

Abstract
Long pulse saturation recovery electron paramagnetic resonance spectroscopy is applied to the investigation of spin-labeled side chains placed along a regular helix extending from 128 to 135 in T4 lysozyme. Under an argon atmosphere, analysis of the exponential saturation recovery curves gives the spin-lattice relaxation rates of the nitroxides, which depend on the nitroxide side-chain dynamics. In the presence of the fast-relaxing paramagnetic reagents O2 or NiEDDA, global analysis of the saturation recovery provides the spin-lattice relaxation rates as well as the Heisenberg exchange rates of the nitroxide with the reagents. As previously shown with power saturation methods, such exchange rates are direct measures of the solvent accessibility of the nitroxide side chains in the protein structure. The periodic dependence of the spin-lattice relaxation rates and the exchange rates along the 128–135 sequence reveal the presence of the helical structure, demonstrating the use of these parameters in structure determination. In general, multiple exponentials are required to fit the saturation recovery data, thus identifying multiple states of the side chain. In one case, multiple conformations detected in the spectrum are not evident in the saturation recovery, suggesting rapid exchange on the timescale of spin-lattice relaxation.
INTRODUCTION
SDSL has become a powerful tool for determination of structure and conformational dynamics in both water-soluble and membrane proteins of arbitrary molecular weight (1–5). The basic strategy of SDSL involves the site-specific introduction of a paramagnetic nitroxide side chain in a protein sequence, and analysis of the EPR spectrum of the spin-labeled protein in terms of a set of parameters that characterize the site in the protein fold. The most commonly used nitroxide side chain is shown in Fig. 1, and is designated R1 for convenience.
FIGURE 1.

Structure of the R1 side chain.
The most useful parameters have proven to be those related to R1 side-chain dynamics and solvent accessibility. The dynamic mode of a nitroxide side chain can in principle be quantitatively described using spectral simulation techniques (6), and this has been accomplished for the R1 side chain at noninteracting α-helix surface sites (7,8) and at solvent exposed sites in β-sheets (9). The inverse spectral second moment and inverse central linewidth of the EPR spectrum (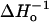 ) are semiempirical measures of nitroxide “mobility” that have proven to be useful in identifying regular secondary structure. For example, the sequence dependence of mobility measured by these quantities clearly reveals the sequence-specific secondary structure by virtue of the periodic variation with sequence position (9–14).
) are semiempirical measures of nitroxide “mobility” that have proven to be useful in identifying regular secondary structure. For example, the sequence dependence of mobility measured by these quantities clearly reveals the sequence-specific secondary structure by virtue of the periodic variation with sequence position (9–14).
By definition, the “accessibility” of an R1 side chain in a macromolecule is proportional to the Heisenberg exchange frequency (Wex) of the nitroxide with a paramagnetic exchange reagent in solution (15–17). It has been found that the accessibility measured in this way and expressed by Π, the “accessibility parameter”, is correlated with structure-based measures of accessibility of the corresponding native side chain, as computed from the crystal structure (14,18). Thus, accessibilities can be used to map structure and serve to compare the structure of a protein in solution and in the crystal (19).
The paramagnetic exchange reagents employed in the measurement of accessibility may be metal ion complexes, molecular oxygen, or organic radicals, such as another nitroxide. For O2 and complexes of Ni(II) and certain other metal ions, the spin-lattice relaxation time of the reagent is much shorter than the spin-lattice relaxation time of the nitroxide (T1e). In these cases, HE is an effective spin-lattice relaxation mechanism for the nitroxide (20), and Wex can be measured as
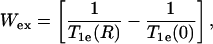 |
where 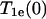 is the nitroxide intrinsic relaxation time, and
is the nitroxide intrinsic relaxation time, and 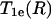 is the effective spin-lattice relaxation time in the presence of an exchange reagent. Thus, any method that gives a parameter proportional to the effective
is the effective spin-lattice relaxation time in the presence of an exchange reagent. Thus, any method that gives a parameter proportional to the effective 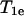 can be used to measure relative values of Wex. One such method is continuous wave (CW) power saturation, provided that
can be used to measure relative values of Wex. One such method is continuous wave (CW) power saturation, provided that 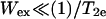 , where
, where 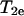 is the transverse spin relaxation time, and that the nitroxide saturates in a homogeneous fashion. These conditions are met when the correlation time of the nitroxide is much shorter than T1e (21), a common situation for R1 in proteins at room temperature. Due to the instrumental simplicity, CW power saturation has been the method of choice for most studies. Multiquantum EPR is another recently developed method that provides a T1e sensitive display that could be used in the same way (22), although it has not yet been exploited for measuring relative accessibility in proteins. When nitroxides are employed as exchange reagents, ELDOR may be employed to measure absolute values of Wex, provided T1e is known (23–25).
is the transverse spin relaxation time, and that the nitroxide saturates in a homogeneous fashion. These conditions are met when the correlation time of the nitroxide is much shorter than T1e (21), a common situation for R1 in proteins at room temperature. Due to the instrumental simplicity, CW power saturation has been the method of choice for most studies. Multiquantum EPR is another recently developed method that provides a T1e sensitive display that could be used in the same way (22), although it has not yet been exploited for measuring relative accessibility in proteins. When nitroxides are employed as exchange reagents, ELDOR may be employed to measure absolute values of Wex, provided T1e is known (23–25).
SR-EPR provides a means to measure T1e directly. In this method, saturating microwave pulses are delivered to the sample, and the recovery of the z-magnetization is monitored in real time. Analysis of the recovery curve in the absence and presence of an exchange reagent provides both T1e and the exchange rate (15,16,26,27). The T1e of a nitroxide in solution at room temperature is determined primarily by the correlation time of the nitroxide via various mechanisms (28–31). Thus, measurements of T1e for R1 in a protein in the absence of exchange reagents should provide a direct measure of nitroxide side-chain dynamics. As mentioned above, R1 dynamics are strongly correlated with protein structure, and one purpose of this study is to evaluate T1e measured by SR-EPR as a parameter for mapping protein structure and dynamics.
A second purpose of this study is to evaluate SR-EPR as a method for determining absolute values of Wex between R1 residues in proteins and fast-relaxing paramagnetic reagents in solution. In this approach, no assumptions regarding the magnitude of T2e or the homogeneous nature of the resonance line are necessary. Moreover, R1 accessibility can be expressed in a quantitative, instrument-independent fashion without recourse to calibrations of the microwave power in the resonator, as is necessary in CW power saturation methods. Finally, in the common situation of multicomponent spin populations, both T1e values and accessibilities for the individual components can be determined.
The protein selected for investigation is T4 lysozyme (T4L), a protein of accurately known structure that has been the subject of previous investigations with SDSL. Saturation recovery data are obtained for R1 residues along a continuous nitroxide scan through a short helix (128–135; Fig. 2) in the absence and presence of O2 or NiEDDA as exchange reagents. In the absence of exchange reagents, T1e is found to vary periodically along the sequence, apparently due to a corresponding variation in R1 dynamics. Accessibilities measured from absolute exchange rates in the presence of O2 or NiEDDA vary in a manner expected from the crystal structure of the protein, and are in excellent agreement with relative accessibilities determined by CW power saturation methods that are reported in a companion article (18). Collectively, the results show that SR-EPR provides a quantitative means for determination of R1 accessibility, and suggest that T1e measurements may provide an alternative method for investigation of R1 dynamics.
FIGURE 2.

Ribbon model of T4 lysozyme. The spheres mark the positions where R1 side chains were introduced, one at a time. The spheres are coded according to solvent accessibility. Highly accessible (gray); solvent inaccessible (black).
METHODS
All EPR measurements were performed using a homebuilt continuous-wave (CW) and SR-EPR spectrometer (32) equipped with a 1-mm loop-gap resonator (33). The spectrometer is controlled by a PC computer through a GPIB 488 interface. All CW-EPR spectra were recorded at 140 μW of incident microwave power at a frequency of ≈9.3 GHz. The magnetic field modulation frequency was 20 kHz, with peak-to-peak amplitude of 1 G. SR-EPR measurements were performed using a 1-μs pump pulse width and a pump power of 95 mW. The observe power was set at 140 μW at the same microwave frequency as used for the pump pulse. Each SR curve was digitized with 1024 points at a rate of either 100, 50, or 25 MHz. The analog bandwidth was limited to 50, 25, or 12.5 MHz, respectively. The number of accumulations for each measurement was either 1.28 or 2.56 million.
The spin-labeled protein sample (≈5 μL of ≈100 μM) was placed in a capillary (i.d., 0.6 mm) made of gas-permeable methylpentene (TPX) polymer. The concentration of oxygen was controlled by equilibrating the sample with the same gas that was used for temperature control, either argon or air. The concentration of NiEDDA was 3 mM. All SR-EPR measurements were made on the central 14N hyperfine line. Three independent SR-EPR curves were recorded for each sample.
Analysis of SR-EPR data
The amplitude of the saturation recovery EPR signal, f, observed on the central line of a nitroxide spin label (mI = 0) of a single population in the presence of an exchange reagent, can be expressed as:
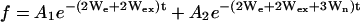 |
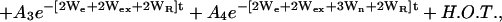 |
(1) |
where We is the electron spin-lattice relaxation rate, Wn is the 14N nuclear spin-lattice relaxation rate, Wex is the bimolecular exchange rate of the exchange reagent (oxygen or NiEDDA) with the nitroxide, WR is the rotational diffusion rate of the nitroxide, and H.O.T. stands for higher order terms (21,27). The relaxation rates are related to the corresponding relaxation times according to 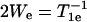 ,
, 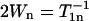 , and
, and 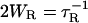 , where τR is the rotational correlation time of the nitroxide.
, where τR is the rotational correlation time of the nitroxide.
The first term represents the sum of the electron relaxation rate and exchange rate alone, whereas the rate for other components is increased by the combination of nuclear relaxation and rotational correlation rates. To extract the rate values of all relaxation processes, Haas et al. (21) performed both SR-EPR and SR-ELDOR on the same sample. In SR ELDOR, the signs of amplitudes of some components are inverted, making possible a unique determination of relaxation rates. In these studies, only SR-EPR experiments were performed, and it is not possible to extract all relaxation rates.
The correlation times for the nitroxides in T4L lie in the range of 1–6 ns, the upper bound corresponding to the rotational diffusion of the entire protein in solution at room temperature. Thus, WR for R1 is of the order 108 s−1. For correlation times in the range of 1–6 ns, Wn for nitroxides is of the order 107 s−1 (28). Thus, saturation recovery due to rotational diffusion and nuclear relaxation is essentially complete within the effective dead time of data collection set by the bandwidth of the spectrometer. On the other hand, Wex and We are of the order 105 s−1 and are the only processes observed in the recovery. Under these conditions, single exponentials reflecting only the first term in Eq. 1 are expected for a single population of R1. Indeed, the saturation recovery signals from some of the sites investigated are best characterized by single exponentials. However, it is common for the EPR spectrum of the R1 side chain in proteins to reflect two dynamic states. Thus, the SR data are analyzed in terms of single and double exponential curves.
The analysis of the experimental data was performed in stages. First, single or double exponential curves were fitted to each SR curve. From these fits, the first approximations to Wex and We were obtained. A second stage using a global analysis of the data was performed (34) using the initial fit parameters as inputs. For each residue, theoretical parameters were simultaneously fit to the set of six SR-EPR curves, two each for the sample in Ar, O2, and NiEDDA. The χ2 function
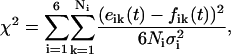 |
(2) |
was used for minimization, where the 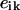 are experimental curves and the
are experimental curves and the 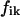 are theoretical model functions. For experimental data sets, values of σi were calculated by fitting single exponentials to the tail ends of the curves. Ni is the number of points in the ith curve. For each SR curve, the last 974 points were taken for analysis. Minimizations of Eq. 2 were performed with the use of routines based on the Minuit package (CN/ASD group, MINUIT User Guide, program library D506; CERN, 1993). The Minuit program was designed as a tool to find the minimum value of a multiparameter χ2 function and analyze the shape of the function around the minimum. The program contains several minimization methods, and the Davidson-Fletcher-Powell variable-metric method was used in the analysis of the SR data. The SR curves for a single exponential (model I) are described by
are theoretical model functions. For experimental data sets, values of σi were calculated by fitting single exponentials to the tail ends of the curves. Ni is the number of points in the ith curve. For each SR curve, the last 974 points were taken for analysis. Minimizations of Eq. 2 were performed with the use of routines based on the Minuit package (CN/ASD group, MINUIT User Guide, program library D506; CERN, 1993). The Minuit program was designed as a tool to find the minimum value of a multiparameter χ2 function and analyze the shape of the function around the minimum. The program contains several minimization methods, and the Davidson-Fletcher-Powell variable-metric method was used in the analysis of the SR data. The SR curves for a single exponential (model I) are described by
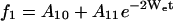 |
(3) |
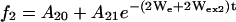 |
(4) |
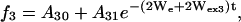 |
(5) |
where f1, f2, and f3 represent SR curves under argon, in equilibrium with air, or in the presence of 3 mM NiEDDA under argon, respectively. Wex2 and Wex3 are the corresponding exchange rates for oxygen and NiEDDA, respectively. In this model, We, Wex2, Wex3, the Aio, and the Ai1 were simultaneously fit to the set of experimental curves for each residue.
Preliminary analysis showed that some of the SR curves clearly had a biexponential character, suggesting the existence of two dynamic populations of nitroxide. In most of these situations, the two populations were also recognizable in the CW-EPR spectrum, and the SR curves could be described by the set of equations (model II),
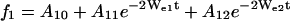 |
(6) |
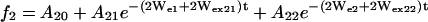 |
(7) |
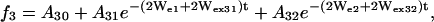 |
(8) |
where the additional subscripts 1 and 2 in the exponentials refer to the two populations. In this model, the rate parameters Wei, Wex2i, Wex3i, the baselines Ai0, and the amplitudes Ai1 and Ai2 were fitted to the set of experimental SR curves for each residue.
RESULTS
CW-EPR measurements
The CW-EPR spectra of the spin-labeled proteins in buffer with R1 at eight consecutive positions, from residue 128 to 135, are presented in Fig. 3 (solid traces). The EPR spectra reflect the motion of the nitroxide, which has contributions from both internal motions within the protein and from rotational diffusion of the protein as a whole. The contribution from rotational diffusion can be largely removed by recording the spectra in a viscous medium, such as 30% (w/v) sucrose. Such spectra, which reflect the motion of the side chain relative to the protein, have been previously reported and analyzed in terms of side-chain motion, and are included in Fig. 3 for reference (dotted traces) (10). The site-dependent variation in spectral lineshape in either buffer or in sucrose solution reflects a periodic variation in nitroxide mobility due to the helical structure of the 128–135 sequence (Fig. 2) (10). For example, residues 128, 131,132, and 135 are on the solvent-exposed face of the helix (gray spheres, Fig. 2), and the EPR spectra correspond to single or dominant populations of nitroxide with high mobility (τR ≈ 1–2 ns). At the other extreme, residues 129 and 133 are solvent inaccessible and buried in the protein interior (black spheres, Fig. 2). The spectra corresponding to 129R1 or 133R1 are dominated by a component that has a correlation time essentially that of the protein rotational diffusion. Residues 130 and 134 are on the solvent-exposed surface of the helix, but make contact with residues in the adjacent helix. The contact interaction is reflected in multiple components in the spectrum of 130 (arrows; dotted trace) that correspond to different dynamic modes of the side chain, and in the broad spectral features of 134R1 that reflect a relatively immobilized state compared to 131R1.
FIGURE 3.
CW-EPR spectra for R1 at the investigated sites in T4 lysozyme. The solid traces are the spectra recorded in buffer at ambient temperature, and the dashed traces are the spectra recorded in 30% sucrose in the same buffer and temperature. The sucrose decreases the rotational diffusion rate by about a factor of three. The arrows on the dotted traces of 130 identify the position of spectral components corresponding to different dynamic modes.
Saturation recovery measurements
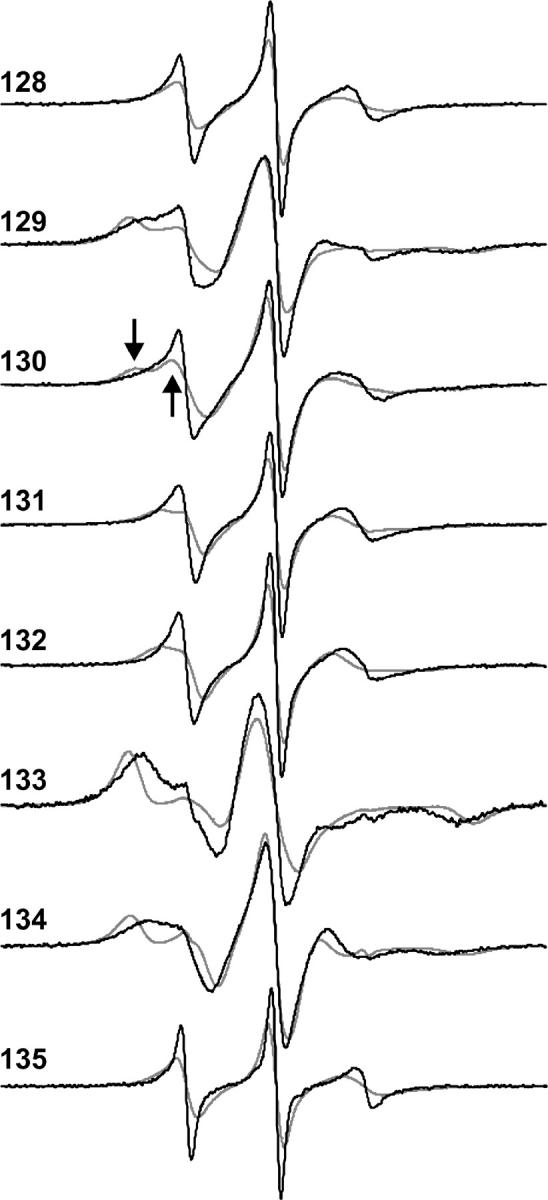
Example saturation recovery curves collected at the central resonance line (mI = 0) under an argon atmosphere for the highly mobile residue 128R1 and the immobilized residue 129R1 are shown in Fig. 4, A and B, respectively. It is evident that the more mobile spin label gives faster saturation recovery (note the difference in timescale in the two figures). Moreover, the faster decay for 128R1 can be represented very well by a single exponential function, whereas the slower decay for 129R1 is better represented by a double exponential function. This is evident in the plots of the residuals for residue 128 (Fig. 4 A, trace 2, from fit to model I) and residue 129 (Fig. 4 B, traces 2 and 3, from fits to models I and II, respectively).
FIGURE 4.
Saturation recovery for R1 at residues 128 (A) and 129 (B) under an argon atmosphere. In each case, trace 1 is the experimental recovery curve and trace 2 is the difference between the experimental curve and a single exponential fit (×5). In panel B, trace 3 is the difference between the experimental curve and a double exponential fit (×5).
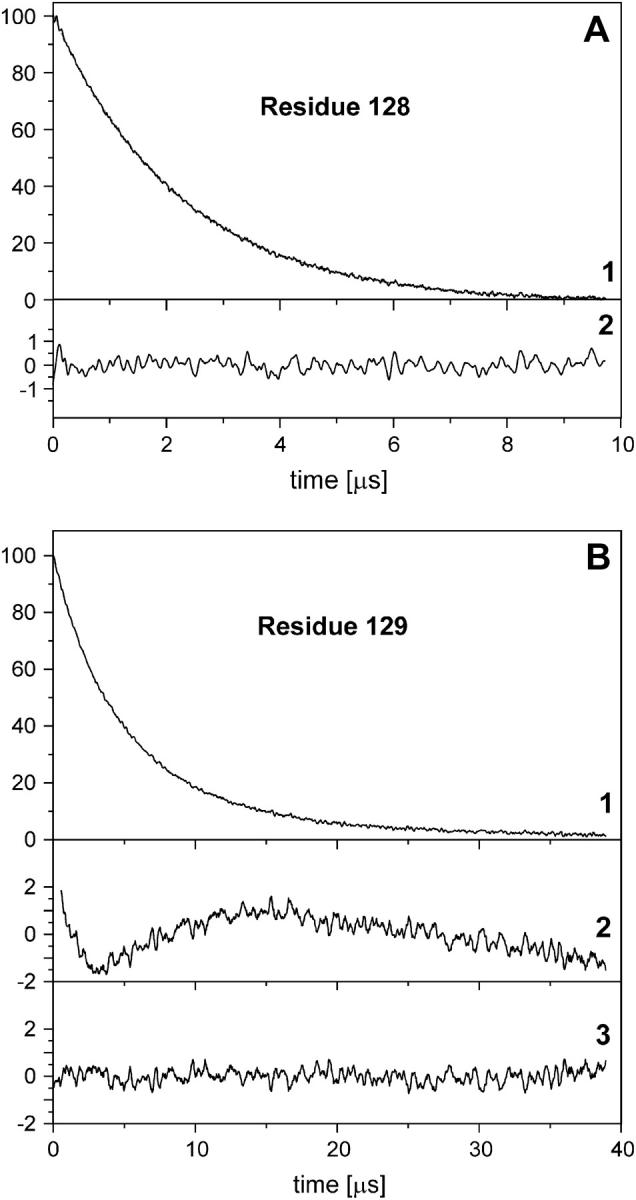
The presence of dissolved oxygen or NiEDDA in the protein sample dramatically shortens the decay times for residue 131R1, as shown in Fig. 5 A. However, for 133R1 O2 has a reduced effect compared to 131, and NiEDDA has essentially no effect (Fig. 5 B). As will be discussed below, these effects result from the different solvent accessibility of the two residues in the protein structure.
FIGURE 5.
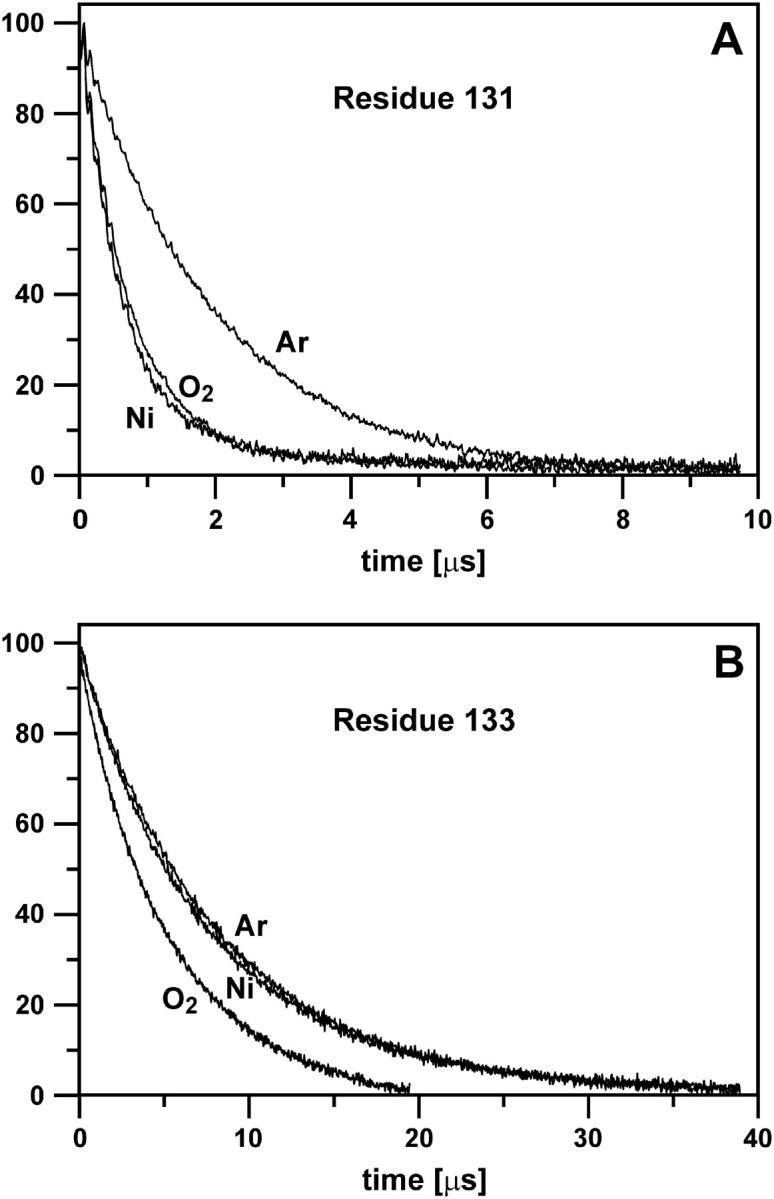
Saturation recovery curves for R1 attached to residues 131 (A) and 133 (B). Curves were recorded under an argon atmosphere (Ar), in the presence of oxygen in equilibrium with air (O2), and in the presence of 3 mM NiEDDA (Ni).
Recovery curves under argon atmosphere
As a starting point for analysis of the SR-EPR data, the curve for each residue under an argon atmosphere was fitted with a single and double exponential function. The ratio of χ2 values obtained for double and single exponential fits was calculated for each residue and plotted in Fig. 6 (dotted trace). This ratio is unity if the data is adequately described by a single exponential function (model I). This is the case for 128R1, 130R1, and 131R1. Residue 132 is also reasonably well fit with a single exponential. For small values of the ratio, the double exponential fit is much better, and this is the case for the buried residues 129R1 and 133R1. Evidently, there is a periodicity in the χ2 ratio that is roughly correlated with the spin-label mobility, measured by 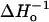 for the same residues (solid trace, Fig. 6). Apparently, interactions that immobilize the nitroxide also give rise to multiple dynamic modes of the side chain.
for the same residues (solid trace, Fig. 6). Apparently, interactions that immobilize the nitroxide also give rise to multiple dynamic modes of the side chain.
FIGURE 6.

A plot of the ratio of χ2 values obtained from single and double exponential fits, χ2(single)/χ2(double), versus sequence position. The data were obtained under an argon atmosphere. Also plotted are values for the inverse central linewidth, 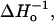 determined from the CW-EPR spectra.
determined from the CW-EPR spectra.
Recovery curves in the presence of exchange reagents
In the next step, the SR-EPR curves for each residue in the absence and presence of the exchange reagents, oxygen and NiEDDA, were globally fit to the single exponential functions of model I, despite the fact that recoveries for some residues were better fit by a double exponential. The motivation was to obtain a single best-fit value for comparison of the data with that obtained by CW saturation techniques (18). Analysis according to model I provided an electron relaxation rate, We, and the exchange rates with oxygen and NiEDDA, Wex, for each residue. As will be shown below, We and Wex obtained from fits to model I are useful for secondary structure analysis.
For a meaningful comparison of exchange rates between O2 and NiEDDA, the exchange rates are formally expressed as Wex = kexcR, where kex and cR are the exchange rate constant and the concentration of exchange reagent, respectively. The exchange rate constants are given in Table 1, and plotted together with We versus sequence number in Fig. 7. As is apparent from the figure, We, kex(O2) and kex(NiEDDA) all have a periodic character of approximately the same period and phase. Also shown for reference in Fig. 7 are the sequence dependencies of 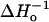 and the fractional solvent accessibility (fsa) of the native residue computed from the crystal structure of T4L (Protein Data Bank, 3lzm). As can be seen, these measures of R1 mobility and accessibility of the native side chain are closely correlated to We and kex.
and the fractional solvent accessibility (fsa) of the native residue computed from the crystal structure of T4L (Protein Data Bank, 3lzm). As can be seen, these measures of R1 mobility and accessibility of the native side chain are closely correlated to We and kex.
TABLE 1.
Parameters from global fits to model I
| Residue* | We (MHz) | T1e (μs) | kex2(O2) (MHz/mM) | kex3 (NiEDDA) (MHz/mM) | χ2† |
|---|---|---|---|---|---|
| 128 | 0.23 | 2.1 | 1.75 | 0.13 | 1.94 |
| 129 | 0.09 | 5.5 | 0.28 | 0.008 | 2.15 |
| 130 | 0.21 | 2.4 | 1.52 | 0.11 | 2.34 |
| 131 | 0.25 | 2.0 | 1.55 | 0.17 | 2.22 |
| 132 | 0.26 | 1.9 | 1.75 | 0.22 | 1.70 |
| 133 | 0.06 | 7.9 | 0.15 | 0.0008 | 2.13 |
| 134 | 0.13 | 3.9 | 1.13 | 0.12 | 3.04 |
| 135 | 0.24 | 2.1 | 0.53 | 0.06 | 2.78 |
The specific exchange rate constants (kex) are obtained by dividing Wex values obtained from fitting by the concentration of exchange reagent, 3 mM for NiEDDA, and 0.26 mM for O2 (38).
From global fits.
FIGURE 7.

The sequence dependence of 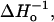 fsa, We, kex (NiEDDA), and kex (O2) obtained from a global fit of the saturation recovery data to a single exponential (model I).
fsa, We, kex (NiEDDA), and kex (O2) obtained from a global fit of the saturation recovery data to a single exponential (model I).
In the final stage of the analysis, the SR-EPR data were globally fit to model II, and the results given in Table 2. The values for electron relaxation rates and collision rate constants for exchange with oxygen and NiEDDA corresponding to the dominant component of the double exponential fits are plotted in Fig. 8. There is an excellent correlation of all three rates with the same sinusoidal function as in Fig. 7, and the correlation is improved for residue 135R1. In contrast, a similar plot for the rates corresponding to the minor components of the two-exponential fit show no regular dependence on the spin-label position (not shown). Again, data for 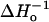 and fsa are shown for reference.
and fsa are shown for reference.
TABLE 2.
Parameters from global fits to model II
| Argon
|
Oxygen
|
NiEDDA
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Residue*†‡ | A11 | We1 | A12 | We2 | A21 | kex21 | A22 | kex22 | A31 | kex31 | A32 | kex32 | χ2§ |
| 128 | 90 | 0.235 | 11 | 0.235 | 11 | 0.0 | 70 | 2.19 | 25 | 0.0 | 66 | 0.265 | 1.66 |
| 129 | 58 | 0.065 | 40 | 0.155 | 62 | 0.21 | 30 | 0.289 | 80 | 0.01 | 17 | 0.108 | 1.03 |
| 130 | 4 | 0.21 | 97 | 0.21 | 1 | 0 | 96 | 1.58 | 14 | 0 | 85 | 0.14 | 2.00 |
| 131 | 1 | 0.25 | 99 | 0.25 | 8 | 0.00 | 94 | 1.96 | 7 | 0.0 | 93 | 0.19 | 1.57 |
| 132 | 8 | 0.16 | 92 | 0.27 | 41 | 3.21 | 57 | 1.23 | 5 | 0.0 | 83 | 0.25 | 1.58 |
| 133 | 41 | 0.095 | 58 | 0.05 | 90 | 0.00 | 7 | 0.635 | 5 | 0.025 | 94 | 0.002 | 1.25 |
| 134 | 52 | 0.105 | 45 | 0.165 | 2 | 0.135 | 74 | 1.61 | 8 | 0.00 | 85 | 0.132 | 2.11 |
| 135 | 48 | 0.16 | 47 | 0.31 | 28 | 0.00 | 57 | 1.69 | 25 | 0.00 | 46 | 0.3 | 1.61 |
We1 and We2 are in MHz; kex is in MHz/mM.
The specific exchange rate constants (kex) are obtained by dividing Wex values from fitting by the concentration of exchange reagent; 3 mM for NiEDDA and 0.26 mM for O2 in equilibrium with air (38).
Standard deviation from global fits is <0.1% and 1% for We and Wex, respectively.
From global fits.
FIGURE 8.

The sequence dependence of 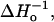 fsa, We, kex (NiEDDA), and kex (O2) obtained from a global fit of the saturation recovery data to two exponentials (model II). The rate constants correspond to the dominant components from Table 2.
fsa, We, kex (NiEDDA), and kex (O2) obtained from a global fit of the saturation recovery data to two exponentials (model II). The rate constants correspond to the dominant components from Table 2.
DISCUSSION
Correlation of We with side-chain dynamics and protein structure
Due to the relatively rapid motion of the spin label at each site studied (1 ns < τR < 6 ns), saturation recovery due to spectral diffusion mechanisms (rotational diffusion, nitrogen nuclear relaxation) is faster than the spectrometer bandwidth employed in these experiments (12.5–50 MHz), and the observed recovery process is determined only by spin-lattice relaxation and Heisenberg exchange mechanisms. Thus, according to Eq. 1, single exponential recovery curves are expected in the absence and presence of exchange reagents, if there is a single population of the R1 side chain. In the presence of multiple states of the side chain in slow exchange on the T1e timescale, there will be, in general, an equal number of exponentials in the saturation recovery curve. Previous EPR studies found that the spectra of R1 at solvent-exposed helix surface sites, where the nitroxide makes no contacts with other parts of the protein, can be accounted for by a single dynamic mode. On the other hand, at least two dynamic states are generally required to account for the spectra of R1 at partially solvent exposed (contact) or buried sites (10). Accordingly, it was found that the saturation recovery data for R1 at sites 128–135 in the T4L helix could be best fit to either one (model I) or two (model II) exponentials.
In the absence of an exchange reagent, the recovery curves reported here are determined only by the spin-lattice relaxation rate. For a nitroxide in aqueous solution at room temperature, We is a function of the rotational diffusion rate of the nitroxide (18,19,31). As shown in Fig. 4 and the results from global analysis according to model II (Table 2), recovery data for 128R1, 130R1, and 131R1 in the absence of exchange reagents were well fit with single exponentials. Although the data for 132R1 under argon are somewhat better fit with two exponentials, one is strongly dominant (Table 2), and that population has the same value for We as that obtained in the single exponential fit (Table 1). The values of We for these sites lay in the narrow range 0.21–0.27 MHz, corresponding to T1e values of 2.4–1.9 μs, respectively.
Thus, the relaxation data suggest essentially a single dynamic population of R1 at sites 128, 130, 131, and 132. For residues 128, 131, and 132, this conclusion is consistent with their location on the solvent-exposed face of the helix where they do not make tertiary contacts (Fig. 2, gray spheres; Fig. 7), and with the single-component CW-EPR spectral lineshapes recorded in 30% sucrose (viscosity ≈ 3 cP; Fig. 3). For example, the EPR spectrum of 131R1 in 30% sucrose has been previously shown to arise from a weakly anisotropic motion with a correlation time for internal motion of ∼2 ns (7). With the superposed rotational motion of the T4L molecule under the conditions where saturation recovery experiments were conducted (viscosity ≈ 1 cP, τ ≈ 6 ns), the approximate effective correlation time of the nitroxide would be τR ≈ 1.5 ns. From the published dependence of τR on We (28,29), a correlation time of 1.5 ns corresponds to a We ≈ 0.21 MHz, close to the value of 0.25 MHz observed here (Tables 1 and 2). The lineshape of 132R1 is similar to that of 131R1, suggesting a similar dynamic mode. Site 128 lies at the N-terminal end of the helix, and the EPR spectrum has narrow features characteristic of a rapid, essentially isotropic motion, consistent with a dominant dynamic mode with a single spin-lattice relaxation rate.
Sites 130 and 135 are interesting exceptions to the general correlation between CW-EPR lineshape, protein structure, and We. As seen in the T4L model of Fig. 2 and the fractional solvent accessibility plot in Fig. 7, site 130 is a partially solvent-exposed site where the nitroxide is expected to make tertiary contacts with an adjacent helix. Consistent with this expectation, the CW-EPR spectrum has two well-resolved components corresponding to two dynamic modes of motion. However, only a single spin-lattice relaxation rate is obtained from a global analysis (Table 2). This result strongly suggests that the two populations evident in the spectrum are in fast exchange on the T1e timescale. Thus, the site-exchange rate between the two populations, ωex, must be such that ωex > 1/T1e ≈ 0.4 MHz, but <∼10 MHz, where the upper limit is set by the requirement that the two populations be resolved in the X-band CW spectrum. This result illustrates the potential use of SR-EPR to investigate exchange rates in this time window.
Site 135 is highly solvent exposed (Figs. 2 and 7), yet appears to have two dynamic modes with spin-lattice relaxation rates of 0.16 and 0.31 MHz, respectively, corresponding to T1e values of 3.1 and 1.6 μs. A crystal structure of the 135R1 mutant is not available, and the structural origin of these components is not clear.
The saturation recovery curves of R1 at sites 129, 133, and 134 in the absence of exchange reagents require two exponentials for the best fit (Fig. 7 and Table 2), consistent with the location of these residues at sites of tertiary interaction (Fig. 2) with low solvent accessibility (Fig. 8). The EPR spectra of 129 and 134 clearly reveal two spectral components corresponding to two dynamical modes (arrows, Fig. 3).
The plots of We versus sequence number along 128R1–135R1 from fits to model I or from the dominant component from fits to model II reveal the periodicity of the α-helix (Figs. 7 and 8). The periodicity presumably arises from the variation in side-chain mobility as the nitroxide is scanned along the periodic structure. This interpretation is supported by the similarity of the plots to those for 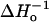 (Figs. 7 and 8). A similar plot for the minor component of We from model II fits reveals no consistent pattern that is correlated with the structure, supporting the view that the dominant state reflects the global structural features. This result supports the validity of using We obtained from single exponential fits in experiments to map simple periodic structural features of proteins. However, this does not imply that the detection of a second component at some of the sites is without value. On the contrary, relaxation properties of a second component will likely be of use in analyzing the interaction of the nitroxide with the environment to provide details of the local structure.
(Figs. 7 and 8). A similar plot for the minor component of We from model II fits reveals no consistent pattern that is correlated with the structure, supporting the view that the dominant state reflects the global structural features. This result supports the validity of using We obtained from single exponential fits in experiments to map simple periodic structural features of proteins. However, this does not imply that the detection of a second component at some of the sites is without value. On the contrary, relaxation properties of a second component will likely be of use in analyzing the interaction of the nitroxide with the environment to provide details of the local structure.
Mapping residue accessibility with SR-EPR
The sequence dependence of side-chain solvent accessibility provides a wealth of information on protein structure, including sequence-specific secondary structure and the location of surfaces of tertiary contact. The exchange rate of a nitroxide side chain due to collision with paramagnetic species in solution appears to be a valid experimental measure of side-chain solvent accessibility, and thus is an important analytical tool for structural analysis as well as an efficient means of comparing the structure of a protein in solution and in the crystalline state (14). As mentioned above, the accessibility of an R1 side chain to collision with fast-relaxing paramagnetic species in solution is directly measured by changes in the apparent T1e of the nitroxide, and this in turn is most directly determined by SR-EPR.
The results of global analysis of recovery data according to model II given in Table 2 show that the best fits in the presence of O2 or NiEDDA generally require two components. In each case, the exchange rate constant for the dominant component for O2 exceeds that for NiEDDA, presumably due to the larger size of the latter, as has been previously noted based on CW power saturation studies (35). The dominant component in each case mirrors the helical pattern of the fractional solvent accessibility of the native side chain computed from the crystal structure (Fig. 8), except at 132R1 for NiEDDA, where the relative value for kex is higher than that expected from the solvent accessibility. The minor component of kex, as for that of We, does not show a regular pattern (not shown). Again, these results support the view that the dominant component best reflects the protein structure.
Global analysis of the recovery data according to a single exponential process (model I) gives kex values for O2 and NiEDDA that closely track the sequence dependence of the dominant component from the global analysis of model II, except at 135R1, where the value is significantly smaller in the latter case. This is due to the existence of a substantial population with an apparent inaccessibility for exchange with either reagent when using the model II analysis (see below).
For 130, 131, 133, and 134, one component is sufficiently dominant that the site may be considered to have a single population available for exchange. For 130, there are clearly two components in the EPR spectrum, but a single value for We. As argued above, this is likely to result from a site exchange fast on the T1e timescale. Because the exchange rates are on the order of 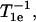 the site exchange would give rise to a single Wex for the two populations. For the solvent-exposed site 131, the single component of high exchange rate is consistent with the single dynamic mode reflected in the CW-EPR spectra, with the single value of We, and with the structure of the protein. For 133R1, the single component of extremely low exchange rate for both reagents is consistent with the buried location of the site and the CW-EPR spectrum that suggests a dominant component of very slow internal motion. Apparently, there are two dynamic states to this side chain, judging from the two populations of We, but both are inaccessible to exchange with paramagnetic reagents. For 134R1, the intermediate values for O2 and NiEDDA exchange rates are consistent with the location at a partially solvent-exposed site.
the site exchange would give rise to a single Wex for the two populations. For the solvent-exposed site 131, the single component of high exchange rate is consistent with the single dynamic mode reflected in the CW-EPR spectra, with the single value of We, and with the structure of the protein. For 133R1, the single component of extremely low exchange rate for both reagents is consistent with the buried location of the site and the CW-EPR spectrum that suggests a dominant component of very slow internal motion. Apparently, there are two dynamic states to this side chain, judging from the two populations of We, but both are inaccessible to exchange with paramagnetic reagents. For 134R1, the intermediate values for O2 and NiEDDA exchange rates are consistent with the location at a partially solvent-exposed site.
For the remaining sites 128, 129, 132, and 135, the amplitudes of the two components, for either O2 and/or NiEDDA, are sufficiently large to be individually considered. For the two solvent-exposed sites 128 and 135, the smaller of the two exchange components for both O2 and NiEDDA have kex ≈ 0. There is no evidence of a strongly immobilized component in the spectra of these sites that would be expected for such an inaccessible state, and the values of We do not correspond to slowly moving nitroxides. Moreover, the structure of T4L does not suggest a possible origin for such a protected population. Thus, the interpretation of these data is unclear and must await further analysis. For the solvent-exposed site 132, there is a single high rate constant detected for NiEDDA, but two components of comparable amplitude that differ by about a factor of two are resolved for O2 exchange, one of which is more than an order of magnitude larger than for NiEDDA. This may indicate a highly ordered environment around 132, but a specific interpretation must await elucidation of the mechanisms that determine exchange rates at protein surfaces. Finally, residue 129R1 clearly has two dynamic populations, reflected both in values of We and in the EPR spectrum. The dominant population in both O2 and NiEDDA exchange rates reflects a relatively buried population corresponding to the component in the EPR spectrum with well-resolved outer hyperfine extrema and the low spin-lattice relaxation rate. The minor component of higher exchange rates is then assigned to the more mobile state evident in the EPR spectrum and corresponding to the higher relaxation rate.
SUMMARY AND CONCLUSIONS
The results presented above indicate that spin-lattice relaxation (We) and Heisenberg exchange rates (Wex) for R1 determined from saturation recovery curves are measures of R1 side-chain mobility and side-chain accessibility, respectively. Thus, relative values of We are closely correlated with 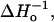 the inverse of the central linewidth of the EPR spectrum that has been used as a semiempirical measure of side-chain mobility in earlier work.
the inverse of the central linewidth of the EPR spectrum that has been used as a semiempirical measure of side-chain mobility in earlier work.
In a companion article, it is shown that R1 accessibility to collision with O2 or NiEDDA, determined by CW power saturation and expressed as Wex, is highly correlated with the Wex values determined here from saturation recovery (18). The correlation coefficient is 0.99 and 0.88 for NiEDDA and O2, respectively, which is much higher than observed by Nielsen et al. (31) in similar experiments. Although quantitative measurements of Wex can be made by either SR or CW saturation (18), the SR method has a distinct advantage in that it can resolve multiple populations with different accessibilities, as shown above. On the other hand, CW saturation cannot easily distinguish between two homogeneously saturating components with different accessibility and a single component with inhomogeneous saturation. Disadvantages of the saturation recovery method include the specialized instrumentation needed, and difficulty in determination of Wex > 10 MHz, where saturation recovery occurs within the effective dead time of the instrument. Although SR-EPR measurements to date on biological systems have been made on homebuilt instrumentation, Bruker BioSpin has recently introduced a commercial spectrometer system capable of SR-EPR (Bruker BioSpin, Billerica, MA).
Although the best fits of the saturation recovery data in the presence of exchange reagents requires multiple exponentials corresponding to multiple states of the spin label, single exponential fits are adequate for estimation of Wex and We in experiments designed for mapping protein structure via the periodic behavior along a sequence.
An important future use of SR-EPR may be in determining the exchange rate of R1 between two populations with microsecond lifetimes. The populations may represent two states of the protein or of the side chain. Such site exchange is indicated in this study by the saturation recovery data of residue 130R1, where two populations are resolved in the CW-EPR spectrum, but only one in the saturation recovery, presumably due to exchange rapid on the We, Wex timescales. The dependence of Wex on the concentrations of exchange reagents may allow the determination of the exchange rate between the two populations (36). Together with pulsed ELDOR (37), T1e-based relaxation EPR spectroscopy may become a powerful tool in monitoring conformational fluctuations of proteins.
Acknowledgments
We thank Hassane Mchaourab, Linda Columbus, and Ralf Langen for supplying T4L mutants used in the experiments.
This work was supported by State Committee for Scientific Research, Poland (grant 2P04A04323 to W.F.), National Institutes of Health (grant EY05216 to W.L.H), Fogarty International Center (TW00456 to W.L.H.), and the Jules Stein Professorship endowment (W.L.H.).
Abbreviations used: SDSL, site-directed spin labeling; CW, continuous wave; ELDOR, electron-electron double resonance; EPR, electron paramagnetic resonance; HE, Heisenberg exchange; NiEDDA Ni (II) ethylenediaminediacetate; SR, saturation recovery; 44R1, the mutant with the R1 side chain at site 44 (mutants with a single spin label are given the sequence number for the spin-labeled position followed by R1 designates).
References
- 1.Hubbell, W. L., H. S. Mchaourab, C. Altenbach, and M. A. Lietzow. 1996. Watching proteins move using site-directed spin labeling. Structure. 4:779–783. [DOI] [PubMed] [Google Scholar]
- 2.Hubbell, W. L., A. Gross, R. Langen, and M. A. Lietzow. 1998. Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 8:649–656. [DOI] [PubMed] [Google Scholar]
- 3.Hubbell, W. L., D. S. Cafiso, and C. Altenbach. 2000. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7:735–739. [DOI] [PubMed] [Google Scholar]
- 4.Columbus, L., and W. L. Hubbell. 2002. A new spin on protein dynamics. Trends Biochem. Sci. 27:288–295. [DOI] [PubMed] [Google Scholar]
- 5.Feix, J. B., and C. S. Klug. 1998. Spin labeling: the next millennium. In Biological Magnetic Resonance, Vol. 14. L. J. Berliner, editor. Plenum Press, New York, NY. 251–281.
- 6.Budil, D. E., S. Lee, S. Saxena, and J. H. Freed. 1989. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J. Magn. Reson. Series A. 120:155–189. [Google Scholar]
- 7.Columbus, L., T. Kalai, J. Jeko, K. Hideg, and W. L. Hubbell. 2001. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry. 40:3828–3846. [DOI] [PubMed] [Google Scholar]
- 8.Barnes, J. P., Z. Liang, H. S. Mchaourab, J. H. Freed, and W. L. Hubbell. 1999. A multifrequency electron spin resonance study of T4 lysozyme dynamics. Biophys. J. 76:3298–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lietzow, M. A., and W. L. Hubbell. 2004. Motion of spin labeled side chains in cellular retinol binding protein: correlation with structure and nearest neighbor interactions in an anti-parallel beta sheet. Biochemistry. 43:3137–3151. [DOI] [PubMed] [Google Scholar]
- 10.Mchaourab, H. S., M. A. Lietzow, K. Hideg, and W. L. Hubbell. 1996. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 35:7692–7704. [DOI] [PubMed] [Google Scholar]
- 11.Altenbach, C., K. Yang, D. L. Farrens, Z. T. Farahbakhsh, H. G. Khorana, and W. L. Hubbell. 1996. Structural features and light-dependent changes in the cytoplasmic interhelical E-F loop region of rhodopsin: a site-directed spin-labeling study. Biochemistry. 35:12470–12478. [DOI] [PubMed] [Google Scholar]
- 12.Koteiche, H. A., and H. S. Mchaourab. 1999. Folding pattern of the alpha-crystallin domain in alphaA-crystallin determined by site-directed spin labeling. J. Mol. Biol. 294:561–577. [DOI] [PubMed] [Google Scholar]
- 13.Salwinski, L., and W. L. Hubbell. 1999. Structure in the channel forming domain of colicin E1 bound to membranes: the 402–424 sequence. Protein Sci. 8:562–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isas, J. M., R. Langen, H. T. Haigler, and W. L. Hubbell. 2002. Structure and dynamics of a helical hairpin and loop region in annexin 12: a site-directed spin labeling study. Biochemistry. 41:1464–1473. [DOI] [PubMed] [Google Scholar]
- 15.Altenbach, C., W. Froncisz, J. S. Hyde, and W. L. Hubbell. 1989. Conformation of spin-labeled melittin at membrane surfaces investigated by pulse saturation recovery and continuous wave power saturation electron paramagnetic resonance. Biophys. J. 56:1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altenbach, C., S. L. Flitsch, H. G. Khorana, and W. L. Hubbell. 1989. Structural studies on transmembrane proteins. 2. Spin labeling of bacteriorhodopsin mutants at unique cysteines. Biochemistry. 28:7806–7812. [DOI] [PubMed] [Google Scholar]
- 17.Farahbakhsh, Z. T., C. Altenbach, and W. L. Hubbell. 1992. Spin labeled cysteines as sensors for protein-lipid interaction and conformation in rhodopsin. Photochem. Photobiol. 56:1019–1033. [DOI] [PubMed] [Google Scholar]
- 18.Altenbach, C., W. Froncisz, R. Hemker, H. Mchaourab, and W. L. Hubbell. 2005. Accessibility of nitroxide side chains: absolute Heisenberg exchange rates from power saturation EPR. Biophys. J. 89:2103–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbell, W. L., C. Altenbach, C. M. Hubbell, and H. G. Khorana. 2003. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv. Protein Chem. 63:243–290. [DOI] [PubMed] [Google Scholar]
- 20.Molin, Y. N., K. M. Salikhov, and K. I. Zamaraev. 1980. Spin Exchange. Springer-Verlag, New York, NY.
- 21.Haas, D. A., T. Sugano, C. Mailer, and B. H. Robinson. 1993. Motion in nitroxide spin labels: direct measurements of rotational times by pulsed electron double resonance. J. Phys. Chem. 97:2914–2921. [Google Scholar]
- 22.Mchaourab, H. S., and J. S. Hyde. 1993. Dependence of the multiple-quantum EPR signal on the spin-lattice relaxation time. Effect of oxygen in spin-labeled membranes. J. Magn. Reson. B10:178–184. [Google Scholar]
- 23.Feix, J. B., C. A. Popp, S. D. Venkataramu, A. H. Beth, J. H. Park, and J. S. Hyde. 1984. An electron-electron double-resonance study of interactions between [14N]- and [15N]stearic acid spin-label pairs: lateral diffusion and vertical fluctuations in dimyristoylphosphatidylcholine. Biochemistry. 23:2293–2299. [DOI] [PubMed] [Google Scholar]
- 24.Hyde, J. S., and J. B. Feix. 1989. Spin labeling: theory and applications. In Biological Magnetic Resonance, Vol. 8. L. J. Berliner, editor. Plenum Press, New York, NY. 305–334.
- 25.Shin, Y. K., and W. L. Hubbell. 1992. Determination of electrostatic potentials at biological interfaces using electron-electron double resonance. Biophys. J. 61:1443–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Percival, P. W., and J. S. Hyde. 1975. Pulsed EPR spectrometer. Rev. Sci. Instrum. 46:1522–1529. [Google Scholar]
- 27.Yin, J.-J., M. Pasenkiewicz-Gierula, and J. S. Hyde. 1987. Lateral diffusion of lipids in membranes by pulse saturation recovery electron spin resonance. Proc. Natl. Acad. Sci. USA. 84:964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson, B. H., D. A. Haas, and C. Mailer. 1994. Molecular dynamics in liquids: spin-lattice relaxation of nitroxide spin labels. Science. 263:490–493. [DOI] [PubMed] [Google Scholar]
- 29.Percival, P. W., and J. S. Hyde. 1976. Saturation recovery measurements of the spin-lattice relaxation times of some nitroxides in solution. J. Magn. Reson. 23:249–257. [Google Scholar]
- 30.Koptyug, I. V., S. H. Bossmann, and N. J. Turro. 1996. Inversion recovery of nitroxide spin labels in solution and microheterogeneous environments. J. Am. Chem. Soc. 118:1435–1445. [Google Scholar]
- 31.Nielsen, R. D., S. Canaan, J. A. Gladden, M. H. Gelb, C. Mailer, and B. H. Robinson. 2004. Comparing continuous wave progressive saturation EPR and time domain saturation recovery EPR over the entire motional range of nitroxide spin labels. J. Magn. Reson. 169:129–163. [DOI] [PubMed] [Google Scholar]
- 32.Ilnicki, J., T. Oleś, J. Kostrzewa, W. Galiński, R. J. Gurbiel, and W. Froncisz. 1994. Saturation recovery EPR spectrometer. Molecular Physics Reports. 5:203–207. [Google Scholar]
- 33.Froncisz, W., and J. S. Hyde. 1982. The loop-gap resonator: a new microwave lumped circuit ESR sample structure. J. Magn. Reson. 47:515–521. [Google Scholar]
- 34.Beechem, J. M. 1992. Global analysis of biochemical and biophysical data. Methods Enzymol. 210:37–53. [DOI] [PubMed] [Google Scholar]
- 35.Oh, K. J., H. Zhan, C. Cui, C. Altenbach, W. L. Hubbell, and R. J. Collier. 1999. Conformation of the diphtheria toxin T domain in membranes: a site-directed spin-labeling study of the TH8 helix and TL5 loop. Biochemistry. 38:10336–10343. [DOI] [PubMed] [Google Scholar]
- 36.Kawasaki, K., J.-J. Yin, W. K. Subczynski, J. S. Hyde, and A. Kusumi. 2001. Pulse EPR detection of lipid exchange between protein-rich raft and bulk domains in the membrane: methodology development and its application to studies of influenza viral membrane. Biophys. J. 80:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haas, D. A., C. Mailer, and B. H. Robinson. 1993. Using nitroxide spin labels. How to obtain T1e from continuous wave electron paramagnetic resonance spectra at all rotational rates. Biophys. J. 64:594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilhelm, E., R. Battino, and R. J. Wilcock. 1977. Low pressure solubility of gases in liquid water. Chem. Rev. 77:219–262. [Google Scholar]