

Abstract
Antisense oligonucleotides present a powerful means to inhibit expression of specific genes, but their effectiveness is limited by factors including cellular delivery, biochemical attack, and poor binding to target. We have developed a systems model of the processes required for an antisense oligonucleotide to enter, gain access to its target mRNA, and exert activity in a cell. The model accurately mimics observed trends in antisense effectiveness with the stability of the oligonucleotide backbone and with the affinity/kinetics of binding to the mRNA over the time course of inhibition. By varying the model parameters within the physically realizable range, we note that the major molecular and cellular barriers to antisense effectiveness are intracellular trafficking, oligonucleotide-mRNA binding rate, and nuclease degradation of oligonucleotides, with a weaker dependence on total cellular uptake than might be expected. Furthermore, the model may serve as a predictive tool to design and test strategies for the cellular use of antisense oligonucleotides. The use of integrated mathematical modeling can play a significant role in the development of antisense and related technologies.
INTRODUCTION
Antisense oligonucleotides (AS ONs) are being developed clinically for applications in combating viral diseases, cancer, and inflammation (1). In addition, their ability to serve as transient “knockdowns” of gene expression is being exploited to study gene function and has been proposed as a strategy for systematic use in functional genomics (2,3). However, technical challenges have limited the widespread application of this approach to date. These include the selection of antisense sequences and structures with high affinity for their target, the tendency of oligonucleotides to be degraded by nucleases in cells and in serum, and the delivery of biologically effective doses in a specific and nontoxic fashion (4). In addition, the physical properties of the ON (and any delivery vectors) and several cellular variables affect the activity of AS ONs. These include the ability of the ONs to escape endosomes, the rate of ribonuclease H (Rnase H) cleavage within the cells, and the expression level of the particular mRNA being targeted. The molecular design of oligonucleotides with properties tailored to overcome one of these limitations (e.g., phosphorothioate (PS) backbone modification for enhanced stability) often results in a trade-off whereby other properties of the AS ON are compromised (e.g., binding affinity for target and increased propensity for protein binding by PS ONs).
We present an integrated framework for understanding and improving the effectiveness of AS ONs based on mathematical modeling of the cellular events that an AS ON undergoes as it attempts to reach and block the target. A few mathematical models have been developed previously to describe antisense activity under steady-state (5,6) and dynamic (7) conditions. These models are based on mass action kinetic balances as ONs move through extracellular and intracellular compartments, and our work builds upon these studies. In particular, we consider a greater number of compartments (bulk solution, cell surface, endosomal, and cytoplasmic) and ON states (e.g., adsorbed to cell surfaces, hybridized to target species, hybridized to nontarget species). Our goal is to determine those molecular design variables that can be manipulated to produce more effective AS ONs and to provide new insights for the timescales and conditions necessary for successful antisense effects.
MATERIALS AND METHODS
Systems model of oligonucleotide uptake, trafficking, and binding
The model consists of a set of mass action kinetic equations organized into three modules: cellular uptake, endocytosis and trafficking, and hybridization and catalysis. Except for the bulk concentration of oligonucleotide (Ab, in μM), concentrations are expressed as #/cell, and time is expressed in minutes. Transport between cellular compartments is assumed to occur via first-order processes in the concentration of the species in the upstream compartment. Binding and catalytic events are assumed to occur in accordance with their reaction stoichiometry. The binding and reaction steps, variables, and key parameters are depicted in Fig. 1.
FIGURE 1.

Schematic diagram of major interactions in the model for antisense delivery and activity. AS ONs (shown in red throughout) in the extracellular environment (Ab) bind to cellular surface molecules (R) and are internalized by endocytosis. The oligonucleotides that are internalized (Ai) must escape the endosome to enter the cytoplasm (Ac) before routing and degradation in lysosomes (X). Once in the cytoplasm, AS ONs can find their complementary target mRNA (M) and hybridize to form a duplex (D). The duplex is incapable of translation, and the mRNA portion may be degraded by Rnase H. Hybridization of cytoplasmic oligonucleotides to nontarget mRNA (N) is also possible, and these duplexes would also serve as substrates for Rnase H. Definition of all variables can be found in the main text.
Uptake
An oligonucleotide (or a ligand carrying an oligonucleotide) (Ab) binds to a receptor (R) on the cell surface, and the resulting complex (Cs) is internalized. It is also possible for the oligonucleotide to dissociate from the receptor. The second-order rate constant for oligonucleotide-receptor binding is k1, the first-order rate constant for dissociation is k−1, and the first-order rate constant for internalization is ki. In addition, degradation of bulk oligonucleotide occurs via first-order decay (with rate constant δb). The phase ratio, φ, serves as a conversion factor between bulk solution concentration (in μM) and cellular concentration (molecules/cell):
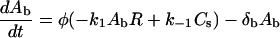 |
(1) |
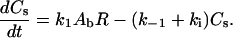 |
(2) |
Endocytosis and trafficking
Once the complex is internalized via endocytosis, the receptor dissociates to form an internalized receptor (Ri), which is capable of recycling (with rate constant kr) to the cell surface. The internalized oligonucleotide (Ai) is either routed for degradation (with rate constant δi) or escapes into the cytoplasm (with rate constant kt) to form cytoplasmic oligonucleotide (Ac):
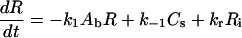 |
(3) |
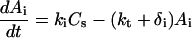 |
(4) |
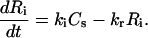 |
(5) |
Binding and catalysis
A cytoplasmic oligonucleotide may hybridize to either its target mRNA (M) or possibly to a nontarget mRNA (N) to form target (D) and nontarget (E) duplexes, respectively. The rate constant for association to target mRNA is ka and to nontarget mRNA is 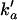 . Duplexes of ON-mRNA are recognized by Rnase H, which cleaves the RNA portion of the duplex to produce degraded target or nontarget mRNA. Because the amount of substrate (i.e., duplex) available for Rnase H is likely to be significantly smaller than the Michaelis constant of 0.2 μM (8), Michaelis-Menten kinetics reduces to a pseudo-first-order expression with rate constant veff, assumed to be identical for target and nontarget duplexes. The oligonucleotide is released intact after mRNA degradation and is available for further hybridization events. Because nontarget mRNA is far in excess to target mRNA, the total concentration of nontarget mRNA can be taken as a constant, N0. Although there are many possible nontarget mRNAs, they are lumped together as a single entity. The presence or absence of nontarget mRNA did not exert a substantial effect on model results. In competition with these hybridization events, oligonucleotide is degraded by cellular nucleases (with rate constant δc). In addition to antisense-mediated degradation, the target mRNA undergoes constitutive synthesis (with constant rate σm) and degradation (with first-order rate constant δm)
. Duplexes of ON-mRNA are recognized by Rnase H, which cleaves the RNA portion of the duplex to produce degraded target or nontarget mRNA. Because the amount of substrate (i.e., duplex) available for Rnase H is likely to be significantly smaller than the Michaelis constant of 0.2 μM (8), Michaelis-Menten kinetics reduces to a pseudo-first-order expression with rate constant veff, assumed to be identical for target and nontarget duplexes. The oligonucleotide is released intact after mRNA degradation and is available for further hybridization events. Because nontarget mRNA is far in excess to target mRNA, the total concentration of nontarget mRNA can be taken as a constant, N0. Although there are many possible nontarget mRNAs, they are lumped together as a single entity. The presence or absence of nontarget mRNA did not exert a substantial effect on model results. In competition with these hybridization events, oligonucleotide is degraded by cellular nucleases (with rate constant δc). In addition to antisense-mediated degradation, the target mRNA undergoes constitutive synthesis (with constant rate σm) and degradation (with first-order rate constant δm)
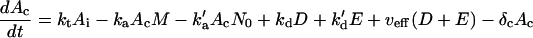 |
(6) |
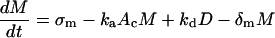 |
(7) |
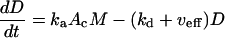 |
(8) |
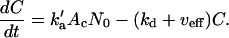 |
(9) |
Applicable ranges for parameter values were determined from the literature and/or our own measurements (Table 1). The equations were solved numerically using the ode15s solver for stiff differential equations in MATLAB 6.1 (The MathWorks, Natick, MA).
TABLE 1.
Values of model parameters
| Parameter | Meaning | Base value | References |
|---|---|---|---|
| k1 | Adsorption of antisense to receptor (saturable) | 0.15 μM−1 min−1 | (10,12) |
| k−1 | Desorption of antisense from receptor | 0.03 min−1 | (11) |
| ki | Internalization | 0.03 min−1 | (11) |
| kr | Recycling of receptors | 0.06 min−1 | (42) |
| kt | Trafficking to mRNA locale (cytoplasm) | 0.003 min−1 | (11) |
| ka | Antisense-mRNA association (target) | 0.1–10 μM−1 min−1 | (17) |
| kd | Antisense-mRNA dissociation (target) | 0.001 min−1 | (17) |
 |
Antisense-nontarget association | 10−3μM−1 min−1 | Estimated |
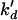 |
Antisense-nontarget dissociation | 0.001 min−1 | (17) |
| veff | RNase H cleavage | 0.003 min−1 | (17) |
| σm | mRNA synthesis rate | 15 cell−1 min−1 | (20) |
| δm | Natural decay of target mRNA | 0.03 min−1 | (20,28) |
| δb | Decay of antisense in bulk | 0.003 min−1 | (11,16) |
| δi | Degradation or efflux of internalized antisense | 0.06 min−1 | (11,43) |
| δc | Decay of antisense in cytoplasm | 0.12 min−1 | (11,16) |
| Ab0 | Initial bulk concentration | 600 nM | Chosen |
| R0 | Free receptors on surface | 1 × 105 cell−1 | (10,12,44) |
| M0 | Target mRNA | 500 cell−1 | (20,28) |
| N0 | Nontarget mRNA | 7 × 106 cell−1 | (20) |
| φ | Extracellular phase ratio | 5 × 10−9 cell μM molecule−1 | Estimated |
Experimental determination of uptake and activity in CHO cells
CHO-K1 cells (ATCC, Manassas, VA) were stably transfected with the pd1EGFP plasmid by a protocol consisting of 4 h transfection in serum-free medium with Lipofectamine 2000 (Invitrogen, Carlsbad, CA), followed by selection in medium containing serum and G418 (Sigma, St. Louis, MO), clonal dilution, expansion, and flow cytometric analysis (Barnes et al., unpublished). An AS ON effective against pd1EGFP in this cell line was identified by screening a number of candidates (B. M. Dunham, L. K. Lee, Z. Li, and C. M. Roth, unpublished). A modified form of this PS oligonucleotide with the Alexa647 fluorophore conjugated at the 5′-end (A-ODN157) was purchased from Integrated DNA Technologies (Coralville, IA). In addition, the identical sequence was obtained without the fluorescent tag (ODN157), as well as a tagged, scrambled ON sequence (A-ODNcontrol).
Each oligonucleotide was combined with Lipofectamine 2000 at a weight ratio of 2.5 (Lipofectamine/ON) at an ON concentration of 100 nM and incubated with the CHO-pd1EGFP cells for varying durations. At each time, replicate wells of cells were washed, trypinized, pelleted by centrifugation, suspended in phosphate-buffered saline maintained on ice, and subjected as quickly as possible to flow cytometry. Forward and side scatter characteristics were used to remove dead cells from the analysis, and the remaining cells were analyzed for Alexa uptake and pd1EGFP inhibition. Each was determined from the geometric mean of the fluorescence intensity for its channel, with pd1EGFP values normalized to those for untreated pd1EGFP cells.
Experimental measurement of gene expression dynamics in H35 cells
Antisense inhibition experiments against gp130 were performed with H35 rat hepatoma cells in 24 well plates under serum-free conditions as previously described (9).
RESULTS
We have developed an integrated mathematical model of the cellular events encountered by an AS ON to provide a framework for interpreting quantitative molecular and cellular measurements and to evaluate strategies for improving the success of AS ONs. The AS ONs, cellular uptake receptors and target and nontarget mRNAs are tracked through various cellular locations and molecular states via mass conservation equations (Fig. 1).
Cellular uptake and trafficking of oligonucleotides
It is commonly believed that the uptake of DNA by cells is low due to the electrostatic repulsion between the anionic charges residing on DNA and those residing on cell membranes from their glycocalyces and phospholipids membranes. ONs, however, are small enough to find local regions on cell surface proteins (which may be cationic) to which favorable binding occurs, followed by adsorptive endocytosis. As a result, significant quantities of ONs are delivered to cells, and reasonably accurate measurements of uptake are available from radiolabel measurements (10–12). Indeed, these studies have demonstrated that significant quantities of oligonucleotides are taken up by cells, much more than would be required for stoichiometric inhibition of target mRNAs expressed on the levels of hundreds to thousands per cell. Although there is not a receptor with a known functional role in mediating ON uptake, cell membrane proteins have been identified as mediating uptake via adsorptive endocytosis, resulting in saturation of uptake at moderate ON concentrations (a nonsaturating component of uptake, believed to be mediated by fluid-phase endocytosis, has also been observed at high ON concentrations) (10–13).
Given that over the course of a few hours, hundreds of thousands of oligonucleotides can enter a cell, what is their fate? After internalization, oligonucleotides must escape vesicular compartments, avoid degradation and efflux, find their complementary target mRNA, hybridize, and remain associated long enough to mediate Rnase H-catalyzed destruction of the target mRNA or to inhibit translation sterically. The intracellular trafficking characteristics are the most difficult to quantify experimentally, but sufficient studies on the endosomal escape of oligonucleotides and the dynamics of endosomal and lysosomal progression permit order-of-magnitude estimation of their rates (14,15) (Table 1). After internalization by endosomes, oligonucleotides may either be routed to lysosomes or achieve escape into the cytoplasm. Significant endosomal concentrations are reached rapidly, peaking at 30 min (results not shown), and the levels of cytoplasmic oligonucleotide peak later, in the range 330–360 min (Fig. 2 A). Because of the saturable nature of receptor-mediated internalization, the maximum level of ON in the cytoplasm varies by a little over twofold as the bulk ON concentration increases fivefold (Fig. 2 A). In contrast, the maximum endosomal and cytoplasmic levels are much more sensitive to the rates of lysosomal routing versus cytoplasmic escape. Specifically, increased endosomal escape leads to a dramatic increase in the cytoplasmic level of oligonucleotide (Fig. 2 B), whereas increased lysosomal routing causes a reciprocal decrease in the cytoplasmic level of oligonucleotide (results not shown). However, the time at which maximal cytoplasmic concentration is reached is relatively insensitive to the values of these parameters. Thus, from the parameter variations associated with uptake and trafficking, the escape from endosomes before lysosomal routing and degradation appears to be the critical step in achieving high levels of oligonucleotide in the cytoplasm.
FIGURE 2.

Effect of model parameters on cytoplasmic concentrations. (A) Effect of bulk oligonucleotide on cytoplasmic concentration, Ac. Curves correspond to bulk concentrations (Ab) of {200, 400, 600, 800, and 1000} nM. (B) Effect of endosomal escape rate on Ac. Curves correspond to escape rate constants (kt) of {0.001, 0.003, 0.010, and 0.030} min−1.
DNA-RNA hybridization and mRNA cleavage
After escape from endosomes, cytoplasmic oligonucleotides are free to find and associate with their target mRNA, and oligonucleotide-mRNA duplexes may be subsequently degraded by Rnase H. Because ONs bound to mRNA are presumably protected themselves from nuclease digestion, the model predicts that slightly greater and longer-lasting ON presence in the cytoplasm will result for a complementary as opposed to noncomplementary ON (Fig. 3 A). To test this prediction, we delivered Alexa647-tagged ONs of both complementary (versus pd1EGFP mRNA and termed A-ON157) and noncomplementary (a scrambled permutation of A-ON157 termed A-ODNcontrol) sequences to pd1EGFP-CHO-K1 cells using the commercial reagent, Lipofectamine 2000. We indeed found a modest extended delivery profile of the complementary ON as opposed to noncomplementary ON (Fig. 3 B), in excellent agreement with the model. Furthermore, the model predicts accurately the dynamics of the uptake. It should be noted that the model parameters in Fig. 3 A are the same as those in the base case except that the intracellular degradation rate was reduced from 0.012 min−1 to 0.003 min−1. The time course of pd1EGFP inhibition by the Alexa-labeled ON-157 was also monitored by flow cytometry. The time course is very consistent with the model predictions, again using base case parameters and a degradation rate of 0.003 min−1 (Fig. 3, C and D). The pd1EGFP is a destabilized form of GFP with a half-life of ∼1 h, minimizing the time lag between changes in mRNA and protein expression. Thus, one set of model parameters reproduces faithfully both the dynamics of uptake and dynamics of inhibition, suggesting that we have captured the essential relationship between these processes in the model.
FIGURE 3.

Interplay of uptake and activity dynamics. (A) Predicted difference in cytoplasmic concentration of a fast binding (ka = 10.0 μM−1 min−1) versus nonbinding (ka = 0) oligonucleotide. Base case parameters were used in the model except for a cytoplasmic degradation rate of 0.003 min−1. (B) Experimental results for uptake of fluorescently labeled oligonucleotides that are complementary (A-ODN157) or noncomplementary (A-ODNcontrol) as a function of time after delivery to CHO-pd1EGFP cells, as determined by flow cytometry. (C) Predicted dynamics of antisense activity for the same parameter set as in A. (D) Experimentally determined geometric mean pd1EGFP fluorescence as determined by flow cytometry for treatment of the same samples analyzed in B for the complementary oligonucleotide A-ODN157. Experimental values shown are mean ± SE, with the error bars in D being mostly smaller than the symbols.
The time course of target mRNA inhibition was explored further for the base case parameters in Fig. 4. In addition, a set of model simulations was performed in which each of the model parameters was allowed to vary randomly in the range of 10% or 20% of the base case value. This variation in parameters did not change appreciably the time course of the antisense effect, but it did result in a modest variability in the extent of inhibition at any time. Since the kinetic rate parameters and numbers of species (e.g., target mRNA molecules) is likely to vary to this extent, a distribution of inhibition levels is expected to occur. Indeed, we observed a substantial spreading of fluorescence levels in the flow cytometry of antisense-inhibited CHO-pd1EGFP cells (data not shown). The range depicted in Fig. 4 is the maximum variation observed over 100 simulation runs. The standard error would be considerably less but would require efficient sampling over the high-dimensional space spanned by the parameters.
FIGURE 4.

Effect of random variation in model parameters on predictions. The antisense activity (change in target mRNA levels) was simulated for the base case parameters and is shown in the solid line. The values of all model parameters were each varied randomly within 10% (dotted lines) or 20% (dashed lines), and the range of mRNA dynamics resulting from 100 runs are shown.
As expected from their effects on cytoplasmic levels, bulk concentration of oligonucleotide has only a modest effect on the maximum extent and dynamics of antisense inhibition observed (Fig. 5 A), whereas endosomal escape has a more significant effect (Fig. 5 B). The time of maximum inhibition did not vary beyond the range of 210–240 min for any bulk concentration or endosomal escape rate tested. The oligonucleotide-mRNA hybridization rate and the oligonucleotide stability exhibited the most profound influences on antisense activity. Under the normal range observed experimentally for hybridization rate (see Table 1), outputs vary from no antisense effect to substantial effect (Fig. 6). When the half-life of the ON is very short (∼20 min), the model predicts that little inhibition will be observed, regardless of hybridization rate (Fig. 6 A). For an ON half-life on the order of hours, the association rate between ON and target mRNA dictates the extent of inhibition but does not significantly affect the timescale over which inhibition occurs (Fig. 6 B). A very stable ON (half-life ∼40 h) could result in extended duration of inhibition; however, a sufficiently fast hybridization rate would still be required (Fig. 6 C).
FIGURE 5.

Effect of bulk concentration and trafficking on antisense inhibition. The minimum target mRNA levels, normalized to the initial values, as a function of (A) bulk oligonucleotide concentration and (B) endosomal escape rate. In both cases, the minima occurred at times between 210 and 240 min.
FIGURE 6.

Interplay of hybridization rate and stability on antisense activity. (A) Predicted mRNA dynamics as a function of hybridization rate for (A) unstable (δm = 0.03 min−1), (B) moderately stable (δm = 0.003 min−1), and (C) very stable (δm = 0.0003 min−1) oligonucleotides. In each case (A–C), the hybridization rate constants are ka = {0.1, 1.0, and 10.0} μM−1 min−1. (D) Experimentally observed dynamics, measured using real-time polymerase chain reaction of the rat gp130 mRNA in H35 cells treated with PS (moderate stability) oligonucleotides selected for fast binding and slow binding, respectively.
The intracellular half-lives of phosphodiester (PO) ONs have been estimated to be tens of minutes and those of PS ONs on the order of several hours, with some variation depending on delivery method and cell type (16). Thus, Fig. 6, A and B, corresponds to our expectations for PO and PS ONs, respectively. We can compare these predictions to experiments quantifying the levels of the rat gp130 mRNA targeted by fast binding and slow binding antisense PS ONs (9). The dynamics of inhibition and their dependence on binding rate match well the predictions of the model (Fig. 6 D, cf. Fig. 6 B). The fact that the model describes accurately both the magnitude and dynamics of inhibition, as the critical parameter of hybridization association rate is varied, testifies to the utility of the model in understanding antisense effects at the cellular level.
It is generally assumed that a major factor in the effectiveness of AS ONs is the recruitment of Rnase H to ON-mRNA duplexes, followed by cleavage of the target mRNA. As the concentration of ON-mRNA duplexes should be always significantly less than the Michaelis constant (17), we employ a pseudo-first-order expression for Rnase H cleavage (Eqs. 6, 8, and 9). The effect of the Rnase H cleavage rate on maximum antisense inhibition was relatively minor, both at low (Fig. 7 A) and high (Fig. 7 B) rates of hybridization. The time of maximum inhibition was only slightly affected, ranging 225–270 min for ka = 1.0 μM−1 min−1 and 195–255 min for ka = 10.0 μM−1 min−1.
FIGURE 7.

Effect of Rnase H degradation on antisense activity. Minimum mRNA levels, normalized to the initial values, as a function of the first-order rate constant for Rnase H-mediated target degradation, for moderate oligonucleotide-mRNA binding (ka = 1.0 μM−1 min−1), and for more rapid oligonucleotide-mRNA binding (ka = 10.0 μM−1 min−1).
Finally, we studied the predicted effect of mRNA copy number and stability on antisense effectiveness. The steady-state (i.e., in the absence of antisense administration) mRNA level is the ratio of the synthesis rate to the decay rate of the mRNA. For a given mRNA copy number, the effectiveness of antisense inhibition will depend on the stability of the mRNA, with rapidly turned over mRNA species resulting in lower levels and shorter durations of inhibition (Fig. 8 A). As a corollary, if the synthesis rate is held constant and the decay rate is varied, lower copy number transcripts are more difficult to inhibit, as they more rapidly regenerate their steady-state mRNA level (Fig. 8 B). However, for a set of transcripts with comparable half-lives, the level and dynamics of inhibition are relatively insensitive to copy number (not shown).
FIGURE 8.

Effect of mRNA decay on antisense inhibition. (A) The mRNA decay rate (δm) was varied with the initial mRNA level held constant at 500 copies per cell, i.e., the mRNA synthesis rate (σm) was varied correspondingly. Individual curves correspond to mRNA decay rates of {0.001, 0.003, 0.01, 0.03, and 0.1} min−1. (B) The mRNA decay rate was varied with the mRNA synthesis rate held constant, thus resulting in a decrease in initial mRNA level with increasing decay rate. Individual curves correspond to mRNA decay rates of {0.003, 0.075, 0.03, 0.075, 0.3, and 0.75} min−1.
DISCUSSION
Our model builds upon previous mathematical models of gene and oligonucleotide interactions with cells. The compartmentalization of DNA being delivered to a cell has been considered in previous models of nonviral gene transfer (14,18,19). It is generally assumed that trafficking among compartments occurs via first-order processes. As a consequence, these barriers control to a considerable extent the time required for a biological effect and the efficiency of the process. Previous models of antisense effects focused on the mechanisms (Rnase H versus various modes of steric hindrance) in antisense activity and on the correspondence between down-regulation at the mRNA level and at the protein level (5–7). Our model combines the elements of uptake, trafficking, hybridization, and catalysis to provide a comprehensive, systems-level framework for antisense activity at the cellular level.
Estimates for the parameters in the model are taken from the accumulated AS ON literature. The cellular events—uptake, trafficking, and degradation—have all been monitored in a variety of experiments but typically not all simultaneously in the same cells under the same conditions. Although the parameters used are thus from disparate experimental protocols, in most cases they should be reasonable order of magnitude estimates. The expected range of variability of these parameters was explored to determine the sensitivity of antisense activity to them, both individually to see how one parameter affects the new antisense activity but also through random variation in each parameter (Fig. 4). In addition, the mRNA levels were varied, as specific transcripts may have abundances ranging from a few copies per cell to ∼104 per cell (20). Modifications to AS ON sequence and structure are designed to influence the kinetics of hybridization, the accessibility of the mRNA portion of ON-mRNA duplex to Rnase H degradation, and the degradation of the ON to nuclease digestion. Quantitative measurements of these processes can be made in vitro and provide relatively precise values for inclusion in the model, to the extent that the rates of these processes in vitro correspond to those in cells.
With this mathematical framework, we can both identify the steps most likely limiting the effectiveness of AS ONs and evaluate the feasibility of potential molecular designs for improving their effectiveness. AS ONs must overcome several barriers to enter cells, gain access to the target mRNA, and inhibit its expression. Upon reaching the cytoplasm, AS ONs can interact with mRNA through hybridization events that are potentially limiting, until they are compartmentalized, degraded, or cleared. The molecular properties of the AS ONs influence many of the events controlling AS effectiveness, thus providing an opportunity for improvement via molecular design.
For example, an oligonucleotide is a highly anionic molecule, and this is presumed to hinder entrance into the cell. Yet, significant quantities of ON do enter cells, achieving intracellular concentration on the order of 10–100 nM, based on 104–105 molecules entering a cell of volume 1 picoliter (10,21). However, high concentrations (hundreds of nanomolar) are required for the highest levels of uptake. The most efficient and selective uptake may be conferred by a delivery system in which one or a few high affinity (e.g., epidermal growth factor) ligands are used to deliver a depot of oligonucleotide, such as in a targeted liposome or vesicle (22–24).
Nonetheless, delivery of ONs, both in naked form and using liposomes or polymeric delivery vectors, appears to be saturable (10–12), and this limits the utility of increasing bulk ON concentration as a means of improving antisense effectiveness (Figs. 2 A and 5 A). A considerable amount of research is being directed toward the development of carriers that will protect DNA and shuttle it into cells. However, the major delivery obstacles are inside the cell, particularly escape from endosomes while avoiding routing to lysosomes and degradation (Figs. 2 B and 5 B). As such, the design of delivery vectors is increasingly focusing on materials that will aid in the escape from endosomes (15,25–27).
Another factor affecting antisense activity is the stability of the particular mRNA target. Genome-wide analyses of mRNA decay have been conducted in some mammalian cells, and these suggest a relatively wide distribution of transcript half-lives in which many mRNAs are stable for hours but some are turned over in as little as 30 min (20,28,29). This particular range of values can have a significant effect on the effectiveness of an antisense treatment (Fig. 8), as those transcripts with short half-lives are already “programmed” to maintain their baseline levels of expression in the presence of the cell's natural degradation processes. As such, it is likely that those genes that are rapidly turned over, such as “early response genes” induced in inflammatory responses, may prove recalcitrant to antisense treatment.
The most easily controlled design variable of an AS ON is its sequence. The ON sequence chosen can have a dramatic effect on its antisense effectiveness, as many target regions of the mRNA are thermodynamically or kinetically inaccessible. Experimental techniques for identifying accessible regions of mRNA, such as oligonucleotide array hybridization and Rnase H mapping, have emerged (30,31). Furthermore, computational design approaches based on the structure-dependent thermodynamics of ON-mRNA interaction can be used to select effective AS ONs (9,32,33). As shown by our model simulations and the experimental data of numerous researchers, antisense activity is quite dynamic, and thus hybridization association rate rather than equilibrium governs activity. However, affinity is proportional to association (“on”) rate for oligonucleotide-mRNA hybridization (17), and so algorithms that select for high affinity also select for high association rate. The measured numerical values of association rate are such that they span a range over which little to no activity (at low association rates) to strong and potentially prolonged antisense activity (at high association rate) are predicted (Fig. 6).
The antisense molecule can also be manipulated via its chemistry. Indeed, most efforts to improve the performance of AS ONs have focused on chemical modification, with particular emphasis on nuclease resistance. As can be seen in Fig. 6, ONs with short half-lives have no opportunity to inhibit the target mRNA, and the duration of the antisense effect is dependent on the ON exhibiting extended intracellular stability (in conjunction with the turnover rate of the mRNA as per previous discussion). For most therapeutic applications, a duration of effect of at least days and preferably longer is required. As such, nuclease resistance is a necessary condition for a strong antisense effect, but it is not sufficient because of the sensitive interplay with the hybridization association rate. Thus, a major challenge is designing ON chemistry with improved stability without any significant loss in hybridization association rate (or specificity). Second- and later-generation oligonucleotides such as 2′-O-methyl oligoribonucleotides, C-5 propynylated oligonucleotides, and locked nucleic acids may meet these criteria (34–36). How these modifications affect delivery and trafficking has been less extensively explored. Modeling frameworks such as the one presented here can be a useful tool in interpreting potential trade-offs among delivery, stability, and hybridization rate.
To be useful, the model should be applicable to a wide variety of cell types and antisense AS ONs. We specifically studied two systems experimentally in our work. Both utilized PS oligodeoxynucleotides, but in one case gp130 was the target mRNA in H35 rat hepatoma cells, and in the other pd1EGFP was the target in CHO cells. A number of parameters in the model may depend on the target and cell type, including the mRNA level and constitutive degradation rate, the adsorption and internalization rates, the lysosomal sorting and endosomal escape rates, and the cytoplasmic degradation/efflux rate. With this number of parameters, the general estimation problem would require a global optimization approach (37), so we sought the simplest explanation for cell-type differences. Based on the observed dynamics being of longer duration in CHO cells, the cytoplasmic degradation rate was the most logical candidate parameter. Indeed, changing the value of this parameter fourfold from the base case (which was used to simulate gp130 in H35 cells) resulted in excellent agreement for both uptake and activity dynamics in the CHO cells. The dynamics that we observed are in the range observed for other cell culture systems, in which maximum inhibition may occur anywhere from 4 h to 48 h, depending on the ON structure and cell type (38,39). Thus, at least for these experimental systems, the model provides a useful construct to understand cell-type differences in antisense behavior.
In summary, our model provides a framework for understanding the interplay of mechanistic events in overall antisense effectiveness. It predicted accurately the timescale and effect of molecular and cellular parameters, including the ON-mRNA hybridization rate and ON degradation rate, on antisense activity. By variation of model parameters, it shows that delivery to the cell is likely not a major barrier to effectiveness, but trafficking, hybridization, and stability within the cell are. AS ONs can be modified primarily through choice of sequence and ON chemistry, and our work provides a framework for rationally designing these. With the advent of RNA interference (RNAi), this framework could be modified to replace ON hybridization and Rnase H-mediated destruction of the mRNA with formation of a RNA-induced silencing complex via short interfering RNA followed by target mRNA degradation (40,41). Through a systems understanding of the molecular and cellular processes affecting the activity of oligonucleotides, both in antisense and in RNAi, it should be possible to accelerate their development in a variety of research and applications areas.
Acknowledgments
The author thanks Yannis Androulakis for helpful discussions as well as Sumati Sundaram and Li Kim Lee for careful reading and for help with figure preparation. The author is particularly grateful to Jennifer Oddo and Sandra Viriyayuthakorn for performing the CHO-pd1EGFP experiments.
This work was supported in part by The Whitaker Foundation (TF 02-002), the Charles and Johanna Busch Memorial Fund, and National Institutes of Health grant 5 R01 GM65913.
References
- 1.Opalinska, J. B., and A. M. Gewirtz. 2002. Nucleic-acid therapeutics: basic principles and recent applications. Nat. Rev. Drug Discov. 1:503–514. [DOI] [PubMed] [Google Scholar]
- 2.Dean, N. M. 2001. Functional genomics and target validation approaches using antisense oligonucleotide technology. Curr. Opin. Biotechnol. 12:622–625. [DOI] [PubMed] [Google Scholar]
- 3.Taylor, M. F., K. Wiederholt, and F. Sverdrup. 1999. Antisense oligonucleotides: a systematic high-throughput approach to target validation and gene function determination. Drug Discov. Today. 4:562–567. [DOI] [PubMed] [Google Scholar]
- 4.Lee, L. K., and C. M. Roth. 2003. Antisense technology in molecular and cellular bioengineering. Curr. Opin. Biotechnol. 14:505–511. [DOI] [PubMed] [Google Scholar]
- 5.Ramanathan, M., R. D. MacGregor, and C. A. Hunt. 1993. Predictions of effect for intracellular antisense oligodeoxyribonucleotides from a kinetic model. Antisense Res. Dev. 3:3–18. [DOI] [PubMed] [Google Scholar]
- 6.Fennell, D. A. 1997. Quantitative antisense dose-response relationships: mathematical modeling of antisense action under steady-state conditions. Antisense Nucleic Acid Drug Dev. 7:49–53. [DOI] [PubMed] [Google Scholar]
- 7.Fennell, D. A., and F. E. Cotter. 2001. A dynamical systems model to simulate the perturbation kinetics of gene expression by antisense oligonucleotides. J. Theor. Biol. 209:103–112. [DOI] [PubMed] [Google Scholar]
- 8.Haruki, M., Y. Tsunaka, M. Morikawa, S. Iwai, and S. Kanaya. 2000. Catalysis by Escherichia coli ribonuclease HI is facilitated by a phosphate group of the substrate. Biochemistry. 39:13939–13944. [DOI] [PubMed] [Google Scholar]
- 9.Jayaraman, A., S. P. Walton, M. L. Yarmush, and C. M. Roth. 2001. Rational selection and quantitative evaluation of antisense oligonucleotides. Biochim. Biophys. Acta. 1520:105–114. [DOI] [PubMed] [Google Scholar]
- 10.Yakubov, L. A., E. A. Deeva, V. F. Zarytova, E. M. Ivanova, A. S. Ryte, L. V. Yurchenko, and V. V. Vlassov. 1989. Mechanism of oligonucleotide uptake by cells: involvement of specific receptors? Proc. Natl. Acad. Sci. USA. 86:6454–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakai, D., T. Seita, T. Terasaki, S. Iwasa, Y. Shoji, Y. Mizushima, and Y. Sugiyama. 1996. Cellular uptake mechanism for oligonucleotides: involvement of endocytosis in the uptake of phosphodiester oligonucleotides by a human colorectal adenocarcinoma cell line, HCT-15. J. Pharmacol. Exp. Ther. 278:1362–1372. [PubMed] [Google Scholar]
- 12.Beltinger, C., H. U. Saragovi, R. M. Smith, L. LeSauteur, N. Shah, L. DeDionisio, L. Christensen, A. Raible, L. Jarett, and A. M. Gewirtz. 1995. Binding, uptake, and intracellular trafficking of phosphorothioate-modified oligodeoxynucleotides. J. Clin. Invest. 95:1814–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Diesbach, P., F. N'Kuli, C. Berens, E. Sonveaux, M. Monsigny, A. C. Roche, and P. J. Courtoy. 2002. Receptor-mediated endocytosis of phosphodiester oligonucleotides in the HepG2 cell line: evidence for nonconventional intracellular trafficking. Nucleic Acids Res. 30:1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varga, C. M., K. Hong, and D. A. Lauffenburger. 2001. Quantitative analysis of synthetic gene delivery vector design properties. Mol. Ther. 4:438–446. [DOI] [PubMed] [Google Scholar]
- 15.Zelphati, O., and F. C. Szoka Jr. 1996. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. USA. 93:11493–11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maksimenko, A. V., M. B. Gottikh, V. Helin, Z. A. Shabarova, and C. Malvy. 1999. Physico-chemical and biological properties of antisense phosphodiester oligonucleotides with various secondary structures. Nucleosides Nucleotides. 18:2071–2091. [DOI] [PubMed] [Google Scholar]
- 17.Walton, S. P., G. N. Stephanopoulos, M. L. Yarmush, and C. M. Roth. 2002. Thermodynamic and kinetic characterization of antisense oligodeoxynucleotide binding to a structured mRNA. Biophys. J. 82:366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ledley, T. S., and F. D. Ledley. 1994. Multicompartment, numerical model of cellular events in the pharmacokinetics of gene therapies. Hum. Gene Ther. 5:679–691. [DOI] [PubMed] [Google Scholar]
- 19.Banks, G. A., R. J. Roselli, R. Chen, and T. D. Giorgio. 2003. A model for the analysis of nonviral gene therapy. Gene Ther. 10:1766–1775. [DOI] [PubMed] [Google Scholar]
- 20.Lodish, H., D. Baltimore, A. Berk, S. L. Zipursky, P. Matsudaira, and J. Darnell. 1995. Molecular Cell Biology. W. H. Freeman and Company, New York.
- 21.Wu-Pong, S. 1996. The role of multivalent cations in oligonucleotide cellular uptake. Biochem. Mol. Biol. Int. 39:511–519. [DOI] [PubMed] [Google Scholar]
- 22.Leonetti, J. P., P. Machy, G. Degols, B. Lebleu, and L. Leserman. 1990. Antibody-targeted liposomes containing oligodeoxyribonucleotides complementary to viral RNA selectively inhibit viral replication. Proc. Natl. Acad. Sci. USA. 87:2448–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zelphati, O., J. L. Imbach, N. Signoret, G. Zon, B. Rayner, and L. Leserman. 1994. Antisense oligonucleotides in solution or encapsulated in immunoliposomes inhibit replication of HIV-1 by several different mechanisms. Nucleic Acids Res. 22:4307–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semple, S. C., S. K. Klimuk, T. O. Harasym, N. Dos Santos, S. M. Ansell, K. F. Wong, N. Maurer, H. Stark, P. R. Cullis, M. J. Hope, and P. Scherrer. 2001. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta. 1510:152–166. [DOI] [PubMed] [Google Scholar]
- 25.Lee, H., J. H. Jeong, and T. G. Park. 2001. A new gene delivery formulation of polyethylenimine/DNA complexes coated with PEG conjugated fusogenic peptide. J. Control. Release. 76:183–192. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Chaumont, C., O. Seksek, J. Grzybowska, E. Borowski, and J. A. Bolard. 2000. Delivery systems for antisense oligonucleotides. Pharmacol. Ther. 87:255–277. [DOI] [PubMed] [Google Scholar]
- 27.Roth, C. M., and S. Sundaram. 2004. Engineering synthetic vectors for DNA delivery: insights from intracellular pathways. Annu. Rev. Biomed. Eng. 6:397–426. [DOI] [PubMed] [Google Scholar]
- 28.Raghavan, A., R. L. Ogilvie, C. Reilly, M. L. Abelson, S. Raghavan, J. Vasdewani, M. Krathwohl, and P. R. Bohjanen. 2002. Genome-wide analysis of mRNA decay in resting and activated primary human T lymphocytes. Nucleic Acids Res. 30:5529–5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang, E., E. van Nimwegen, M. Zavolan, N. Rajewsky, M. Schroeder, M. Magnasco, and J. E. Darnell Jr. 2003. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13:1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho, S. P., Y. Bao, T. Lesher, R. Malhotra, L. Y. Ma, S. J. Fluharty, and R. R. Sakai. 1998. Mapping of RNA accessible sites for antisense experiments with oligonucleotide libraries. Nat. Biotechnol. 16:59–63. [DOI] [PubMed] [Google Scholar]
- 31.Milner, N., K. U. Mir, and E. M. Southern. 1997. Selecting effective antisense reagents on combinatorial oligonuculeotide arrays. Nat. Biotechnol. 15:537–541. [DOI] [PubMed] [Google Scholar]
- 32.Mathews, D. H., M. E. Burkard, S. M. Freier, J. R. Wyatt, and D. H. Turner. 1999. Predicting oligonucleotide affinity to nucleic acid targets. RNA. 5:1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walton, S. P., G. N. Stephanopoulos, M. L. Yarmush, and C. M. Roth. 1999. Prediction of antisense oligonucleotide binding affinity to a structured RNA target. J. Biochem. Microbiol. Technol. Eng. 65:1–9. [PubMed] [Google Scholar]
- 34.Elayadi, A. N., D. A. Braasch, and D. R. Corey. 2002. Implications of high-affinity hybridization by locked nucleic acid oligomers for inhibition of human telomerase. Biochemistry. 41:9973–9981. [DOI] [PubMed] [Google Scholar]
- 35.Barnes 3rd, T. W., and D. H. Turner. 2001. Long-range cooperativity in molecular recognition of RNA by oligodeoxynucleotides with multiple C5-(1-propynyl) pyrimidines. J. Am. Chem. Soc. 123:4107–4118. [DOI] [PubMed] [Google Scholar]
- 36.Zhou, W., and S. Agrawal. 1998. Mixed-backbone oligonucleotides as second-generation antisense agents with reduced phosphorothioate-related side effects. Bioorg. Med. Chem. Lett. 8:3269–3274. [DOI] [PubMed] [Google Scholar]
- 37.Esposito, W. R., and C. A. Floudas. 2000. Global optimization for the parameter estimation of differential-algebraic systems. Ind. Eng. Chem. Res. 39:1291–1310. [Google Scholar]
- 38.Dheur, S., N. Dias, A. van Aerschot, P. Herdewijn, T. Bettinger, J. S. Remy, C. Helene, and E. T. Saison-Behmoaras. 1999. Polyethylenimine but not cationic lipid improves antisense activity of 3′-capped phosphodiester oligonucleotides. Antisense Nucleic Acid Drug Dev. 9:515–525. [DOI] [PubMed] [Google Scholar]
- 39.Vickers, T. A., S. Koo, C. F. Bennett, S. T. Crooke, N. M. Dean, and B. F. Baker. 2003. Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents. A comparative analysis. J. Biol. Chem. 278:7108–7118. [DOI] [PubMed] [Google Scholar]
- 40.Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 411:494–498. [DOI] [PubMed] [Google Scholar]
- 41.Scherr, M., M. A. Morgan, and M. Eder. 2003. Gene silencing mediated by small interfering RNAs in mammalian cells. Curr. Med. Chem. 10:245–256. [DOI] [PubMed] [Google Scholar]
- 42.Lauffenburger, D. A., and J. Linderman. 1993. Receptors: Models for Binding, Trafficking, and Signalling. Oxford University Press, New York.
- 43.Temsamani, J., M. Kubert, J. Tang, A. Padmapriya, and S. Agrawal. 1994. Cellular uptake of oligodeoxynucleotide phosphorothioates and their analogs. Antisense Res. Dev. 4:35–42. [DOI] [PubMed] [Google Scholar]
- 44.Hawley, P., and I. Gibson. 1996. Interaction of oligodeoxynucleotides with mammalian cells. Antisense Nucleic Acid Drug Dev. 6:185–195. [DOI] [PubMed] [Google Scholar]