

Abstract
Narrow hydrophobic regions are a common feature of biological channels, with possible roles in ion-channel gating. We study the principles that govern ion transport through narrow hydrophobic membrane pores by molecular dynamics simulation of model membranes formed of hexagonally packed carbon nanotubes. We focus on the factors that determine the energetics of ion translocation through such nonpolar nanopores and compare the resulting free-energy barriers for pores with different diameters corresponding to the gating regions in closed and open forms of potassium channels. Our model system also allows us to compare the results from molecular dynamics simulations directly to continuum electrostatics calculations. Both simulations and continuum calculations show that subnanometer wide pores pose a huge free-energy barrier for ions, but a small increase in the pore diameter to ∼1 nm nearly eliminates that barrier. We also find that in those wider channels the ion mobility is comparable to that in the bulk phase. By calculating local electrostatic potentials, we show that the long range Coulomb interactions of ions are strongly screened in the wide water-filled channels. Whereas continuum calculations capture the overall energetics reasonably well, the local water structure, which is not accounted for in this model, leads to interesting effects such as the preference of hydrated ions to move along the pore wall rather than through the center of the pore.
INTRODUCTION
Ion transport across membranes, mediated by ion-channel proteins, is central to many biological processes (1,2), such as neural signal transmission and electrical excitability of muscle. Recent progress in the expression, purification, and crystallization of membrane proteins has led to a rapidly growing number of high-resolution structures (3,4), including those of several ion channels (5–8). Channel structures, together with detailed biochemical and biophysical characterizations using, e.g., amino-acid mutation studies and single-channel conductance measurements, provide key insights into the energetics, selectivity, and gating of ion channels. The availability of atomic structures has also enabled increasingly detailed computational studies that relate ion-channel structure and dynamics to functional mechanisms (9–21).
Understanding ion selectivity has been a major focus of both structural and computational studies, and enormous progress has been made in particular for potassium channels, which are highly permeable for K+ ions but not Na+ ions. Structural studies of potassium channels have also provided new insight into ion-channel gating, relating ion permeability and impermeability to differences in the channel structures between the “open” and “closed” states, respectively. The first high-resolution x-ray structures showed the potassium channel in a closed state, but recently the structure of the MthK potassium channel in the open state was reported (22). Remarkably, the open pore of the MthK channel is lined predominantly by nonpolar amino acids. One of the questions we address in this article is a functional role of the nonpolar pore region of potassium channels.
Narrow and relatively hydrophobic pore regions are a repeatedly observed feature of biological channels facilitating the transport of polar species such as water, protons, and ions. They are present in the potassium channels, e.g., KcsA, MthK, or the Kir family (7,22,23). The mechanosensitive channels (e.g., MscS (24)), and the nicotinic acetylcholine receptor nAChr (25) have similar hydrophobic regions, which may function as gating regions that control the ion permeability (26,27). Narrow, mostly hydrophobic cavities are also involved in the regulation of water or proton transport in several biological systems, such as aquaporins (28,29), cytochrome c oxidase (30), and bacteriorhodopsin (31).
The presence of nonpolar pores as conduits for ions seems at first sight rather surprising for electrostatic reasons. From a simple dielectric model of ion permeation through a hydrophobic pore, one might expect a substantial barrier for displacing a charged particle from the high dielectric solution into a channel surrounded by a low dielectric medium, uncompensated by strong interactions with polar residues lining the channel wall (32). The high conductivity of potassium channels in the open state thus raises the question whether hydrophobic channels are more permeable for ions than one may think based on this simple dielectric argument. To address this question, we will use a combination of computer simulations and continuum electrostatics calculations.
Biomolecular systems as large and complex as membrane channels are becoming accessible to computer simulations thanks to advances both in the raw computational power and in the simulation methodology (9–18,33). However, one problem in theoretical ion-channel research is the complexity of the resulting systems of membrane-embedded proteins, making it difficult to identify the principles underlying the functional mechanisms in the wealth of detailed information provided by molecular simulations. The irregularly shaped pores are lined by both hydrophilic and hydrophobic regions of various widths, and single amino-acid residues can play a crucial role at specific sites. Separating the overall free energies for ion transport into individual contributions to quantify the roles of, e.g., “electrostatics”, “hydrogen bonds”, “desolvation”, or “conformational fluctuations” thus proves difficult, with results that can depend on the separation procedure.
Therefore, to answer the general question about a functional relevance of hydrophobic pores for gating we pursue a different strategy. We avoid the complexity of the biological systems by studying simpler model systems designed to capture specific properties. In addition to being more easily interpretable, such systems are also computationally less expensive than the more realistic membrane-protein systems. For ion translocation through channels, this is of particular relevance, because long-range electrostatics plays an important role in the energetics. To avoid surface effects, molecular dynamics simulations of molecular systems are usually performed either with periodic boundary conditions or for solvent droplets, possibly “embedded” in a dielectric continuum. In either case, electrostatic artifacts seem possible because of the long range of the Coulomb interaction, the strong dielectric discontinuity at the membrane, and the weakly screened interactions within the low-dielectric membrane region. By using a comparatively simple system, we can explicitly explore the energetic contributions coming from the simulation boundary conditions. Moreover, using a simple channel system enables us also to perform direct and fully quantitative comparisons of explicit molecular dynamics simulations to continuum electrostatics calculations. Normally, the motions of protein and membrane, and the inhomogeneous dielectric environment permit only qualitative comparisons. Here, we will study a system with a relatively rigid pore and a well-defined dielectric constant of the entire membrane region.
We focus on the basic principles that govern ion transport through narrow hydrophobic pores by simulation of model membranes formed of hexagonally packed carbon nanotubes (CNTs), as displayed in Fig. 1. Central questions of our study are: Which factors determine the energetics of ion translocation through such nonpolar nanopores? How large is the free-energy barrier for ion translocation through a hydrophobic pore with a diameter similar to that of the KcsA channel in the closed-state structure (7)? How much does that barrier drop if the pore is widened to a diameter corresponding to that of the open-state structure of the MthK potassium channel (22), and how mobile are ions in wide channels?
FIGURE 1.

Schematic of simulation setup. (A) Snapshot of the channel simulation system with eight copies of the periodic unit cell. Each unit cell contains one open and three capped CNTs, which are periodically replicated in the x, y plane, resulting in a hexagonally packed membrane. In the z direction, the periodic copies of the CNT membrane are separated by a water layer. (B and C) Illustration of the channel and sieve setups, respectively. The content of one unit cell is indicated with gray atoms. (D) Schematic of the simulation system, indicating the translocation of the Na+ ion through the open pore in a “channel” system.
Our model system also allows us to explore methodological issues in computational studies of ion translocation (34). By comparing continuum electrostatics calculations to results from molecular dynamics simulations, we will address the question which level of theory/simulation is required to understand various aspects of the translocation process.
The article is organized as follows. We first present different computational approaches to investigate ion transport through membrane channels and describe the simulation methods used in this study. Then, we present our results on the translocation of a Na+ ion through model membranes formed by carbon nanotubes. Finally, based on our simulation results, we discuss the role of hydrophobic cavities and the water phase inside them in the process of charge transport through biological membranes.
METHODS
Overview
Theoretical ion-channel research includes a large variety of simulation techniques, ranging from implicit-solvent simulations that rely on continuum electrostatics to computationally much more expensive classical (explicit-solvent) or ab initio molecular dynamics (MD) simulations. Continuum descriptions using Poisson-Nernst-Planck theory as well as explicit descriptions of ions in Brownian dynamics or MD simulations are used to study ion transport (20,35–38). In (all-atom) MD simulations, the inclusion of solvent greatly increases the computational cost, without even considering electronic degrees of freedom. Typically, large system sizes and long simulations are required to obtain reliable results in simulations of membrane proteins. In continuum models, one faces several well-known limitations (39–42): 1), bulk properties of water such as density and dielectric constant are applied uniformly to the water phase in the system, even though the properties of water might locally deviate strongly from those of bulk water; 2), the assumption of a uniformly distributed polarizable medium is problematic for cavities that contain few and inhomogeneously distributed water molecules; and 3), surface polarization effects cannot be accounted for in simple continuum models since they are a result of surface ordering of water molecules without an external electric field.
The carbon-nanotube model membranes used in our study result in systems that are small compared to biological membrane channels and thus computationally relatively inexpensive. The small size and relative simplicity of our system allow us to investigate the thermodynamics and kinetics of ion translocation through a hydrophobic pore with explicit-solvent MD simulations. We used an umbrella sampling technique (43,44) to obtain the free-energy for moving an ion from bulk solvent into a nanopore embedded in a membrane. For reference, we also computed the free energy of ion translocation using a dielectric-continuum model. By comparing the free energies obtained from all-atom MD simulations and continuum electrostatics, we identify those aspects of the ion translocation energetics determined specifically by the local properties of solvent molecules. This comparison is supported by the analysis of several structural properties of the ion and the solvent in the MD simulations, among which the local electrostatic potential is of particular interest for charge translocation.
MD simulations
MD simulations were performed with the sander module of AMBER 6.0 (45). The periodically replicated rectangular unit cell contains four CNTs that are hexagonally packed in the x, y plane and between 1183 and 1188 TIP3P water molecules (46). Two types of CNTs of (10,10) armchair type were used: water-permeable tubes with open ends, and impermeable tubes with capped ends. The tubes consist of 360 (open) and 340 (capped) sp2 carbon atoms (47,48) and are 20.7 (open) and 20.4 Å (capped) long with a radius of 6.7 Å (tube axis to carbon nuclei). This setup results in system sizes of ∼5000 atoms. We used two types of membranes, one consisting exclusively of open tubes (“sieve” geometry), and the other combining three capped and one open tube within a unit cell in the x, y plane. The resulting system mimics a channel in an otherwise sealed membrane and is denoted as “channel” geometry. The two setups are illustrated in Fig. 1. Each unit cell contains a single Na+ ion (49). For comparison and to complement the results of the simulations with a single ion, three additional types of systems were simulated: 1), the above (10,10) CNT membranes without ions; 2), the (10,10) CNT membranes with five pairs of Na+ and Cl− ions (initially placed randomly in the bulk water phase); and 3), sieve and channel systems consisting of narrower (6,6) CNTs (4 Å radius) with a single Na+ ion and 516 TIP3P water molecules. Simulations of systems with five ion pairs corresponding to an ionic concentration of ∼0.2 mol/L will allow us on one hand to compare free energies from an umbrella sampling procedure to unbiased simulations. On the other hand, the comparison of “infinite dilution” and “finite concentration” simulations will also allow us to assess possible ion-concentration dependences in the low-concentration regime. Finally, simulations at finite concentration will give us insights into the rate of ion translocation across the membrane.
In all simulations reported here, the preassembled CNT membranes remained stable with only sub-Ångstrom fluctuations out of plane. All simulations were carried out with a time step of 2 fs; the simulation temperature was kept constant at 300 K by weak coupling to a temperature bath with a relaxation time of 0.2 ps (50). The cutoff distance for Lennard-Jones interactions was set to 10 Å. Particle-mesh Ewald summation was used for electrostatic interactions (51). For an equilibration period of 4 ns, the systems were simulated at a constant pressure of 1 atm using the weak coupling algorithm with a relaxation time of 0.5 ps. Anisotropic pressure scaling was applied, i.e., all three box lengths were allowed to fluctuate independently. After equilibration, production simulations and free-energy calculations were carried out at constant volume. Simulation lengths for analysis of equilibrium properties of the free simulations (without umbrella potentials) were 10 ns (with ions) and 12 ns (without ions), respectively.
Umbrella sampling
The free energy of an ion as a function of the axial distance z from the membrane center and the radial distance r from the tube axis was determined by umbrella sampling (43). A harmonic biasing potential Kr,i(r – ri)2/2 + Kz,i(z – zi)2/2 was applied to the ion, with counter forces distributed uniformly over all carbon atoms of the CNT membranes. r and z are the axial distance from the membrane center and the radial distance from the tube axis respectively; ri and zi are the target positions; and Kr,i and Kz,i are the corresponding force constants in the ith umbrella-sampling window. In simulations with channel geometry, the ion was pulled into the single open tube. In the sieve geometry, one specific tube was selected. The ion-membrane target position zi was varied from 20 to 0 Å in 0.5-Å increments with a force constant Kz,i of 2.0 kcal mol−1 Å−2. In the radial direction, three types of restraints were used. Restraints with a target position ri = 0 Å and two different force constants Kr,i of 0.1 and 0.07 kcal mol−1 Å−2 were applied to all zi windows. Additionally, restraints with a target position ri of 5 Å and a force constant of 0.1 kcal mol−1 Å−2 were applied to windows with 12 Å ≤ zi ≤ 20 Å (where z = 0 corresponds to the membrane center). Altogether, this results in 99 overlapping windows with 390-ps sampling time in each window (38.6 ns total). The weighted histogram analysis method (44,52,53) was used for a collective analysis of the data.
The (unbiased) probabilities of the ion position P(z, r) that are directly obtained from the weighted histogram analysis method are radial distribution functions in the r direction and thus weighted by the size of the volume element 2πr. We compute the “local” free energy G(x, y, z) = –kBT ln(P(z, r)/(2πr)), where the distances x and y from the tube center in the direction of the corresponding axes satisfy r = (x2 + y2)1/2. kB is Boltzmann's constant, and T is the absolute temperature.
To obtain a one-dimensional potential of mean force (PMF), i.e., the free-energy profile G(z) along the pore axis, we integrate P(z, r) in the x, y plane. In the Results section, we compare the resulting PMFs, where P(z, r) is integrated from r = 0 to R = 5 Å, i.e.,
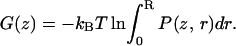 |
To analyze the hydration structure of ions in the bulk phase and in the center of the nanotubes, additional simulations were carried out with ions restrained only in the z direction (Kz,i = 2.0 kcal mol−1 Å−2, zi = 0 and 20 Å).
Computing local electrostatic potentials
Local electrostatic potentials play a central role in the translocation of ions through molecular pores. In explicit-solvent simulations, those potentials are determined by the fluctuating molecular structure of water in the bulk phase, the membrane interface, and the pore, as well as the position of ions and other charged or polarizable groups. In our simulations, we determine an average local charge-density distribution ρ(r) by binning atomic charges of the solvent and ions into a three-dimensional grid. The local electrostatic potential (and field) generated by ρ(r) is determined by the Poisson equation (54)
 |
(1) |
Since our simulations are performed in a periodic system with a rectangular box, we solve Eq. 1 on a discrete grid using fast Fourier transforms (FFTs). In reciprocal space, Eq. 1 reads
 |
(2) |
The procedure to compute average local electrostatic potentials using Eq. 2 is described elsewhere (I. C. Yeh, C. Peter, and G. Hummer, unpublished data). In this work, a grid of Nx = 180, Ny = 150, and Nz = 288 was used, resulting in a grid spacing of ∼0.2 Å. We tested the accuracy of the method by varying the grid size. In addition, we also included local dipole densities (relative to the grid centers) in the Poisson equation, with essentially unchanged results at the small grid sizes used. A related method has recently been used by Aksimentiev and Schulten (55), with the difference that in their method the atomic charges are smeared out by Gaussians and the corresponding potential is obtained directly on the relatively coarse grid of the particle-mesh Ewald FFT of the MD simulations before being averaged. Here, we can afford a fine grid because we average the charges and solve the Poisson equation only once. Since the Poisson equation is a linear equation, this leads to the same result as computing the potential at every time step and averaging the potential at the end.
Continuum electrostatics
A continuum electrostatics description approximating the system in the channel-type simulations consists of a rectangular periodic box filled with high-dielectric (εs = 78) medium and a low-dielectric (εi = 1) slab with a cylindrical (high-dielectric) pore. The ion is treated as a sphere with an internal permittivity of εi = 1 and a single charge at its center. We use the iterative, FFT-based method to solve the Poisson equation for such a system (56,57). The region of low dielectric permittivity (εi = 1) consists of the interior of the ion and the low-dielectric slab. Geometric parameters are the size of the periodic box (32 × 28.8 × 51.2 Å, corresponding to the simulated system), the thickness of the low dielectric (εi = 1) slab (20 Å), the pore radius (5.3 Å), and the ionic radius (1.5 Å). Algorithmic parameters are applied as described before (56,57). A grid spacing of 0.2 Å was used.
The solvation free energy of the ion was computed for several ion positions inside the box. As indicated in Fig. 4, we systematically varied the distance from the membrane center in the z direction and the distance from the channel axis in the x, y direction. The absolute solvation free energies obtained by this grid-based algorithm depend somewhat on the position of the ion center relative to the grid. However, since the aim of the study was not to compute absolute solvation free energies of the ion, these dependences could be largely eliminated. Grid effects were suppressed by subtracting from the solvation free energy of the ion in the system with membrane the solvation free energy of the ion at the same coordinate in an otherwise identical system without membrane.
FIGURE 4.

(A) Setup for continuum-electrostatics calculations: rectangular periodic box with high-dielectric (εs = 78) bulk medium and low-dielectric (εi = 1) slab with a cylindrical (high-dielectric) pore (10.6-Å diameter). Ion positions are varied from z = 0 to z = 25 Å at distances from the tube axis of r = 0 Å (black), 2 Å (red), 3 Å (green), 3.8 Å (blue), and 5.3 Å (magenta). (B) Continuum-electrostatics free-energy profiles along the z direction for several radial distances from the tube axis (color code as in A) and PMF obtained from radially integrated probabilities (dashed cyan line). Note that the free-energy curves were shifted to zero at the center of the bulk phase. (C) Comparison of free energies from umbrella sampling (thin lines) and continuum electrostatics (thick lines).
RESULTS AND DISCUSSION
In this section, we first determine the free energy for pore translocation of ions by umbrella sampling simulations. Then, we compare the energetics from all-atom simulations to those obtained from continuum electrostatics calculations. An analysis of the local water structure, electrostatic potentials, and internal energies will provide insights into the observed thermodynamics of ion translocation. Finally, the mobility of the ion in the pore and in bulk solution will be compared.
Free energies of pore translocation
Fig. 2 shows the two-dimensional free-energy surface G(z, r) of a sodium ion in a carbon nanotube as a function of the axial distance z of the ion from the membrane center and the radial distance r from the tube axis, as defined in the Methods section. We find that the free-energy difference between the bulk water phase and a narrow cylindrical region in the channel interior is small overall. Inside the tube, a radial distance of ∼2 Å from the tube axis is energetically favored for both geometries (channel and sieve). For the channel geometry, this region is slightly higher in free energy than the bulk water phase; for the sieve geometry, this region is slightly lower (and thus favored) compared to the bulk. In contrast, ion positions near the tube axis (r ∼ 0) are highly unfavorable. (Note that the data in Fig. 2 have been corrected for the “Jacobian” 2πr.)
FIGURE 2.

Free-energy surfaces for ion and water penetration into membrane pores. (Upper panels) Two-dimensional free-energy surface G(z, r) of a sodium ion in a CNT as a function of the axial distance of the ion from the membrane center z and the radial distance from the tube axis r as obtained from umbrella sampling (left, sieve geometry; right, channel geometry). (Lower left panel) Free-energy surface for water determined from the average density as –kBT ln(ρ/ρ0) + 4 kJ/mol (where ρ0 = 0.033 Å−3; same scale as the ion free energies). The white bar indicates the positions of the carbon atom centers. (Lower right panel) Radial ion, water, and carbon distribution in the tube (averages z = −5 to 5 Å; ion and water densities presented relative to the corresponding density in the bulk phase; carbon density in arbitrary units). As discussed in the Methods section, the “Jacobian” 2πr has been removed, so that uniform ion and water distributions would produce flat free energy profiles.
Fig. 3 shows the one-dimensional PMF of the ion projected onto the z coordinate by integrating the probability in the radial dimension from r = 0 to r = 5 Å. We see that the resulting apparent free-energy barrier for the ion to pass through a nanotube with an effective diameter of ∼10 Å is remarkably low (∼3–6 kJ/mol).
FIGURE 3.

Free energy of ion translocation through membrane pore. The two-dimensional PMF of the ion was projected onto the z coordinate by integrating the probability in the radial dimension from r = 0 to r = 5 Å. Results are shown for MD simulations of the wide nanotubes (black solid line, umbrella sampling/channel system; red solid line, umbrella sampling/sieve system; red dashed line, free simulation/sieve system) and continuum electrostatics (cyan dashed line). (Purple line and inset) MD simulations of narrow tubes in channel system.
Since the difference in the two-dimensional free energy between points in the bulk water phase and some regions inside the nanotube is small, a major contribution to the PMF is the restriction of the accessible space in the x, y direction, once the ion enters the tube. Not accounted for in Fig. 3 is an additional entropic contribution of kBT ln 4 = 3.5 kJ/mol that makes entering a tube more likely in a simulation with sieve geometry (four open tubes) than in one with channel geometry (one open tube).
Beyond this trivial entropy contribution, we find that it is slightly more favorable for the ion to enter a tube in a sieve system than in a channel system. As Fig. 3 shows, the free-energy barrier is ∼2 kJ/mol lower. This barrier reduction can be explained by the fact that the neighboring nanotubes in the sieve geometry are filled with water. The presence of this high-dielectric medium inside the membrane decreases the dielectric penalty for the ion to go into the pore by increasing the effective dielectric constant of the surrounding membrane.
For comparison we also analyzed 10 ns of unrestrained simulations (i.e., with a freely moving ion without umbrella potential). As predicted by the umbrella sampling simulations, the barrier for the ion to pass though the nanotube membrane is low and several passages can be observed for the sieve systems. In a simulation with a single Na+ ion, the ion enters one of the tubes once; in a simulation with five Na+/Cl− ion pairs, one of the Na+ ions enters a tube once and stays inside for ∼1.5 ns, and later in the course of the simulation one Na+ ion passes through a tube. We find at most one ion at a time in the membrane pores. A crude estimate for the PMF based on the simulation with five ion pairs in a sieve geometry is included in Fig. 3. It reproduces the magnitude of the free-energy barrier obtained from umbrella sampling. No passages are observed for the free simulations of the channel system, confirming that the free-energy barrier is considerably higher because of the lower number of open pores and the higher free-energy barrier of the single pore, as discussed. We note further that we did not observe any Cl− ions penetrating into the nanotube pores.
Also included in Fig. 3 is the PMF for the corresponding systems with narrower pores formed by (6,6) nanotubes (pore radius 4 Å with respect to carbon nuclei, corresponding to a ∼2.5 Å “effective” radius accessible to water, as obtained by subtracting the “radius” of the carbon atoms lining the pore). Here, the free-energy barrier estimated by umbrella sampling is considerably higher (∼120 kJ/mol instead of 5 kJ/mol). As in the wider pores, the barrier for the sieve geometry is only marginally lower (by ∼3 kJ/mol). In summary, a hydrophobic pore with an effective radius of ∼5–5.5 Å does not pose a high energetic barrier for a Na+ ion. In contrast, a narrow pore with an effective radius of ∼2.5 Å poses a very high barrier.
This dramatic change in ion permeability with a relatively small change in the pore diameter provides an explanation for potassium-channel gating by pore-width modulation (58). The hydrophobic gating region of the KcsA-channel structure in the closed form has a diameter of ∼4 Å at its narrowest point, whereas the gating region of the MthK structure in the open form has widened considerably—with a diameter of 12 Å at its narrowest point (22). Based on our estimates for a much simpler pore, such a change lowers the barrier for ion translocation from >100 kJ/mol (40 kBT) to near zero.
Continuum electrostatics
In this section, we explore to what extent a continuum electrostatics description of the system captures the essence of the all-atom free-energy calculations. A comparison of continuum calculations and explicit-solvent simulations will also allow us to explore the role of the solvent molecular structure for channel electrostatics. Fig. 4 shows the continuum electrostatic contribution to the solvation free energy of an ion (single charge; radius 1.5 Å) for several positions of the ion inside the box. As sketched in Fig. 4 A, the distance from the membrane center in the z direction and the radial distance from the channel axis in the x, y plane is systematically varied. The free-energy profiles in the z direction are shown in Fig. 4 B for several radial distances. The free energies are given relative to the solvation free energy of the ion at the center of the bulk water phase, where no dependence from the position relative to the channel axis (x and y coordinates) is observed. General observations are as expected: 1), the free-energy increases as the ion enters the channel and gets closer to the membrane center, thus getting deeper into the low-dielectric slab; 2), the free-energy barrier increases as the ion approaches the channel wall, i.e., as the layer of high dielectric, polarizable medium between the ion and the pore wall gets thinner. In Fig. 4 C, we compare the free energies from umbrella sampling and continuum electrostatics for a two-dimensional cut through the membrane (as a function of z and r). Overall, the free energies from the continuum electrostatics calculations are in a range comparable to those determined from explicit-solvent MD simulations. However, as a function of the distance from the channel axis, the free energy of the ion in the explicit-solvent simulations differs significantly from the continuum-electrostatics predictions. As expected by symmetry, the continuum electrostatics calculation finds the pore axis to be the most favorable location for the ion inside the pore. In explicit water, the ion almost exclusively stays in a cylindrical layer ∼2 Å from the channel axis. To “coarse grain” such molecular effects, we also compare radially integrated probabilities from continuum electrostatics and MD simulations (from r = 0 to 5 Å). The resulting PMFs are shown in Fig. 4 B and Fig. 3. The overall barrier is approximately twice as high in a continuum electrostatics description as in the simulations with explicit solvent, but with an absolute difference of only ∼4–5 kJ/mol (∼2 kBT). The free-energy profiles obtained from continuum electrostatics also have a somewhat different shape than those from explicit-solvent simulations. The latter level off after the ion has entered the pore, corresponding to a flat free-energy barrier, whereas the profiles from continuum electrostatics have a clear maximum at the membrane center. It should be noted, however, that a quantitative comparison of the two free-energy profiles requires some caution, since the continuum electrostatics results depend on model parameters such as tube and ion radius (i.e., the definition of the dielectric boundary), and the dielectric permittivity of the high dielectric medium. For example, the PMF obtained from the continuum model assuming an ionic radius of 2 Å is ∼1.5 kJ/mol lower, and is thus in better agreement with the MD data than the barrier height obtained for an ionic radius of 1.5 Å. Similarly, the barrier height obtained assuming a dielectric constant of 94, which corresponds to the TIP3P water model (59), is 0.4 kJ/mol lower than with a dielectric constant of 78 (experimental dielectric constant of water). For comparison, lowering the dielectric constant to 50 increases the barrier by ∼1.5 kJ/mol.
The shape of the free-energy profile obtained from explicit-solvent simulations of the membranes with narrow pores agrees well with that obtained from continuum electrostatics (not shown). However, the height of the barrier is substantially lower in the continuum case. This difference reflects mostly difficulties in defining a “correct” continuum system for a narrow molecular pore. Naively taking the parameters of the narrow-pore system (effective tube radius 2.5 Å, ion radius 1.5 Å) and solving the continuum electrostatics problem results in a much too low free-energy barrier of ∼40 kJ/mol compared to ∼120 kJ/mol from MD simulations. The reason is that in this simple approach the thin space between ion and membrane is filled by high-dielectric medium, which leads to dielectric screening that is not present in the “real” system. There, the space between ion and carbon wall is empty (except for a single water molecule). We realize that such small, empty volumes are more accurately accounted for in some continuum electrostatics simulations, for example through the rolling probe algorithm (60). This discussion highlights possible problems of continuum models for narrow cavities.
In summary, continuum electrostatics correctly predicts that a hydrophobic pore of ∼10 Å in diameter poses a comparatively low free-energy barrier for an ion, although not as low as was found in the explicit-solvent simulations. Continuum electrostatics also predicts that ions face a much higher barrier for the narrow pores. However, without optimizing the boundaries between high and low dielectric, that barrier is substantially lower than that of the explicit-solvent simulations. Moreover, for the wide pores continuum electrostatics does not explain the preferred path of ions through the channel—not at the center but in a narrow, cylindrical shell close to the pore wall. To explain this behavior, we next study the local water structure inside the pore and analyze the hydration shell of the ion.
Solvation structure, electrostatics, and energetics
In the lower right panel of Fig. 2, we compare the ion and water densities inside a nanotube as a function of the distance r from the tube axis. The ion densities are obtained from the umbrella sampling simulations (after unbiasing). Water (and nanotube carbon) densities are obtained from free simulations of the nanotube systems without ions. We find that water forms cylindrical shells, with a core of high water density in the tube center and a high-density layer along the tube wall (there is no difference in the water density profiles inside the nanotubes between sieve and channel systems). Similarly layered water structure inside narrow hydrophobic pores (61–64) or small cavities (65) has been observed in previous studies. We find that the ion is located predominantly in the cylindrical space between the two regions of high water density. This finding reproduces results by Lynden-Bell and Rasaiah (61), who investigated ion diffusion in hydrophobic tubes of different radii (61) using a rather different simulation setup (an infinite tube generated by periodically replicating a tube segment in one dimension; a repulsive cylindrical wall; minimum-image convention; and a different water model).
Fig. 5 shows the average local electrostatic potential on a cut through two nanotube centers of a sieve system parallel to the xz plane. (All observations made in the following section are qualitatively identical for the channel system.) In the simulation, the ion is restrained in the z direction so that it remains close to the membrane center. Note that by adding the restraint, only the z position of the ion deviates from equilibrium. From the restraint simulations, we can thus evaluate the equilibrium averages of the potential (and later ion hydration structure) conditional on the ion being at the membrane center. The left panel of Fig. 5 shows the total electrostatic potential, in contrast to the electrostatic potential generated by the solvent charges, shown on the right. In Fig. 5, the left tube contains the ion, which generates a region of high positive potential (on average) at the preferred location of the ion, a ring around the tube axis of radius ∼2 Å. The right tube is filled solely with water, and the local potential inside is a good representation for the local potential inside a nanotube in a simulation without any ions (not shown). Similar to the structure in the water density (Fig. 2, lower panels), one also observes a layer structure in the electrostatic potential. A one-dimensional potential profile along the x and z axes respectively, is displayed in Fig. 6 (calculated from simulations without ions), together with the distribution of oxygen and hydrogen atoms. We see that the layers of low (negative) electrostatic potential coincide with the regions of high oxygen and hydrogen density, and the layers of higher electrostatic potential coincide with the regions of low water density and with the regions of the low dielectric membrane interior. A small excess of hydrogen atoms is pointing toward the carbon walls and toward the low water density region, leading to a net polarization and the observed structure in the local electrostatic potential. The structure of the hydrogen bond network at inert or hydrophobic surfaces and the fraction of hydrogen atoms that point toward this surface has been analyzed extensively (66–70), leading to an electrostatic surface potential of ∼+0.5 V (70,71), consistent with the results shown in Figs. 5 and 6. This surface potential results from the ordering of water molecules without any external field and cannot be accounted for by continuum electrostatics.
FIGURE 5.

Average local electrostatic potential on a cut through two nanotube centers of a sieve system parallel to the xz plane with the ion inside the left tube (restrained to the membrane center in the z direction). (Left panel) Total electrostatic potential. (Right panel) Electrostatic potential generated by the solvent charges.
FIGURE 6.

Average total local electrostatic potential (A and B) and water atom distribution (C and D; solid lines, oxygen; dashed lines, hydrogen) of free MD simulations of the sieve system without ions. (A and C) Profile along the x direction through the centers of two CNTs. (B and D) Profile along the z direction through the low-dielectric/carbon-rich region of the membrane between two nanotubes. The coordinates of the cuts and the width over which the potential was averaged are indicated in A and B.
Does the electrostatic potential in the absence of the ion determine the location of the ion in the pore? With the given layer structure, a positively charged Na+ ion should occupy the tube region with negative electrostatic potential. Instead, as shown in Figs. 2 and 5, the ion is preferentially found in a region with initially positive potential but low water density. This result suggests that it is thermodynamically more favorable for the ion to move to a region of low water density but unfavorable positive potential, thus forcing a reorientation of the surrounding water molecules, than to move to the region of high water density, where it displaces water and disturbs its structure. This observation illustrates that conclusions based on average electrostatic potentials, without consideration of fluctuations, need to be drawn with caution, especially in the case of potentials due to solvent polarization without external field or ionic charge. Introducing a charged particle amounts to a large perturbation that may alter the solvent polarization qualitatively. This polarization effect can be seen on the right side of Fig. 5, where the electrostatic potential of the polarized solvent is displayed. Water reorientation leads to a highly negative potential at the site of the ion and the water structure is considerably perturbed compared to the nanotube without ion. The solvent polarization leads to an almost perfect screening of the ionic charge, so that in contrast to the electrostatic potential generated by the solvent, the (average) total electrostatic potential remains almost unperturbed by the ion except for its immediate vicinity (compare panels in Fig. 5).
A closer inspection of the water structure around the ion and the energetics of the ion in the tube and in the bulk solution may help to explain why the ion so readily moves into the tube. Fig. 7 presents the radial distribution function of the water oxygen atoms around the ion in a channel geometry. In the simulation, the ion is restrained in the z direction, with reference distances z0 to the membrane center of 0 (inside the tube) and 20 Å (outside the tube/in the bulk water phase). Fig. 7 shows that the extent of the first solvation shell—defined as the number of oxygen atoms that have a distance to the ion of ≤3 Å—is practically identical in the tube and in the bulk phase. The cumulative distribution function in the inset shows that the first solvation shell contains between five and six water molecules, which is slightly higher than the average coordination number for Na+ of 4.6 predicted by ab initio calculations (72). The average coordination numbers are given in Table 1, together with the fraction of structures that have five and six water molecules, respectively, in the first solvation shell. (In all cases the fraction of structures where the ion is coordinated by <5 or >6 water molecules is <2%.) Inside the nanotube, the fraction of ions with six water molecules in the first solvation shell is lower than in the bulk water phase. Fig. 8 shows the radial distribution of ions inside the nanotube with five- and six-membered solvation shells. We find that ions with five-membered solvation shells are preferentially found closer to the tube wall. Ions with six-membered solvation shells are only found at distances from the wall that are large enough to fit a water molecule between the ion and carbon wall. In Fig. 8, snapshots of the ion and its first solvation shell inside the nanotube are presented for both five- and six-coordinated ions. The spatial separation of the two states of the ion with six- and five-membered solvation shells is reflected in the correlation time of the fluctuation between the two states. As can be seen in Fig. 9 and Table 1, the interconversion between five- and six-coordinated states is much slower in the pore than in the bulk water phase (∼5 vs. 1 ps relaxation time). With a two-state analysis, we find that the dominant effect is a ∼5-fold slowdown of the transition from five to six water molecules in the pore compared to the bulk. One possible explanation, which combines this result and the radial distribution of the two states shown in Fig. 8, is that the ion with a five-membered solvation shell “sticks” to the tube wall, and needs to “dissociate” to bind to a sixth water molecule.
FIGURE 7.

Radial distribution function of the water oxygen atoms around the ion inside (solid line) and outside (dashed line) the tube (normalized by the density of TIP3P water of 0.033 Å−3) in the simulation of the channel system. (Inset) Cumulative distribution of oxygen atoms around ion.
TABLE 1.
Fraction of Na+ ions with coordination numbers of 5 and 6, average coordination number, and correlation time of coordination number
| n = 5 (%) | n = 6 (%) | 〈n〉 | τ (ps) | ||
|---|---|---|---|---|---|
| Channel | Tube | 48.5 | 49.7 | 5.49 | 4.8 |
| Bulk | 32.0 | 67.4 | 5.67 | 1.3 | |
| Sieve | Tube | 58.5 | 40.0 | 5.39 | 4.9 |
| Bulk | 35.0 | 64.2 | 5.64 | 1.1 |
n is the cordination number (water oxygen atoms closer than 3 Å); 〈n〉 is the average coordination number of Na+ ion; and τ is the correlation time for transitions between solvation shells with n ≤ 5 and n ≥ 6 members.
FIGURE 8.

Distribution of ions inside the nanotube as a function of the distance r from the tube axis for ions with five (solid black bars) and six (striped bars) water molecules in their first solvation shell. In the channel-geometry simulation, the ion was harmonically restrained near the membrane center (along the z direction, but not radially). Insets show representative structures.
FIGURE 9.

Normalized correlation function of fluctuations between five- and six-coordinated ions. The ions were harmonically restrained near the membrane center (z0 = 0 Å) and outside the tube (z0 = 20 Å). Solid and dashed lines are for channel and sieve simulations, respectively.
To investigate the energetics of ion permeation into the pores, we calculate their average internal energy 〈U〉 (electrostatic and Lennard-Jones interactions of the ion with the rest of the system). Table 2 shows the means and the variances of the internal energy of the ion inside the tube and in the bulk water phase for the channel and sieve systems of wide (10,10) and narrow (6,6) CNTs. The average internal energy of the ion is lower in the bulk phase than inside the pore, the difference being ∼10 kJ/mol for the wide tubes and ∼300 kJ/mol for the narrow tubes. Following perturbation theory (73,74), higher-order terms of the energy distribution can be used to obtain a rough estimate of the free energy:
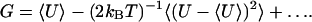 |
(3) |
TABLE 2.
Means and variances of the internal energy of the ion inside and outside the tube for the channel and sieve systems of wide and narrow CNTs
| 〈U〉 (kJ/mol) | (2kBT)−1(〈U2〉−〈U〉2) (kJ/mol) | ||
|---|---|---|---|
| Channel (wide) | Tube | −841.4 | 352.2 |
| Bulk | −852.6 | 381.9 | |
| Sieve (wide) | Tube | −838.1 | 355.0 |
| Bulk | −848.5 | 378.7 | |
| Channel (narrow) | Tube | −537.9 | 61.6 |
| Bulk | −850.5 | 361.6 | |
| Sieve (narrow) | Tube | −536.8 | 56.0 |
| Bulk | −846.0 | 360.3 |
U is the total potential energy of the ion, including ionic self interactions; 〈U〉 is the mean interaction energy; and (2kBT)−1(〈U2〉−〈U〉2) represents the energy variance.
We find that in the wide pores, the slightly higher average energies of the ion inside the pore are compensated in part by a narrowing of the energy distribution. A similar compensating effect was observed for water confined in nanotubes (47). For the narrow pores, the average energy lost by an ion entering the pore is large, ∼300 kJ/mol, and that loss cannot be compensated by the narrower distribution. However, the simple second-order perturbation theory is not quantitatively reliable (75) for such broad energy distributions.
Ion mobility in the pore
We also estimated local diffusion coefficients of the ion inside the pore along z direction and in the bulk fluid. From the positional correlation of ions in the umbrella sampling simulations we can estimate the diffusion coefficient as
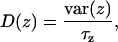 |
(4) |
where var(z) is the variance of the z-position of the ion, and τz is the correlation time of z. As shown in a previous study (76), this relation is the exact analytic limit of the Laplace-transform expression by Woolf and Roux (77). The diffusion coefficient inside the nanotube was determined as 2.3 × 10−5 cm2/s (± 0.2 × 10−5 cm2/s) by averaging the diffusion coefficients for all umbrella windows with a target position z0 from the membrane center between 0 and 5 Å. The diffusion coefficient of the ion outside the nanotube was determined as 1.5 × 10−5 cm2/s (± 0.2 × 10−5 cm2/s) by averaging all umbrella windows with a target position z0 from the membrane center between 15 and 20 Å. These results can be compared to diffusion coefficients reported by Lynden-Bell and Rasaiah (61) of 1.2 × 10−5 cm2/s for a Na+ ion in SPC/E water and ∼0.8 × 10−5 cm2/s for a Na+ ion inside a nanotube of ∼5-Å radius. This qualitative difference (of slower diffusion of the ion inside a nanotube compared to the faster diffusion found here) is probably due to the different setup. Lynden-Bell and Rasaiah investigate an infinite nanotube obtained by periodic replication, where diffusion requires correlated motion of essentially all particles of the system. Considering the large periodicity effects even in three dimensions (78), a one-dimensional periodic tube may likely result in slower diffusion compared to our open pore, where water can freely exchange with the bulk phase between the periodic copies of the membrane and the pore. The observation of higher diffusion inside the nanotube than in the bulk water phase seems to contradict other studies on diffusion in ion channels (79,80) where, in general, a slower diffusion inside the channel is observed. However, this seeming discrepancy can be explained by the dependence of the diffusion coefficient inside a water-filled nanotube on the tube radius. As shown in in Fig. 7 of Lynden-Bell and Rasaiah (61), a drastic increase in the diffusion coefficient is observed once the tube radius is larger than a critical value. This critical radius can be linked to the change in water structure, as can be seen in Fig. 5 of Lynden-Bell and Rasaiah (61). A likely factor in the fast diffusion here is also the hydrophobic pore walls that provide little resistance for water flow (47).
CONCLUSIONS
The main results of our simulation study are that narrow nonpolar pores (less than ∼5 Å effective diameter at a length of ∼21 Å) block ions, but only slightly wider pores (∼10-Å effective diameter) are highly permeable. In the narrow pore, ions get stripped off their solvation shell, resulting in a large energetic penalty; in the wider pore, the first shell remains intact, and the energetic penalty for translocation reduces essentially to the entropic loss for confining an ion in a pore. Remarkably, in the wide pore, we find not only a low barrier, but also a high ionic mobility along the pore axis, comparable to that of an ion in bulk water.
This strong dependence of the pore permeability on the pore diameter can provide a powerful gating mechanism in biological ion channels. Based on our simulations, a relatively modest change in the pore diameter is sufficient to convert a nonpolar pore between conducting and nonconducting states. Indeed, recent crystallographic studies of potassium channels showed hydrophobic pores of less than ∼5-Å diameter in the closed state (7) and ∼12-Å diameter in the open state (22).
With our simple model system, we could also explore to what degree continuum electrostatics calculations capture the energetics of ion translocation through nonpolar membrane pores. We find that the continuum calculations correctly predict that a hydrophobic pore of ∼10-Å effective diameter poses only a comparatively low free-energy barrier for an ion. Whereas the absolute barrier is somewhat higher than that found in the explicit-solvent simulations, relatively modest changes in the atom radii can improve the agreement.
However, probed in more detail, continuum electrostatics does not explain the preferred path of the ions through the channel—not at the center but in a narrow, cylindrical shell close to the pore wall. To explain this behavior, one needs to take the water structure inside the hydrophobic pore into account. As has been suggested elsewhere, the confined water phase inside narrow hydrophobic cavities has unique structural (61,62,65) and dynamic (27,47,64,81–83) properties that seem to affect water, proton, and ion transport through membrane channels.
Two studies on ion permeation through nanopores by Beckstein et al. (84,85), which came to our attention only after completion of this work, come to the similar conclusion that the molecular structure of the water phase inside the nanopore is crucial to the understanding of the ion permeation process. At a qualitative level, the results are similar, but Beckstein et al. (84) appear somewhat more pessimistic about the continuum results. They report only one-dimensional free-energy profiles, similar to the G(z) obtained here by integration in the radial direction, and find that the continuum barrier is dramatically higher for narrow pores and slightly lower for wide pores. These qualitative differences from our results (of a continuum barrier that is higher than the MD barrier for the wide pore, but lower for the narrow pore) can probably be explained by differences in the continuum calculations. In particular, we use periodic boundary conditions in both MD and continuum calculations; we do not use a “rolling probe” correction to define the high-dielectric region in the narrow tube; and we do not use the Poisson-Boltzmann equation with a finite ion concentration (neither for the ion in the pore nor in the bulk water phase).
We find that water inside the nanotube forms cylindrical shells of high and low water density, and that the Na+ ion is predominantly found in an interstitial cylindrical layer of low water density at a distance of ∼2 Å from the pore axis. This can be seen, for instance, in the radial distribution profiles shown in Fig. 2. Between the two water shells, the ion maintains an intact first hydration shell. The ion needs to replace fewer water molecules in the low-density region inside the tube than in the bulk water phase, which is an energetic advantage that partly compensates for the loss of the solvation contributions of the second hydration shell and beyond. Other studies of ions at the water/vapor interface have shown that ions can be found at interfaces, as long as they can maintain their first hydration shell (86).
As reported in a study by Dzubiella et al. (87), the transport properties of an ion through a nanopore are strongly linked to the water structure and the water density in the pore. Qualitative differences between the results of Dzubiella et al. (87) and those of our study in terms of ion permeability of a nanopore can be explained by the smaller effective pore radius in (87), which leads to a different water structure inside the nanopore (61,64,87). This observation further illustrates the sensitivity of ion permeability to modifications in the pore width and the resulting changes in the water structure. (However, in this context it is worth noting that comparisons of pore radii can be difficult because of differences in the definition, and are best performed by using the radial water density profiles.)
The results from continuum-electrostatics calculations, which correctly predict the magnitude of the free-energy barrier for ion translocation through a hydrophobic pore, confirm that a major contribution to this free-energy barrier is due to electrostatic interactions and the different dielectric environment outside and inside the pore. However, atomistic simulations show that the relationship between pore radius, structure of the water phase inside the pore, and ion permeability is rather complex. The local water structure—which cannot be easily accounted for in a simple dielectric model—potentially affects gating mechanisms in biological ion channels.
In conclusion, we want to emphasize that we have explored various methodologies to simulate ion translocation through membrane channels, in particular the treatment of long-range electrostatic forces (lattice-sum or reaction-field techniques) and the influence of system size and periodic boundary conditions. We found that different methodologies do not change the results qualitatively. A detailed study of these aspects will be presented elsewhere (C. Peter and G. Hummer, unpublished data).
Acknowledgments
The authors thank In-Chul Yeh for many helpful discussions. This study used the BIOWULF PC/LINUX cluster at the National Institutes of Health.
C.P. was supported through a fellowship of the German Academic Exchange Service (project No. D/03/18673), which is gratefully acknowledged.
Christine Peter's present address is Max-Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany.
References
- 1.Hille, B. 2001. Ionic Channels of Excitable Membranes, 3rd ed. Sinauer Associates,Sunderland, MA.
- 2.DeCoursey, T. E. 2002. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 83:475–579. [DOI] [PubMed] [Google Scholar]
- 3.White, S. H. 2004. The progress of membrane protein structure determination. Protein Sci. 13:1948–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Domene, C., S. Haider, and M. S. P. Sansom. 2003. Ion channel structures: A review of recent progress. Curr. Opin. Drug Discov. Dev. 6:611–619. [PubMed] [Google Scholar]
- 5.Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. L. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 6.Chang, G., R. H. Spencer, A. T. Lee, M. T. Barclay, and D. C. Rees. 1998. Structure of the MscL homolog from mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 282:2220–2226. [DOI] [PubMed] [Google Scholar]
- 7.Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- 8.Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a CIC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- 9.Chung, S. H., and S. Kuyucak. 2002. Recent advances in ion channel research. Biochim. Biophys. Acta. Biomemb. 1565:267–286. [DOI] [PubMed] [Google Scholar]
- 10.Ash, W. L., M. R. Zlomislic, E. O. Oloo, and D. P. Tieleman. 2004. Computer simulations of membrane proteins. Biochim. Biophys. Acta. 1666:158–189. [DOI] [PubMed] [Google Scholar]
- 11.Bond, P. J., and M. S. P. Sansom. 2004. The simulation approach to bacterial outer membrane proteins. Mol. Membr. Biol. 21:151–161. [DOI] [PubMed] [Google Scholar]
- 12.Åqvist, J., and V. Luzhkov. 2000. Ion permeation mechanism of the potassium channel. Nature. 404:881–884. [DOI] [PubMed] [Google Scholar]
- 13.Guidoni, L., V. Torre, and P. Carloni. 2000. Water and potassium dynamics inside the KcsA K+ channel. FEBS Lett. 477:37–42. [DOI] [PubMed] [Google Scholar]
- 14.Berneche, S., and B. Roux. 2001. Energetics of ion conduction through the K+ channel. Nature. 414:73–77. [DOI] [PubMed] [Google Scholar]
- 15.de Groot, B. L., and H. Grubmüller. 2001. Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science. 294:2353–2357. [DOI] [PubMed] [Google Scholar]
- 16.Tajkhorshid, E., P. Nollert, M. O. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud, and K. Schulten. 2002. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 296:525–530. [DOI] [PubMed] [Google Scholar]
- 17.Allen, T. W., O. S. Andersen, and B. Roux. 2004. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA. 101:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noskov, S. Y., S. Berneche, and B. Roux. 2004. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 431:830–834. [DOI] [PubMed] [Google Scholar]
- 19.Burykin, A., M. Kato, and A. Warshel. 2003. Exploring the origin of the ion selectivity of the KcsA potassium channel. Proteins. 52:412–426. [DOI] [PubMed] [Google Scholar]
- 20.Eisenberg, B. 2003. Proteins, channels and crowded ions. Biophys. Chem. 100:507–517. [DOI] [PubMed] [Google Scholar]
- 21.Lopez, C. F., S. O. Nielsen, P. B. Moore, and M. L. Klein. 2004. Understanding nature's design for a nanosyringe. Proc. Natl. Acad. Sci. USA. 101:4431–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- 23.Kuo, A., J. M. Gulbis, J. F. Antcliff, T. Rahman, E. D. Lowe, J. Zimmer, J. Cuthbertson, T. Ashcroft, F. M. Ezaki, and D. A. Doyle. 2003. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 300:1922–1926. [DOI] [PubMed] [Google Scholar]
- 24.Bass, R. B., P. Strop, M. Barclay, and D. C. Rees. 2002. Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science. 298:1582–1587. [DOI] [PubMed] [Google Scholar]
- 25.Miyazawa, A., Y. Fujiyoshi, and N. Unwin. 2003. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 423:949–955. [DOI] [PubMed] [Google Scholar]
- 26.Beckstein, O., P. C. Biggin, P. Bond, J. N. Bright, C. Domene, A. Grottesi, J. Holyoake, and M. S. B. Sansom. 2003. Ion channel gating: insights via molecular simulations. FEBS Lett. 555:85–90. [DOI] [PubMed] [Google Scholar]
- 27.Anishkin, A., and S. Sukharev. 2004. Water dynamics and dewetting transitions in the small mechanosensitive channel MscS. Biophys. J. 86:2883–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agre, P., L. S. King, M. Yasui, W. B. Guggino, and O. P. Ottersen. 2002. Aquaporin water channels—from atomic structure to clinical medicine. J. Physiol. 542:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sui, H. X., B. G. Han, J. K. Lee, P. Walian, and B. K. Jap. 2001. Structural basis of water-specific transport through the AQP1 water channel. Nature. 414:872–878. [DOI] [PubMed] [Google Scholar]
- 30.Wikström, M., M. I. Verkhovsky, and G. Hummer. 2003. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta. 1604:61–65. [DOI] [PubMed] [Google Scholar]
- 31.Wikström, M. 1998. Proton translocation by bacteriorhodopsin and heme-copper oxidases. Curr. Opin. Struct. Biol. 8:480–488. [DOI] [PubMed] [Google Scholar]
- 32.Parsegian, A. 1969. Energy of an ion crossing a low dielectric membrane: solutions to four relevant electrostatic problems. Nature. 221:844–846. [DOI] [PubMed] [Google Scholar]
- 33.Baudry, J., E. Tajkhorshid, F. Molnar, J. Phillips, and K. Schulten. 2001. Molecular dynamics study of bacteriorhodopsin and the purple membrane. J. Phys. Chem. 105:905–918. [Google Scholar]
- 34.Miloshevsky, G. V., and P. C. Jordan. 2004. Permeation in ion channels: the interplay of structure and theory. Trends Neurosci. 27:308–314. [DOI] [PubMed] [Google Scholar]
- 35.Kurnikova, M. G., R. D. Coalson, P. Graf, and A. Nitzan. 1999. A lattice relaxation algorithm for three-dimensional Poisson-Nernst-Planck theory with application to ion transport through the gramicidin A channel. Biophys. J. 76:642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung, S. H., M. Hoyles, T. Allen, and S. Kuyucak. 1998. Study of ionic currents across a model membrane channel using Brownian dynamics. Biophys. J. 75:793–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Im, W., S. Seefeld, and B. Roux. 2000. A grand canonical Monte Carlo-Brownian dynamics algorithm for simulating ion channels. Biophys. J. 79:788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Im, W., and B. Roux. 2002. Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J. Mol. Biol. 322:851–869. [DOI] [PubMed] [Google Scholar]
- 39.Moy, G., B. Corry, S. Kuyucak, and S. H. Chung. 2000. Tests of continuum theories as models of ion channels. I. Poisson-Boltzmann theory versus Brownian dynamics. Biophys. J. 78:2349–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corry, B., S. Kuyucak, and S. H. Chung. 2000. Tests of continuum theories as models of ion channels. II. Poisson-Nernst-Planck theory versus Brownian dynamics. Biophys. J. 78:2364–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards, S., B. Corry, S. Kuyucak, and S. H. Chung. 2002. Continuum electrostatics fails to describe ion permeation in the gramicidin channel. Biophys. J. 83:1348–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin, J. H., N. A. Baker, and J. A. McCammon. 2002. Bridging implicit and explicit solvent approaches for membrane electrostatics. Biophys. J. 83:1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torrie, G. M., and J. P. Valleau. 1974. Monte Carlo free energy estimates using non-Boltzmann sampling: Application to the sub-critical Lennard-Jones fluid. Chem. Phys. Lett. 28:578–581. [Google Scholar]
- 44.Roux, B. 1995. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 91:275–282. [Google Scholar]
- 45.Pearlman, D. A., D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham III, S. DeBolt, D. Ferguson, G. Seibel, and P. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91:1–41. [Google Scholar]
- 46.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 47.Hummer, G., J. C. Rasaiah, and J. P. Noworyta. 2001. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 414:188–190. [DOI] [PubMed] [Google Scholar]
- 48.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 49.Straatsma, T. P., and H. J. C. Berendsen. 1988. Free energy of ionic hydration: analysis of a thermodynamic integration technique to evaluate free energy differences by molecular dynamics simulations. J. Chem. Phys. 89:5876–5886. [Google Scholar]
- 50.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 51.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 52.Ferrenberg, A. M., and R. H. Swendsen. 1988. New Monte Carlo technique for studying phase transitions. Phys. Rev. Lett. 61:2635–2638. [DOI] [PubMed] [Google Scholar]
- 53.Kumar, S., D. Bouzida, R. H. Swendsen, P. A. Kollman, and J. M. Rosenberg. 1992. The weighted histogram analysis method for free-energy calculations on biomolecules.1. The method. J. Comput. Chem. 13:1011–1021. [Google Scholar]
- 54.Jackson, J. D. 1999. Classical. Electrodynamics. 3rd ed. John Wiley, NY.
- 55.Aksimentiev, A., and K. Schulten. 2005. Imaging alpha-hemolysin with molecular dynamics: Ionic conductance, osmotic permeability and the electrostatic potential map. Biophys. J. 88:3745–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peter, C., P. H. Hünenberger, and W. F. van Gunsteren. 2002. Solving the Poisson equation for solute-solvent systems using fast Fourier transforms. J. Chem. Phys. 116:7434–7451. [Google Scholar]
- 57.Peter, C., P. H. Hünenberger, and W. F. van Gunsteren. 2003. A fast-Fourier transform method to solve continuum-electrostatics problems with truncated electrostatic interactions: algorithm and application to ionic solvation and ion-ion interaction. J. Chem. Phys. 119:12205–12223. [Google Scholar]
- 58.Chung, S. H., T. Allen, and S. Kuyucak. 2002. Modeling diverse range of potassium channels with Brownian dynamics. Biophys. J. 83:263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yeh, I. C., and G. Hummer. 2004. Diffusion and electrophoretic mobility of single-stranded RNA from molecular dynamics simulations. Biophys. J. 86:681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richards, F. M. 1977. Areas, volumes, packing, and protein-structure. Annu. Rev. Biophys. Bioeng. 6:151–176. [DOI] [PubMed] [Google Scholar]
- 61.Lynden-Bell, R. M., and J. C. Rasaiah. 1996. Mobility and solvation of ions in channels. J. Chem. Phys. 105:9266–9280. [Google Scholar]
- 62.Sansom, M. S., I. D. Kerr, J. Breed, and R. Sankararamakrishnan. 1996. Water in channel-like cavities: structure and dynamics. Biophys. J. 70:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martí, J., and M. C. Gordillo. 2001. Temperature effects on the static and dynamic properties of liquid water inside nanotubes. Phys. Rev. E. 64:021504. [DOI] [PubMed] [Google Scholar]
- 64.Beckstein, O., and M. S. P. Sansom. 2003. Liquid-vapor oscillations of water in hydrophobic nanopores. Proc. Natl. Acad. Sci. USA. 100:7063–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brovchenko, I., D. Paschek, and A. Geiger. 2000. Gibbs ensemble simulation of water in spherical cavities. J. Chem. Phys. 113:5026–5036. [Google Scholar]
- 66.Pratt, L. R., and A. Pohorille. 2002. Hydrophobic effects and modeling of biophysical aqueous solution interfaces. Chem. Rev. 102:2671–2692. [DOI] [PubMed] [Google Scholar]
- 67.Lee, C. Y., J. A. McCammon, and P. J. Rossky. 1984. The structure of liquid water at an extended hydrophobic surface. J. Chem. Phys. 80:4448–4455. [Google Scholar]
- 68.Van Buuren, A. R., S. J. Marrink, and H. J. C. Berendsen. 1995. Characterisation of aqueous interfaces with different hydrophobicities by molecular dynamics. Colloids Surf. A. 102:143–157. [Google Scholar]
- 69.Gordillo, M. C., and J. Martí. 2000. Hydrogen bond structure of liquid water confined in nanotubes. Chem. Phys. Lett. 329:341–345. [Google Scholar]
- 70.Mamatkulov, S. I., P. K. Khabibullaev, and R. R. Netz. 2004. Water at hydrophobic substrates: curvature, pressure, and temperature effects. Langmuir. 20:4756–4763. [DOI] [PubMed] [Google Scholar]
- 71.Wilson, M. A., and A. Pohorille. 1994. Molecular dynamics of a water-lipid bilayer interface. J. Am. Chem. Soc. 116:1490–1501. [DOI] [PubMed] [Google Scholar]
- 72.Rempe, S. B., and L. R. Pratt. 2001. The hydration number of Na+ in liquid water. Fluid Phase Equilibr. 183:121–132. [Google Scholar]
- 73.Levy, R. M., M. Belhadj, and D. B. Kitchen. 1991. Gaussian fluctuation formula for electrostatic free-energy changes in solution. J. Chem. Phys. 95:3627–3633. [Google Scholar]
- 74.Hummer, G., L. R. Pratt, and A. E. Garcia. 1996. Free energy of ionic hydration. J. Phys. Chem. 100:1206–1215. [Google Scholar]
- 75.Hummer, G., L. R. Pratt, and A. E. Garcia. 1997. Ion sizes and finite-size corrections for ionic-solvation free energies. J. Chem. Phys. 107:9275–9277. [Google Scholar]
- 76.Hummer, G. 2005. Position-dependent diffusion coefficients and free energies from Bayesian analysis of equilibrium and replica molecular dynamics simulations. N. J. Phys. 34:1–14. [Google Scholar]
- 77.Woolf, T. B., and B. Roux. 1994. Conformational flexibility of o-phosphorylcholine and o-phosphorylethanolamine: a molecular dynamics study of solvation effects. J. Am. Chem. Soc. 116:5916–5926. [Google Scholar]
- 78.Yeh, I.-C., and G. Hummer. 2004. System-size dependence of diffusion coefficients and viscosities from molecular dynamics simulations with periodic boundary conditions. J. Phys. Chem. B. 108:15873–15879. [Google Scholar]
- 79.Tieleman, D. P., and H. J. C. Berendsen. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J. 74:2786–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith, G. R., and M. S. P. Sansom. 1998. Dynamic properties of Na + ions in models of ion channels: a molecular dynamics study. Biophys. J. 75:2767–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Waghe, A., J. C. Rasaiah, and G. Hummer. 2002. Filling and emptying kinetics of carbon nanotubes in water. J. Chem. Phys. 117:10789–10795. [Google Scholar]
- 82.Allen, R., S. Melchionna, and J. P. Hansen. 2002. Intermittent permeation of cylindrical nanopores by water. Phys. Rev. Lett. 89:175502. [DOI] [PubMed] [Google Scholar]
- 83.Saparov, S. M., and P. Pohl. 2004. Beyond the diffusion limit: water flow through the empty bacterial potassium channel. Proc. Natl. Acad. Sci. USA. 101:4805–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beckstein, O., K. Tai, and M. S. P. Sansom. 2004. Not ions alone: barriers to ion permeation in nanopores and channels. J. Am. Chem. Soc. 126:14694–14695. [DOI] [PubMed] [Google Scholar]
- 85.Beckstein, O., and M. S. P. Sansom. 2004. The influence of geometry, surface character, and flexibility on the permeation of ions and water through biological pores. Phys. Biol. 1:42–52. [DOI] [PubMed] [Google Scholar]
- 86.Jungwirth, P., and D. J. Tobias. 2001. Molecular structure of salt solutions: a new view of the interface with implications for heterogeneous atmospheric chemistry. J. Phys. Chem. B. 105:10468–10472. [Google Scholar]
- 87.Dzubiella, J., R. J. Allen, and J. P. Hansen. 2004. Electric field-controlled water permeation coupled to ion transport through a nanopore. J. Chem. Phys. 120:5001–5004. [DOI] [PubMed] [Google Scholar]