harbor bulky extensions with useful properties (14,36).
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grants 99/03387-4 to I.P. and 03/09361-4 to M.S.S.), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grant 401913/2003-1 to M.S.S.), the Coordenação de Aperfeiçoamento de Pessoal do Nível Superior (CAPES), and the National Institutes of Health (DK41482, DK51281, and DK64148 to J.D.B.) for financial support. J.D.B. has proprietary interests in and serves as a consultant and deputy director to Karo Bio AB, which has commercial interests in this area of research.
References
- 1.Ribeiro, R. C. J., P. J. Kushner, and J. D. Baxter. 1995. The nuclear hormone receptor gene superfamily. Annu. Rev. Med. 46:443–453. [DOI] [PubMed] [Google Scholar]
- 2.Bourguet, W., P. Germain, and H. Gronemeyer. 2000. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci. 21:381–388. [DOI] [PubMed] [Google Scholar]
- 3.Gronemeyer, H., J. A. Gustafsson, and V. Laudet. 2004. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 3:950–964. [DOI] [PubMed] [Google Scholar]
- 4.Apriletti, J. W., R. C. J. Ribeiro, R. L. Wagner, W. Feng, P. Webb, P. J. Kushner, B. L. West, S. Nilsson, T. S. Scanlan, R. J. Fletterick, and J. D. Baxter. 1998. Molecular and structural biology of thyroid hormone receptors. Clin. Exp. Pharmacol. Physiol. Suppl. 25:S2–S11 (Suppl. S.). [DOI] [PubMed] [Google Scholar]
- 5.Laudet, V., and H. Gronemeyer. 2002. The Nuclear Receptor Facts Book, 1st ed. Academic Press, London.
- 6.Wagner, R. L., J. W. Apriletti, M. E. McGrath, B. L. West, J. D. Baxter, and R. J. Fletterick. 1995. A structural role for hormone in the thyroid hormone receptor. Nature. 378:690–697. [DOI] [PubMed] [Google Scholar]
- 7.Nettles, K. W., and G. L. Greene. 2005. Ligand control of coregulator recruitment to nuclear receptors. Annu. Rev. Physiol. 67:309–333. [DOI] [PubMed] [Google Scholar]
- 8.Francis, G. A., E. Fayard, F. Picard, and J. Auwerx. 2003. Nuclear receptors and the control of metabolism. Annu. Rev. Physiol. 65:261–311. [DOI] [PubMed] [Google Scholar]
- 9.Johnson, B. A., E. M. Wilson, Y. Li, D. E. Moller, R. G. Smith, and G. Zhou. 2000. Ligand-induced stabilization of PPARgamma monitored by NMR spectroscopy: implications for nuclear receptor activation. J. Mol. Biol. 298:187–194. [DOI] [PubMed] [Google Scholar]
- 10.Pissios, P., I. Tzameli, P. Kushner, and D. D. Moore. 2000. Dynamic stabilization of nuclear receptor ligand binding domains by hormone or corepressor binding. Mol. Cell. 6:245–253. [DOI] [PubMed] [Google Scholar]
- 11.Gee, A. C., and J. A. Katzenellenbogen. 2001. Probing conformational changes in the estrogen receptor: evidence for a partially unfolded intermediate facilitating ligand binding and release. Mol. Endocrinol. 15:421–428. [DOI] [PubMed] [Google Scholar]
- 12.Kallenberger, B. C., J. D. Love, V. K. Chatterjee, and J. W. Schwabe. 2003. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat. Struct. Biol. 10:136–140. [DOI] [PubMed] [Google Scholar]
- 13.Weatherman, R. V., R. J. Fletterick, and T. S. Scanlan. 1999. Nuclear-receptor ligands and ligand-binding domains. Annu. Rev. Biochem. 68:559–581. [DOI] [PubMed] [Google Scholar]
- 14.Webb, P., N. H. Nguyen, G. Chiellini, H. A. Yoshihara, S. T. Cunha Lima, J. W. Apriletti, R. C. Ribeiro, A. Marimuthu, B. L. West, P. Goede, K. Mellstrom, S. Nilsson, and others. 2002. Design of thyroid hormone receptor antagonists from first principles. J. Steroid Biochem. Mol. Biol. 83:59–73. [DOI] [PubMed] [Google Scholar]
- 15.Glass, C. K., and M. G. Rosenfeld. 2000. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14:121–141. [PubMed] [Google Scholar]
- 16.Renaud, J. P., N. Rochel, M. Ruff, V. Vivat, P. Chambon, H. Gronemeyer, and D. Moras. 1995. Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature. 378:681–689. [DOI] [PubMed] [Google Scholar]
- 17.Blondel, A., J. P. Renaud, S. Fischer, D. Moras, and M. Karplus. 1999. Retinoic acid receptor: a simulation analysis of retinoic acid binding and the resulting conformational changes. J. Mol. Biol. 291:101–115. [DOI] [PubMed] [Google Scholar]
- 18.Egea, P. F., A. Mitschler, N. Rochel, M. Ruff, P. Chambon, and D. Moras. 2000. Crystal structure of the human RXRalpha ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 19:2592–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carlson, K. E., I. Choi, A. Gee, B. S. Katzenellenbogen, and J. A. Katzenellenbogen. 1997. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 36:14897–14905. [DOI] [PubMed] [Google Scholar]
- 20.Gee, A. C., K. E. Carlson, P. G. Martini, B. S. Katzenellenbogen, and J. A. Katzenellenbogen. 1999. Coactivator peptides have a differential stabilizing effect on the binding of estrogens and antiestrogens with the estrogen receptor. Mol. Endocrinol. 13:1912–1923. [DOI] [PubMed] [Google Scholar]
- 21.Darimont, B. D., R. L. Wagner, J. W. Apriletti, M. R. Stallcup , P. J. Kushner, J. D. Baxter, R. J. Fletterick, and K. R. Yamamoto. 1998. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 12:3343–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner, R. L., B. R. Huber, A. K. Shiau, A. Kelly, S. T. Cunha Lima, T. S. Scanlan, J. W. Apriletti, J. D. Baxter, B. L. West, and R. J. Fletterick. 2001. Hormone selectivity in thyroid hormone receptors. Mol. Endocrinol. 15:398–410. [DOI] [PubMed] [Google Scholar]
- 23.Sandler, B., P. Webb, J. W. Apriletti, B. R. Huber, M. Togashi, S. T. Cunha Lima, S. Juric, S. Nilsson, R. Wagner, R. J. Fletterick, and J. D. Baxter. 2004. Thyroxine-thyroid hormone receptor interactions. J. Biol. Chem. 279:55801–55808. [DOI] [PubMed] [Google Scholar]
- 24.Huber, B. R., M. Desclozeaux, B. L. West, S. T. Cunha Lima, H. T. Nguyen, J. D. Baxter, H. A. Ingraham, and R. J. Fletterick. 2003. Thyroid hormone receptor-beta mutations conferring hormone resistance and reduced corepressor release exhibit decreased stability in the N-terminal ligand-binding domain. Mol. Endocrinol. 17:107–116. [DOI] [PubMed] [Google Scholar]
- 25.Huber, B. R., B. Sandler, B. L. West, S. T. Cunha Lima, H. T. Nguyen, J. W. Apriletti, J. D. Baxter, and R. J. Fletterick. 2003. Two RTH mutants with impaired hormone binding. Mol. Endocrinol. 17:643–652. [DOI] [PubMed] [Google Scholar]
- 26.Nolte, R. T., G. B. Wisely, S. Westin, J. E. Cobb, M. H. Lambert, R. Kurokawa, M. G. Rosenfeld, T. M. Willson, C. K. Glass, and M. V. Milburn. 1998. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor gamma. Nature. 395:137–143. [DOI] [PubMed] [Google Scholar]
- 27.Borngraeber, S., M. J. Budny, G. Chiellini, S. T. Cunha-Lima, M. Togashi, P. Webb, J. D. Baxter, T. S. Scanlan, and R. J. Fletterick. 2003. Ligand selectivity by seeking hydrophobicity in thyroid hormone receptor. Proc. Natl. Acad. Sci. USA. 100:15358–15363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elber, R., and M. Karplus. 1990. Enhanced sampling in molecular dynamics: use of time-dependent Hartree approximation for a simulation of carbon monoxide diffusion through myoglobin. J. Am. Chem. Soc. 112:9161–9175. [Google Scholar]
- 29.Quillin, M. L., T. S. Li, J. S. Olson, G. N. Phillips, Y. Dou, M. Ikedasaito, R. Regan, M. Carlson, Q. H. Gibson, H. Y. Li, and R. Elber. 1995. Structural and functional effects of apolar mutations of the distal valine in myoglobin. J. Mol. Biol. 245:416–436. [DOI] [PubMed] [Google Scholar]
- 30.Jensen, M. O., S. Park, E. Tajkhorshid, and K. Schulten. 2002. Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc. Natl. Acad. Sci. USA. 99:6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kosztin, D., S. Izrailev, and K. Schulten. 1999. Unbinding of retinoic acid from its receptor studied by steered molecular dynamics. Biophys. J. 76:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Budhu, A. S., and N. Noy. 2000. On the role of the carboxyl-terminal helix of RXR in the interactions of the receptor with ligand. Biochemistry. 39:4090–4095. [DOI] [PubMed] [Google Scholar]
- 33.Baxter, J. D., P. Goede, J. W. Apriletti, B. L. West, W. Feng, K. Mellstrom, R. J. Fletterick, R. L. Wagner, P. J. Kushner, R. C. Ribeiro, P. Webb, T. S. Scanlan, and S. Nilsson. 2002. Structure-based design and synthesis of a thyroid hormone receptor (TR) antagonist. Endocrinology. 143:517–524. [DOI] [PubMed] [Google Scholar]
- 34.Ye, L., Y. L. Li, K. Mellstrom, C. Mellin, L. G. Bladh, K. Koehler, N. Garg, A. M. Garcia Collazo, C. Litten, B. Husman, K. Persson, J. Ljunggren, and others. 2003. Thyroid receptor ligands. 1. Agonist ligands selective for the thyroid receptor beta1. J. Med. Chem. 46:1580–1588. [DOI] [PubMed] [Google Scholar]
- 35.Ye, H. F., K. E. O'Reilly, and J. T. Koh. 2001. A subtype-selective thyromimetic designed to bind a mutant thyroid hormone receptor implicated in resistance to thyroid hormone. J. Am. Chem. Soc. 123:1521–1522. [DOI] [PubMed] [Google Scholar]
- 36.Baxter, J. D., W. H. Dillmann, B. L. West, R. Huber, J. D. Furlow, R. J. Fletterick, P. Webb, J. W. Apriletti, and T. S. Scanlan. 2001. Selective modulation of thyroid hormone receptor action. J. Steroid Biochem. Mol. Biol. 76:31–42. [DOI] [PubMed] [Google Scholar]
- 37.Grover, G. J., D. M. Egan, P. G. Sleph, B. C. Beehler, G. Chiellini, N. H. Nguyen, J. D. Baxter, and T. S. Scanlan. 2004. Effects of the thyroid hormone receptor agonist GC-1 on metabolic rate and cholesterol in rats and primates: selective actions relative to 3,5,3′-triiodo-L-thyronine. Endocrinology. 145:1656–1661. [DOI] [PubMed] [Google Scholar]
- 38.Baxter, J. D., P. Webb, G. Grover, and T. S. Scanlan. 2004. Selective activation of thyroid hormone signaling pathways by GC-1: a new approach to controlling cholesterol and body weight. Trends Endocrinol. Metab. 15:154–157. [DOI] [PubMed] [Google Scholar]
- 39.Grover, G. J., K. Mellstrom, L. Ye, J. Malm, Y. L. Li, L. G. Bladh, P. G. Sleph, M. A. Smith, R. George, B. Vennstrom, K. Mookhtiar, R. Horvath, and others. 2003. Selective thyroid hormone receptor-beta activation: a strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc. Natl. Acad. Sci. USA. 100:10067–10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webb, P. 2004. Selective activators of thyroid hormone receptors. Expert Opin. Investig. Drugs. 13:489–500. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen, N. H., J. W. Apriletti, S. T. Cunha Lima, P. Webb, J. D. Baxter, and T. S. Scanlan. 2002. Rational design and synthesis of a novel thyroid hormone antagonist that blocks coactivator recruitment. J. Med. Chem. 45:3310–3320. [DOI] [PubMed] [Google Scholar]
- 42.Liu, D. C., and J. Nocedal. 1989. On the limited memory BFGS method for large-scale optimization. Math. Program. 45:503–528. [Google Scholar]
- 43.Ponder, J. W. 2001. TINKER Software Tools for Molecular Design. Version 3. 9. [DOI] [PMC free article] [PubMed]
- 44.Kraulis, P. J. 1991. Molscript—a program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24:946–950, Part 5. [Google Scholar]
- 45.Merrit, E. A., and D. J. Bacon. 1997. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277:505–524. [DOI] [PubMed] [Google Scholar]
- 46.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 47.Verlet, L. 1967. Computer simulations on classical fluids. I. Thermodynamics properties of Lennard Jones Molecules. Phys. Rev. 159:98–103. [Google Scholar]
- 48.Gibson, Q. H., R. Regan, R. Elber, J. S. Olson, and T. E. Carver. 1992. Distal pocket residues affect picosecond ligand recombination in myoglobin—an experimental and molecular dynamics study of position 29 mutants. J. Biol. Chem. 267:22022–22034. [PubMed] [Google Scholar]
- 49.Scott, E. E., Q. H. Gibson, and J. S. Olson. 2001. Mapping the pathways for O2 entry into and exit from myoglobin. J. Biol. Chem. 267:5177–5188. [DOI] [PubMed] [Google Scholar]
- 50.Brunori, M., and Q. H. Gibson. 2001. Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2:674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ulitsky, A., and R. Elber. 1993. The thermal equilibrium aspects of the time dependent Hartree and the locally enhanced sampling approximations: formal properties, a correction, and computational examples for rare gas clusters. J. Chem. Phys. 98:3380–3388. [Google Scholar]
- 52.Straub, J. E., and M. E. Karplus. 1991. Energy equilibrium in the classical time-dependent Hartree approximation. J. Chem. Phys. 94:6737–6739. [Google Scholar]
- 53.Ulitsky, A., and R. Elber. 1994. Application of the locally enhanced sampling (LES) and a mean-field with binary collision correction (cLES) to the simulation of Ar diffusion and NO recombination in myoglobin. J. Phys. Chem. 98:1034–1043. [Google Scholar]
- 54.Kaminski, G. A., R. A. Friesner, J. Tirado-Rives, and W. L. Jorgensen. 2001. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 105:6474–6487. [Google Scholar]
- 55.Sonoda, M. T., N. H. Moreira, L. Martinez, F. W. Fávero, S. M. Vechi. L. R. Martins, M.S. Skaf. 2004. A review on the dynamics of water. Braz. J. Phys. 34:3–16. [Google Scholar]
- 56.Martinez, L. 2003. Computational study fo thyroid hormone dissociation from its nuclear receptor. Master thesis. State University of Campinas, Campinas, SP, Brazil.
- 57.Jorgensen, W. L., D. S. Maxwell, and J. Tirado-Rives. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118:11225–11236. [Google Scholar]
- 58.Blaney, J. M., P. K. Weiner, A. Dearing, P. A. Kollman, E. C. Jorgensen, S. J. Oatley, J. M. Burridge, and C. C. F. Blake. 1982. Molecular mechanics simulation of protein ligand interactions—binding of thyroid-hormone analogs to pre-albumin. J. Med. Chem. Soc. 104:6424–6434. [Google Scholar]
- 59.Frisch, M. J., et al. 2004. Gaussian, Inc., Wallingford CT.
- 60.Fetrow, J. S. 1995. Omega loops: nonregular secondary structures significant in protein function and stability. FASEB J. 9:708–717. [PubMed] [Google Scholar]
- 61.Brzozowski, A. M., A. C. Pike, Z. Dauter, R. E. Hubbard, T. Bonn, O. Engstrom, L. Ohman, G. L. Greene, J. A. Gustafsson, and M. Carlquist. 1997. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 389:753–758. [DOI] [PubMed] [Google Scholar]
- 62.Shiau, A. K., D. Barstad, P. M. Loria, L. Cheng, P. J. Kushner, D. A. Agard, and G. L. Greene. 1998. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 95:927–937. [DOI] [PubMed] [Google Scholar]
- 63.Kauppi, B., C. Jakob, M. Farnegardh, J. Yang, H. Ahola, M. Alarcon, K. Calles, O. Engstrom, J. Harlan, S. Muchmore, A. K. Ramqvist, and S. Thorell, and others. 2003. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J. Biol. Chem. 278:22748–22754. [DOI] [PubMed] [Google Scholar]
- 64.Bledsoe, R. K., V. G. Montana, T. B. Stanley, C. J. Delves, C. J. Apolito, D. D. McKee, T. G. Consler, D. J. Parks, E. L. Stewart, T. M. Willson, M. H. Lambert, J. T. Moore, and others. 2002. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 110:93–105. [DOI] [PubMed] [Google Scholar]
- 65.Downes, M., M. A. Verdecia, A. J. Roecker, R. Hughes, J. B. Hogenesch, H. R. Kast-Woelbern, M. E. Bowman, J. L. Ferrer, A. M. Anisfeld, P. A. Edwards, J. M. Rosenfeld, J. G. Alvarez, and others. 2003. A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol. Cell. 11:1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sack, J. S., K. F. Kish, C. Wang, R. M. Attar, S. E. Kiefer, Y. An, G. Y. Wu, J. E. Scheffler, M. E. Salvati, S. R. Krystek Jr, R. Weinmann, and H. M. Einspahr. 2001. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc. Natl. Acad. Sci. USA. 98:4904–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watkins, R. E., J. M. Maglich, L. B. Moore, G. B. Wisely, S. M. Noble, P. R. Davis-Searles, M. H. Lambert, S. A. Kliewer, and M. R. Redinbo. 2003. 2.1 A crystal structure of human PXR in complex with the St. John's wort compound hyperforin. Biochemistry. 42:1430–1438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}