

Abstract
Penetratin is a short, basic cell-penetrating peptide able to induce cellular uptake of a vast variety of large, hydrophilic cargos. We have reassessed the highly controversial issue of direct permeation of the strongly cationic peptide across negatively charged lipid membranes. Confocal laser scanning microscopy on rhodamine-labeled giant vesicles incubated with carboxyfluorescein-labeled penetratin yielded no evidence of transbilayer movement, in contradiction to previously reported results. Confocal fluorescence spectroscopy on black lipid membranes confirmed this finding, which was also not affected by application of a transmembrane electric potential difference. A novel dialysis assay based on tryptophan absorbance and fluorescence spectroscopy demonstrated that the permeability of small and large unilamellar vesicles to penetratin is <10−13 m/s. Taken together, the results show that penetratin is not capable of overcoming model membrane systems irrespective of the bilayer curvature or the presence of a transmembrane voltage. Thus, direct translocation across the hydrophobic core of the plasma membrane cannot account for the efficient uptake of penetratin into live cells, which is in accord with recent in vitro studies underlining the importance of endocytosis in the internalization process of cationic cell-penetrating peptides.
INTRODUCTION
Penetratin (pAntp) is a 16-amino acid peptide with the sequence RQIKIWFQNRRMKWKK. It corresponds to the third helix (residues 43–58) of the homeodomain of the Antennapedia homeoprotein, a key transcription factor in the development of Drosophila (1). The short, basic sequence mediates efficient cellular internalization of the homeodomain and has also been shown to convey other large, hydrophilic entities into the cell interior (2). It therefore belongs to the group of so-called cell-penetrating peptides (CPPs), a diverse class of short, water-soluble peptides able to translocate and transport cargo across cell membranes with low toxicity (3). Coupling of CPPs to proteins, oligonucleotides, peptide nucleic acids, and other pharmacologically interesting compounds thus provides a promising strategy for cellular delivery of otherwise membrane-impermeant molecules (4,5).
Initial observations suggested that cellular internalization and nuclear localization of penetratin are energy- and receptor-independent, not saturable, and not impeded by low temperature (4°C), arguing against endocytosis as the dominating uptake mechanism (6,7). This seemed to be in agreement with studies on artificial vesicles, which supported direct permeation of penetratin across lipid membranes without permeabilization (8,9). More recently, harsh cell fixation before microscopy and insufficient washing before flow cytometry have been determined to be responsible for artifactual cellular uptake in the early in vitro experiments (10,11), and it is nowadays widely accepted that endocytotic processes play a major role in the translocation of penetratin and penetratin/cargo complexes (12–14).
However, the question as to whether penetratin permeates a pure lipid bilayer without causing membrane permeabilization has remained a debatable and puzzling one because a prohibitively high Born charging energy is expected to hamper passive diffusion of a molecule with a nominal charge number of znom = 7 across a medium of low dielectricity. Whereas fluorescence microscopic studies with giant vesicles (GVs) seemingly unveiled that penetratin rapidly overcomes lipid bilayers (8), no permeation across the membranes of small and large unilamellar vesicles (SUVs and LUVs, respectively) could be detected by fluorescence resonance energy transfer (15,16). Others have speculated that a transbilayer voltage might be necessary for triggering translocation (17,18). The discrepancies between these results have been attributed to differences in experimental conditions, most notably in size and bilayer curvature of the various lipid vesicles used (16).
This led us to resume a systematic assessment of penetratin transport through diverse model membranes with a variety of experimental techniques. We reevaluated the ability of penetratin to translocate across the lipid bilayers of different types of liposomes (GVs, SUVs, and LUVs) and black lipid membranes in the absence or presence of an electric potential difference. Confocal laser scanning microscopy (CLSM) was used to follow the distribution of carboxyfluorescein-labeled penetratin interacting with rhodamine-labeled GVs, and the influence of a transmembrane potential difference on the permeability of a planar lipid bilayer was probed with confocal fluorescence spectroscopy. By subjecting differently prepared solutions containing penetratin and SUVs or LUVs to dialysis, we established a new method for monitoring membrane permeation. All our experiments consistently demonstrate that, under the conditions used here, penetratin does not cross any of these lipid barriers regardless of bilayer curvature and transmembrane voltage.
MATERIALS AND METHODS
Materials
The lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG, sodium salt), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[lissamine rhodamine B sulfonyl] (Rh-DPPE, ammonium salt), 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC), and 1,2-diphytanoyl-sn-glycero-3-[phospho-L-serine] (DPhPS, sodium salt) were purchased from Avanti Polar Lipids (Alabaster, AL). Hexane was from Fluka (Buchs, Switzerland) and 5(6)-carboxyfluorescein-N-hydroxysuccinimide ester (FLUOS) from Boehringer Mannheim (Mannheim, Germany). Glucose, sucrose, and fluorescein diacetate were obtained from Sigma (Deisenhof, Germany) and all other chemicals from Merck (Darmstadt, Germany).
Peptide synthesis
Peptides were synthesized automatically on an ABI 433A instrument (Applied Biosystems, Foster City, CA) following the standard solid-phase Nα-9-fluorenylmethoxycarbonyl protocol in a batchwise mode as described previously (19). A carboxyfluorescein-labeled derivative (CF-penetratin) was prepared by N-terminal coupling of FLUOS and C-terminal amidation to conserve the net charge of the peptide. Both analogs were purified by preparative reversed-phase high-performance liquid chromatography on an LC-10AD system (Shimadzu, Kyoto, Japan) operating at 220 nm. The purities of the final products were >98% by analytical high-performance liquid chromatography, and their expected molar masses were confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry on a Voyager-DE STR (Applied Biosystems).
Confocal microscopy with giant vesicles
Sample preparation
Giant vesicles were prepared by the spontaneous-formation method (20). Briefly, 1 mg POPC/POPG (3:1 mol/mol) and 0.1 μg Rh-DPPE (0.0061 mol %), all dissolved in chloroform at 20 mg/mL for the unlabeled and 1 mg/mL for the labeled lipids, respectively, were mixed in a 10-mL sample tube and dried in a rotary evaporator and subsequently under high vacuum overnight. The dried lipid film was prehydrated at 40°C and 100% relative humidity for 1 h in an argon atmosphere before addition of 5 mL internal medium (pH ∼5.9) consisting of 100 mM sucrose, 10 mM KCl, and 0.1 mM ethylenediaminetetraacetate (EDTA). During overnight incubation at 37°C, vesicles formed and gathered in a viscous cloud, which was harvested with a Pasteur pipette. The lipid concentration was determined with a phosphate assay (21). The vesicle solution was diluted with external medium (pH ∼5.5) containing 100 mM glucose, 10 mM KCl, and 0.1 mM EDTA to a lipid concentration of 200 μM. Water-dissolved CF-penetratin was added to give a final peptide concentration of 5 μM.
Confocal laser scanning microscopy
After gentle vortex mixing, an aliquot of the penetratin/GV mixture was placed onto a window of a two-part quartz cuvette (Hellma, Müllheim, Germany) with a pathlength of 0.1 mm and covered with a coverslip. The density difference between the internal and the external medium made the vesicles settle down on the bottom of the cuvette, which was mounted in a homemade holder. CLSM pictures were taken within 4 h after preparation on an LSM 510 META microscope equipped with an LD Achroplan 40×/0.60 corr. objective, a helium/neon laser with a dichroitic HTF mirror, and an argon laser with a DPS1 mirror (Carl Zeiss, Jena, Germany). Upon excitation at 543 nm with the He/Ne laser, rhodamine emission was registered using an LP 560 cutoff filter in front of the detector at a signal amplification of 18%. Carboxyfluorescein was excited at 488 nm with the argon laser, and CLSM images were taken with a BP 505–530 bandpass filter at an amplification of 74%. The pinhole width was 249 μm for both channels.
Confocal spectroscopy with black lipid membranes
Sample preparation
Black lipid membranes (22) were generated in a homemade Teflon trough (23) divided into two compartments by a horizontal polytetrafluoroethylene (PTFE) membrane (Goodfellow, Cambridge, United Kingdom). The upper (cis, c) and the lower (trans, t) chambers were filled with 500 μL and 2 mL buffer, respectively. A planar lipid bilayer consisting of DPhPC/DPhPS (3:1 mol/mol) was formed by brushing a droplet of the lipid mixture dissolved in n-decane at 20 mg/mL onto the 150-μm-diameter hole in the PTFE septum, which was pretreated with hexadecane/hexane (1:99 v/v). Ag/AgCl electrodes positioned in both compartments allowed for the application of an electric potential difference, Δφ ≡ φt – φc. The voltage and the electric current across the lipid bilayer were controlled simultaneously using an EPC-9 patch-clamp amplifier (HEKA Electronics, Lambrecht, Germany) to make sure that the membrane stayed intact during the experiment. For studying bilayer translocation of penetratin, both chambers were filled with 10 mM phosphate buffer (pH 7.4, 154 mM NaCl). After formation of the black lipid membrane, 5 μL 100 μM CF-penetratin was added to the upper compartment to yield a final peptide concentration of 1 μM.
Control experiment
Experiments with fluorescein diacetate as a positive control were done to ensure the suitability of the experimental setup for detecting transmembrane transport. An aliquot of a freshly prepared solution of 10 mM fluorescein diacetate in dimethyl sulphoxide was added to the upper compartment to give a final concentration of 20 μM. The experimental conditions were the same as for CF-penetratin, except that the pH was adjusted to 8.0 to speed up hydrolysis. The nonfluorescent fluorescein diacetate is known to diffuse across lipid membranes as long as it is kept in its unhydrolyzed form (24). Hydrolysis yields the bilayer-impermeant fluorophore fluorescein.
Confocal fluorescence spectroscopy
The distribution of CF-penetratin or fluorescein between the two compartments was monitored using an LSM 510 META inverted confocal laser scanning microscope equipped with a ConfoCor 2 unit (Carl Zeiss). The fluorophores were excited at 488 nm, and the emitted light was filtered with an LP 505 longpass filter in front of the avalanche photodiode detector. The fluorescence intensity, I, was scanned by moving the confocal volume of 0.7 fL vertically (i.e., in z direction) through both compartments with a stepwidth of 1 μm. The confocal volume was then fixed at a distance of 25 μm above or below the planar membrane, respectively, to record fluctuations in fluorescence intensity, from which normalized autocorrelation functions, G(τ), were derived using the ConfoCor 2 software provided by the manufacturer. For a single diffusing species (25), the autocorrelation function is given by
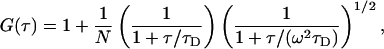 |
(1) |
where τD is the characteristic diffusion time of the fluorophore in the prolate ellipsoidal Gaussian observation volume with a ratio of the axial to the lateral dimension of ω = 5. The confocal volume was calibrated with a solution of rhodamine green. N stands for the mean number of fluorescent particles in the confocal volume and thus yields the concentration of CF-penetratin or fluorescein.
Dialysis with small and large unilamellar vesicles
Sample preparation
POPC and POPG dissolved in chloroform at 20 mg/mL were mixed in a molar ratio of 3:1 and dried in a rotary evaporator and subsequently under high vacuum overnight. For the preparation of pure lipid vesicles, dry lipid films were suspended in 10 mM phosphate buffer (pH 7.4, 154 mM NaF) by vortex mixing for 5 min, yielding large multilamellar vesicles. Bilayers preloaded with penetratin on both leaflets were made in the same manner by suspending lipid films in buffer containing the peptide at the desired final concentration of 25 μM. SUVs were then obtained by ultrasonication in an ice/water bath for 20 min with a Labsonic L instrument (B. Braun, Melsungen, Germany) equipped with a titanium tip, followed by spinning for 5 min at 13 krpm in a 3K30 tabletop centrifuge (Sigma Laborzentrifugen, Osterode am Harz, Germany) to remove titanium debris. LUVs were prepared by 35 extrusion steps through two stacked polycarbonate filters with a pore diameter of 100 nm using a LiposoFast extruder (Avestin, Ottawa, Canada). The vesicle size was checked on an N4 Plus particle sizer (Beckman Coulter, Fullerton, CA) equipped with a 10-mW helium/neon laser with a wavelength of 632.8 nm at a scattering angle of 90° and was narrowly centered at ∼30 nm for SUVs and ∼100 nm for LUVs, respectively. The presence of penetratin during vesicle formation had no effect on the size of the liposomes. The excess of lipid (5 mM) was large enough to ensure virtually complete membrane binding of penetratin, which was confirmed by tryptophan fluorescence titration experiments (data not shown). In addition, no differences in the fluorescence and circular dichroism spectra could be observed between samples with penetratin added before and after vesicle formation, respectively, suggesting similar locations of the peptide in the membrane (data not shown).
Dialysis
Dialysis experiments were performed using Spectra/Por Float-A-Lyzer tubes (Carl Roth, Karlsruhe, Germany) with a volume of 500 μL and a molecular-weight cutoff of 3 × 105. The dialysis tubes were filled with samples consisting of, respectively, 25 μM pure penetratin in phosphate buffer, 25 μM penetratin added externally to 5 mM SUVs or LUVs, and 5 mM vesicles preloaded with a total of 25 μM peptide on both leaflets. Each tube was placed in a covered glass beaker filled with 500 mL buffer, which was replaced every 24 h, and continuously stirred at room temperature for 96 h to attain equilibrium. Differences in peptide concentration upon dialysis were determined by ultraviolet absorbance at 280 nm on a V-550 UV-vis spectrometer (Jasco, Tokyo, Japan) with 1-mm quartz cuvettes (Hellma) and by intrinsic tryptophan fluorescence emission at 300–500 nm upon excitation at 280 nm on a JASCO FP-6500 fluorescence spectrometer using 1-cm2 quartz cuvettes from Hellma. Absorbance values and fluorescence spectra obtained from pure buffer or from 5 mM lipid suspensions without penetratin were subtracted from the corresponding raw data.
RESULTS
Confocal laser scanning microscopy
Translocation of penetratin across the bilayers of giant vesicles was studied by confocal laser scanning microscopy. Fig. 1, A–F, shows CLSM images taken in the equator plane of lipid particles from the same batch containing 200 μM POPC/POPG (3:1 mol/mol) GVs fluorescently labeled with 0.0061 mol % Rh-DPPE in the presence of 5 μM CF-penetratin. The simultaneous use of rhodamine-marked lipid and carboxyfluorescein-labeled peptide allowed for the independent observation of vesicular structures (Fig. 1, A–C) and penetratin distribution (Fig. 1, D–F) in the same sample. From the rhodamine emission images, it is obvious that the spontaneous-formation method led to a variety of different lipid aggregates. Whereas the particle in Fig. 1 A seems to be a simple unilamellar vesicle surrounding an aqueous interior (see Table 1), dense vesicular structures are found in the lumen of the vesicle in Fig. 1 B, and the particle in Fig. 1 C appears to consist largely of lipidic components.
FIGURE 1.

Confocal laser scanning microscopy on giant vesicles composed of POPC/POPG (3:1 mol/mol) and labeled with 0.0061 mol % Rh-DPPE. The images were taken between 5 min and 4 h after addition of 5 μM CF-penetratin to 200 μM lipid. (A–C) Rhodamine fluorescence from lipidic structures was excited at 543 nm and registered above 560 nm. (D–F) Carboxyfluorescein fluorescence was excited at 488 nm and monitored at 505–530 nm. (G) The profile depicts the fluorescence intensities, I, from Rh-DPPE (red line) and CF-penetratin (green line) along the arrow in the equator plane of the vesicle in A and D.
TABLE 1.
Fluorescence characteristics of rhodamine-labeled giant vesicles in the presence of CF-penetratin
| Vesicle | Panels in Fig. 1 | d (μm) | IRh (au) | ICF (au) | 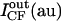 |
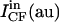 |
|---|---|---|---|---|---|---|
| 1 | A/D | 50 | 90 | 100 | 44 | 25 |
| 2 | B/E | 97 | 250 | 90 | 40 | 21 |
| 3 | C/F | 59 | ≫250* | 70 | 40 | 25 |
| 4 | — | 46 | 80 | 100 | 39 | 25 |
| 5 | — | 72 | 160 | 100 | 50 | 29 |
Summarized are the results obtained for five giant vesicles, three of which are shown in Fig. 1, A–F. d stands for the vesicle diameter. IRh and ICF are the mean values of, respectively, the rhodamine and the carboxyfluorescein fluorescence intensities measured in a 1-μm segment enclosing the membrane along the arrow through vesicle 1 in Fig. 1, A and D. 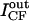 and
and  denote the mean intensities obtained from a 5-μm segment along the same line outside and inside the vesicle, respectively.
denote the mean intensities obtained from a 5-μm segment along the same line outside and inside the vesicle, respectively.
250 au was the upper limit for quantification of fluorescence intensity. See Fig. 1 for experimental conditions.
The strong membrane affinity of penetratin is visualized in Fig. 1 D, where an intense CF-penetratin signal emerges from the bilayer but not from the aqueous solutions outside and inside. This is corroborated by the intensity profile in Fig. 1 G along a projection line through the equator (arrow in Fig. 1, A and D). Note that the fluorescence intensity of ∼25 au observed in the aqueous medium inside the vesicle corresponds to the background signal obtained for a peptide-free liposome suspension and, as such, cannot serve as a measure of penetratin translocation. Interestingly, however, CF-penetratin did not accumulate on the surface of the particles contained in the lumina of the vesicles in Fig. 1, E and F, even after several hours, implying that the latter constituted a tight seal not to be overcome by the peptide. On the other hand, we also observed a few (<5%) vesicles with membrane defects and significant intravesicular CF fluorescence (data not shown, see Discussion, below).
Confocal fluorescence spectroscopy
To assess the influence of membrane curvature and voltage on penetratin transport, we performed translocation experiments on planar lipid bilayers in the absence and in the presence of an external transmembrane electric potential difference. To this end, a negatively charged black membrane composed of DPhPC/DPhPS (3:1 mol/mol) was prepared, across which a voltage of Δφ = −100 mV was applied. Fig. 2 illustrates a fluorescence intensity z-scan taken 10 min after injection of 1 μM CF-penetratin into the upper compartment (z > 0), which exhibited a significant signal due to fluorescent molecules dissolved in the aqueous phase (solid line). The high fluorescence intensity at the position of the lipid bilayer (z = 0) pointed to strong peptide accumulation at the black membrane, whereas no CF-penetratin could be detected in the lower chamber (z < 0). The autocorrelation function, G(τ), recorded 25 μm above the bilayer after 10 min (data not shown) was fitted with Eq. 1, yielding a diffusion time of τD = 61 μs and a mean number of fluorescent particles in the confocal volume (0.7 fL) of N = 190, which corresponds to a fluorophore concentration of 0.45 μM. Thus, the peptide concentration in the upper chamber was reduced by a factor of ∼2 during the first 10 min and further decreased to 0.36 μM after 2 h (dashed line). However, the loss of CF-penetratin in the upper chamber was not accompanied by an intensity gain in the lower compartment and can be explained by peptide adsorption to the Teflon walls of the sample chamber (see Discussion, below). Consequently, no autocorrelation functions could be obtained for the lower compartment. Comparable results indicative of the inability of penetratin to cross planar bilayers were obtained in the absence of an external transmembrane potential (data not shown).
FIGURE 2.

Mean fluorescence intensity, I, along a z-scan through two compartments separated by a black membrane (z = 0) consisting of DPhPC/DPhPS (3:1 mol/mol) in the presence of a transmembrane potential of Δφ ≡ φt – φc = −100 mV. The profiles were recorded at 10 min (solid line) and 2 h (dotted line), respectively, after addition of 1 μM CF-penetratin to the upper chamber (z > 0).
As a positive control, fluorescein diacetate was added to the upper chamber at a final concentration of 20 μM (data not shown). In contrast to CF-penetratin, strong intensities on both sides of the black membrane after 10 min pointed to the presence of the fluorescent hydrolysis product fluorescein in the upper as well as in the lower compartment. As indicated by an increase in the mean fluorescence signal and a concomitant decrease in the corresponding autocorrelation amplitude, the concentration of the fluorescent analog in the lower chamber rose for up to 35 min after the start of the experiment because of progressive hydrolysis of bilayer-permeant fluorescein diacetate. For both CF-penetratin and fluorescein, the absence of an electric current between the two chambers demonstrated that the membranes were not disrupted during the experiments.
Dialysis
Unlike giant vesicles and planar black membranes, small and large unilamellar vesicles with diameters of, respectively, ∼30 nm and ∼100 nm exhibit strong bilayer curvature. We developed a novel dialysis method for the facile monitoring of transbilayer movement in SUVs and LUVs. Fig. 3 A depicts absorbance values measured for three different solutions before (shaded bars) and after (solid bars) dialysis during 96 h. Sample 1 contained 25 μM penetratin only; sample 2 contained 25 μM peptide added externally to 5 mM SUVs composed of POPC/POPG (3:1 mol/mol); and sample 3 contained 5 mM lipid vesicles uniformly preloaded with 25 μM penetratin. Absorbance was virtually completely lost upon dialysis of samples 1 and 2 because the peptide was not hindered from diffusing through the large pores of the dialysis tubes (molecular-weight cutoff 3 × 105). In stark contrast to this, ∼40% of the initial signal was retained in the case of sample 3. Given that 30-nm SUVs have only ∼40% of their lipid molecules in the inner leaflet and that the peptide was fully membrane-bound during vesicle formation, this implies that penetratin could not significantly overcome the lipid bilayer despite the prolonged dialysis time.
FIGURE 3.

Dialysis with a molecular-weight cutoff of 3 × 105 for 96 h of 25 μM penetratin and 5 mM small unilamellar vesicles composed of POPC/POPG (3:1 mol/mol). (A) The absorbance at 280 nm, A, was taken before (shaded bars) and after (solid bars) dialysis of samples initially containing only penetratin (1), penetratin added externally to SUVs (2), or penetratin distributed over both membrane leaflets (3). (B) Irradiation at 280 nm yielded the fluorescence intensity, F, before and after dialysis of penetratin (shaded and solid line, respectively), penetratin on the outside of SUVs (shaded dashed and solid line, respectively), or penetratin on both vesicle leaflets (shaded dotted and solid line, respectively).
A more thorough determination of peptide translocation was possible with fluorescence spectroscopy, as shown in Fig. 3 B. Upon irradiation at 280 nm, pure penetratin in sample 1 (shaded line) displayed a strong peak at 355 nm indicative of a hydrophilic environment of the two tryptophan residues, which was lost during dialysis (solid line). The blue shift to 345 nm and the intensity gain found for peptide added externally to lipid vesicles in sample 2 (shaded dashed line) pointed to a more hydrophobic tryptophan environment because of membrane adsorption. However, peptide desorption and dilution during dialysis lowered the signal to a value near zero (solid dashed line). On the contrary, the fluorescence intensity of sample 3 (shaded dotted line) containing peptide-preloaded vesicles was not reduced below ∼40% (solid dotted line), which is in good accord with the absorbance experiments. Similar results were obtained with LUVs, but the fraction of retained penetratin was raised to ∼50% owing to the larger vesicle diameter (data not shown).
DISCUSSION
Giant vesicles as model systems for translocation studies
The confocal laser scanning microscopy images presented in Fig. 1, A–F, provide evidence that penetratin is not able to translocate across the negatively charged lipid bilayers of giant vesicles under the conditions used in this work. Previous studies on membrane permeability to penetratin have yielded conflicting results, which has been ascribed to differences in experimental conditions, notably in membrane curvature and transbilayer voltage. Our observations are in accordance with accounts reporting no entry of penetratin into SUVs (15) or LUVs (16,18) composed of mixtures of negatively charged and neutral phospholipids in the absence of a transmembrane voltage. However, they are at odds with an early publication claiming the direct visualization by CLSM of penetratin uptake into GVs (8).
In contrast to these authors, we could monitor both lipidic structures and penetratin distribution independently in the same sample owing to the simultaneous but differentiable labeling with rhodamine and carboxyfluorescein, respectively. The lamellarities of the giant vesicles can thus be deduced from the Rh-DPPE fluorescence intensities (20), which are listed in Table 1 for five representative GVs. Compared with the least fluorescent membranes of vesicles 1 (Fig. 1 A) and 4, the fluorescence levels of vesicles 2 (Fig. 1 B) and 5 were elevated by factors of 3 and 2, respectively. Nevertheless, the fluorescence intensities resulting from the excitation of CF-labeled peptide bound to the surface of the undamaged lipid particles 1–5 were independent of vesicle lamellarity.
Importantly, spontaneous self-organization of lipids results in many different types of liposomes and vesicle clumps (16,20), whose membranes are easily disrupted and reannealed in response to mechanic stress even after the addition of peptide (26). The fact that giant vesicles are better comparable in size to live cells than smaller vesicular aggregates should not overshadow that cellular membranes, unlike GVs, are supported by a complex and sophisticated scaffold. Hence, transient membrane disruption might allow free diffusion of penetratin within the aqueous phase without need for transferring the charged peptide across the hydrophobic bilayer core. In fact, we found that a small fraction of the GVs contained penetratin-coated lipid assemblies within their lumen, and this fraction was considerably raised upon vigorous agitation of the sample (data not shown). However, the observation that most vesicles did not permit intravesicular peptide accumulation and exhibited the same CF-penetratin fluorescence intensity on their surface regardless of their lamellarity argues against direct permeation across the lipid bilayer, and instead calls for careful handling of GV solutions.
Influence of a transmembrane voltage
It has been suggested that penetratin (18) and also some positively charged mitochondrial presequences (27) translocate across lipid bilayers only in the presence of a large transmembrane electric potential difference. Unlike assays based on the activity of valinomycin (18), black lipid membrane studies enable direct voltage generation without requiring ionophores. Upon application of a transbilayer potential difference, Δφ, the diffusive flow of an ion across a unit area of a membrane, J, is expected to augment (28) according to
 |
(2) |
where  and
and  denote the interfacial aqueous ion concentrations on the cis and trans sides, respectively (see below), and a ≡ znomeΔφ/kT comprises the nominal charge number, znom; the electronic charge, e; the Boltzmann constant, k; and the absolute temperature, T. For
denote the interfacial aqueous ion concentrations on the cis and trans sides, respectively (see below), and a ≡ znomeΔφ/kT comprises the nominal charge number, znom; the electronic charge, e; the Boltzmann constant, k; and the absolute temperature, T. For 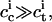 znom = 7, T = 298 K, and for potential differences Δφ < −20 mV, Eq. 2 simplifies to
znom = 7, T = 298 K, and for potential differences Δφ < −20 mV, Eq. 2 simplifies to
 |
(3) |
Thus, the flow increases linearly with the transmembrane voltage and, for znom = 7 in the present case, amounts to J(Δφ = −100 mV) = 27 × J(Δφ = 0). Taking into account an effective charge number of zeff = 5 (29) would further lower this value, implying that the potential difference raises the diffusive permeability of the lipid membrane by only one order of magnitude. In fact, applying confocal fluorescence spectroscopy to a planar bilayer exposed to Δφ = −100 mV, we found no corroboration of voltage-induced transbilayer transport of penetratin.
A rough estimate of the sensitivity of this approach may be gained in terms of the membrane permeability coefficient, P, which is defined as
 |
(4) |
with  and
and 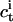 being the penetratin concentrations in the interfacial aqueous phases in immediate vicinity of the membrane on the cis and trans sides of the black lipid membrane, respectively. In general, these values differ from the corresponding bulk concentrations because of electrostatic attraction of the positively charged peptide to the negatively charged membrane. Based on an effective charge number of zeff = 5 and a mole ratio partition coefficient of K = 80 L/mol (see Ref. 29, which is in good agreement with calorimetric data not shown here),
being the penetratin concentrations in the interfacial aqueous phases in immediate vicinity of the membrane on the cis and trans sides of the black lipid membrane, respectively. In general, these values differ from the corresponding bulk concentrations because of electrostatic attraction of the positively charged peptide to the negatively charged membrane. Based on an effective charge number of zeff = 5 and a mole ratio partition coefficient of K = 80 L/mol (see Ref. 29, which is in good agreement with calorimetric data not shown here), 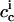 can be calculated with the aid of Gouy-Chapman theory to amount to ∼200 μM, whereas
can be calculated with the aid of Gouy-Chapman theory to amount to ∼200 μM, whereas  remains negligible. The flow of Δn moles of solute across the membrane area A within the time-span Δt is obtained as
remains negligible. The flow of Δn moles of solute across the membrane area A within the time-span Δt is obtained as
 |
(5) |
where r is the radius of the hole in the PTFE septum (∼75 μm), Vt the volume of the lower chamber (2 mL), and Δct the difference in bulk peptide concentration in the lower compartment between the first (t = 10 min) and the last (t = 120 min) measurement. Inserting Eq. 5 into Eq. 4 yields for the membrane permeability coefficient
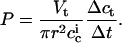 |
(6) |
Because Δct was undetectably low, the high sensitivity of confocal fluorescence spectroscopy imposes Δct ≪ 1 nM, such that the membrane permeability coefficient is estimated to amount to P ≪ 10−7 m/s (see also following section).
In the above considerations, the contribution to the transmembrane potential difference arising from partial compensation of the surface charge by asymmetric penetratin adsorption was not taken into account. However, values of zeff = 5 and K = 80 L/mol (29) predict that the additional transmembrane potential difference cannot exceed −20 mV under the conditions used here and may thus be neglected. Moreover, lipid membranes readily rupture at transmembrane voltages Δφ ≪ −100 mV, which was not borne out experimentally. Much more drastic transbilayer electric field strengths upon asymmetric penetratin adsorption have been suggested to impair membrane stability and permit peptide translocation (17). As reported by these authors, however, the critical threshold of membrane-bound penetratin for this mechanism to be active would necessitate both an elevated percentage (∼50 mol %) of negatively charged lipid headgroups and a high peptide concentration. Since our experiments were conducted with less highly charged membranes, they do not allow for a confirmation of this electroporation-like phenomenon, but the significance of the latter for the in vivo mode of cellular internalization seems questionable.
Hence, the reduction in the CF-penetratin concentration in the cis compartment and at the position of the negatively charged membrane was not due to bilayer permeation because no concomitant rise in concentration was detectable on the trans side. Rather, peptide adsorption to the Teflon walls of the measuring chamber is at the root of this effect, as has recently been demonstrated for a number of various surfaces (29–31).
Dialysis for determining membrane permeability
Area imbalance between the inner and the outer leaflet of a lipid bilayer has been considered an important determinant in transmembrane transport (16). Because giant vesicles and black membranes have only negligible or no net membrane curvature, respectively, we employed small and large unilamellar vesicles as model systems with pronounced curvature stress. Even though SUVs and LUVs are well-characterized tools of great usefulness for numerous biophysical and biochemical assays, transbilayer movement is more difficult to monitor than with GVs or black lipid membranes because the cis and the trans bilayer sides of the former are not observable independently or separable macroscopically. Protocols for assessing membrane translocation therefore often depend on the spectroscopic or isotopic labeling of lipids, peptides, or both (15,16). We aimed at establishing a straightforward permeability assay based on an approach originally developed for microcalorimetric studies (32). In contrast to the latter, the present dialysis procedure does not yield a detailed thermodynamic description of the membrane adsorption process but, in exchange, is applicable also to systems for which no such thermodynamic data are available. In addition, it is, in principle, amenable to a broad spectrum of different detection methods, such as fluorescence, absorbance, circular dichroism, nuclear magnetic resonance, or biochemical assays, and also allows for kinetic data to be derived. Apart from a suitable signal, the sole requirements are that the fraction of initially membrane-bound solute be known, in the simplest case that binding be complete, that the solute suffer no harm during the sometimes harsh treatment necessary for producing liposomes, and that its interactions with the lipid bilayer do not depend on whether it is added before or after vesicle preparation.
The permeability coefficient is again given by Eq. 4, where 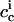 and
and  denote the interfacial aqueous penetratin concentrations inside (cis) and outside (trans) the vesicle, respectively. Whereas
denote the interfacial aqueous penetratin concentrations inside (cis) and outside (trans) the vesicle, respectively. Whereas 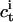 vanishes during dialysis,
vanishes during dialysis, 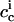 remains virtually constant in the present case. With Rb ≡ cb/cl being the ratio of the concentration of membrane-bound penetratin, cb, to the lipid concentration, cl, a partition equilibrium based on
remains virtually constant in the present case. With Rb ≡ cb/cl being the ratio of the concentration of membrane-bound penetratin, cb, to the lipid concentration, cl, a partition equilibrium based on 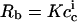 provides
provides
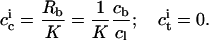 |
(7) |
A lipid concentration of cl = 5 mM is high enough to ensure almost complete binding of the peptide during vesicle preparation, such that cb ≈ 25 μM. The flow, J, can be calculated on the basis of the approximation that the membrane area, A, of nl moles of lipid with a molecular surface area requirement of Al is given by A = NAnlAl/2, with NA denoting Avogadro's number. Thus,
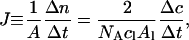 |
(8) |
where Δn and Δc stand for, respectively, the molar amount and the concentration of penetratin that crosses the membrane within the time-span Δt. Inserting Eqs. 7 and 8 into Eq. 4 yields
 |
(9) |
Assuming that the spectroscopic methods employed here are sensitive enough to reduce the error in determining the peptide concentration to <10% with respect to the initial value, Δc < 2.5 μM represents the upper limit of the penetratin concentration lost by permeation during dialysis. For K = 80 L/mol (29), Al = 0.7 nm2, and Δt = 96 h, we obtain a maximum value of P < 10−13 m/s, which is still 2–3 orders of magnitude lower than the permeability coefficients determined for halide anions (33).
CONCLUSIONS
The present work confirms the high affinity of penetratin to negatively charged phospholipid bilayers (29,34). The weak blue shift of the tryptophan emission spectrum from ∼355 nm in aqueous buffer to ∼345 nm in an excess of POPC/POPG (3:1 mol/mol) vesicles points to superficial adsorption of the peptide rather than deep bilayer penetration, which is in agreement with previous studies (35,36). However, we found no evidence of translocation across the bilayers of planar membranes or small, large, and giant vesicles under the conditions used here. These data strongly question observations of penetratin uptake into giant vesicles (8) but are in line with recent studies demonstrating artifactual internalization in vitro (10,11). Similar results have been obtained with different methods for other cationic CPPs, such as TAT (37). Thus, the ability to overcome a lipid bilayer seems to be restricted to a small yet diverse group of peptides including the bee venom melittin (38) and antimicrobial peptides like magainin 2 (39) and buforin 2 (40). On the contrary, penetratin and many other highly charged nonlytic CPPs most likely enter living cells by a mainly endocytotic pathway (12–14), which might be initiated by electrostatic accumulation of the peptides at negatively charged membrane constituents, such as lipid headgroups or heparan sulfate proteoglycans (41–43).
Acknowledgments
We thank Profs. Astrid Gräslund and Yuri Antonenko as well as Drs. Johannes Oehlke and Mazin Magzoub for helpful discussions and comments on the manuscript. Dr. Burkhard Wiesner is gratefully acknowledged for his invaluable support with the confocal microscopic and spectroscopic studies and Heike Nikolenko and Jeannette Müller for excellent technical assistance. We are indebted to Dr. Michael Beyermann, Dagmar Krause, and Bernhard Schmikale for synthesis and purification and to Dr. Michael Schümann for mass-spectrometric characterization of the peptides.
This work was supported by the European Commission with grants No. HPRN-CT-2001-00242 to E.B.W. and No. QLK3-CT-2002-01989 to E.B.W. and S.K.
Elsa Bárány-Wallje and Sandro Keller contributed equally to this work.
References
- 1.Gehring, W. J., Y. Q. Qian, M. Billeter, K. Furukubo-Tokunaga, A. F. Schier, D. Resendez-Perez, M. Affolter, G. Otting, and K. Wüthrich. 1994. Homeodomain-DNA recognition. Cell. 78:211–223. [DOI] [PubMed] [Google Scholar]
- 2.Joliot, A., and A. Prochiantz. 2004. Transduction peptides: from technology to physiology. Nat. Cell Biol. 6:189–196. [DOI] [PubMed] [Google Scholar]
- 3.Langel, Ü. 2002. Cell-Penetrating Peptides. Processes and Applications. CRC Press, Boca Raton.
- 4.Derossi, D., G. Chassaing, and A. Prochiantz. 1998. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 8:84–87. [PubMed] [Google Scholar]
- 5.Järver, P., and Ü. Langel. 2004. The use of cell-penetrating peptides as a tool for gene regulation. Drug Discov. Today. 9:395–402. [DOI] [PubMed] [Google Scholar]
- 6.Derossi, D., A. H. Joliot, G. Chassaing, and A. Prochiantz. 1994. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 269:10444–10450. [PubMed] [Google Scholar]
- 7.Derossi, D., S. Calvet, A. Trembleau, A. Brunissen, G. Chassaing, and A. Prochiantz. 1996. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J. Biol. Chem. 271:18188–18193. [DOI] [PubMed] [Google Scholar]
- 8.Thorén, P. E. G., D. Persson, M. Karlsson, and B. Nordén. 2000. The Antennapedia peptide penetratin translocates across lipid bilayers—the first direct observation. FEBS Lett. 482:265–268. [DOI] [PubMed] [Google Scholar]
- 9.Persson, D., P. E. G. Thorén, and B. Nordén. 2001. Penetratin-induced aggregation and subsequent dissociation of negatively charged phospholipid vesicles. FEBS Lett. 505:307–312. [DOI] [PubMed] [Google Scholar]
- 10.Lundberg, M., and M. Johansson. 2002. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem. Biophys. Res. Commun. 291:367–371. [DOI] [PubMed] [Google Scholar]
- 11.Richard, J. P., K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik, and B. Lebleu. 2003. Cell-penetrating peptides—a reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 278:585–590. [DOI] [PubMed] [Google Scholar]
- 12.Console, S., C. Marty, C. García-Echeverría, R. Schwendener, and K. Ballmer-Hofer. 2003. Antennapedia and HIV transactivator of transcription (TAT) “protein transduction domains” promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J. Biol. Chem. 278:35109–35114. [DOI] [PubMed] [Google Scholar]
- 13.Drin, G., S. Cottin, E. Blanc, A. R. Rees, and J. Temsamani. 2003. Studies on the internalization mechanism of cationic cell-penetrating peptides. J. Biol. Chem. 278:31192–31201. [DOI] [PubMed] [Google Scholar]
- 14.Fischer, R., K. Köhler, M. Fotin-Mleczek, and R. Brock. 2004. A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J. Biol. Chem. 279:12625–12635. [DOI] [PubMed] [Google Scholar]
- 15.Drin, G., H. Déméné, J. Temsamani, and R. Brasseur. 2001. Translocation of the pAntp peptide and its amphipathic analogue AP-2AL. Biochemistry. 40:1824–1834. [DOI] [PubMed] [Google Scholar]
- 16.Persson, D., P. E. G. Thorén, E. K. Esbjörner, M. Goksör, P. Lincoln, and B. Nordén. 2004. Vesicle size-dependent translocation of penetratin analogs across lipid membranes. Biochim. Biophys. Acta Biomembr. 1665:142–155. [DOI] [PubMed] [Google Scholar]
- 17.Binder, H., and G. Lindblom. 2003. Charge-dependent translocation of the Trojan peptide penetratin across lipid membranes. Biophys. J. 85:982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terrone, D., S. L. W. Sang, L. Roudaia, and J. R. Silvius. 2003. Penetratin and related cell-penetrating cationic peptides can translocate across lipid bilayers in the presence of a transbilayer potential. Biochemistry. 42:13787–13799. [DOI] [PubMed] [Google Scholar]
- 19.Beyermann, M., K. Fechner, J. Furkert, E. Krause, and M. Bienert. 1996. A single-point slight alteration set as a tool for structure-activity relationship studies of ovine corticotropin releasing factor. J. Med. Chem. 39:3324–3330. [DOI] [PubMed] [Google Scholar]
- 20.Akashi, K., H. Miyata, H. Itoh, and K. Kinosita. 1996. Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys. J. 71:3242–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Böttcher, C. J. F., C. Pries, and C. M. van Gent. 1961. A rapid and sensitive colorimetric microdetermination of free and bound choline. Recl. Trav. Chim. Pays-Bas—J. R. Neth. Chem. Soc. 80:1169–1178. [Google Scholar]
- 22.Mueller, P., D. O. Rudin, H. T. Tien, and W. C. Wescott. 1963. Methods for formation of single bimolecular lipid membranes in aqueous solution. J. Phys. Chem. 67:534–535. [Google Scholar]
- 23.Serowy, S., S. M. Saparov, Y. N. Antonenko, W. Kozlovsky, V. Hagen, and P. Pohl. 2003. Structural proton diffusion along lipid bilayers. Biophys. J. 84:1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oehlke, J., A. Scheller, B. Wiesner, E. Krause, M. Beyermann, E. Klauschenz, M. Melzig, and M. Bienert. 1998. Cellular uptake of an α-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta Biomembr. 1414:127–139. [DOI] [PubMed] [Google Scholar]
- 25.Elson, E. L., and D. Magde. 1974. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers. 13:1–27. [DOI] [PubMed] [Google Scholar]
- 26.Murray, D., A. Arbuzova, G. Hangyás-Mihályné, A. Gambhir, N. Ben-Tal, B. Honig, and S. McLaughlin. 1999. Electrostatic properties of membranes containing acidic lipids and adsorbed basic peptides: theory and experiment. Biophys. J. 77:3176–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maduke, M., and D. Roise. 1993. Import of a mitochondrial presequence into protein-free phospholipid vesicles. Science. 260:364–367. [DOI] [PubMed] [Google Scholar]
- 28.Evans, D. F., and H. Wennerström. 1999. The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet. Wiley-VCH, New York. 328–333.
- 29.Persson, D., P. E. G. Thorén, M. Herner, P. Lincoln, and B. Nordén. 2003. Application of a novel analysis to measure the binding of the membrane-translocating peptide penetratin to negatively charged liposomes. Biochemistry. 42:421–429. [DOI] [PubMed] [Google Scholar]
- 30.Bellet-Amalric, E., D. Blaudez, B. Desbat, F. Graner, F. Gauthier, and A. Renault. 2000. Interaction of the third helix of Antennapedia homeodomain and a phospholipid monolayer, studied by ellipsometry and PM-IRRAS at the air-water interface. Biochim. Biophys. Acta Biomembr. 1467:131–143. [DOI] [PubMed] [Google Scholar]
- 31.Chico, D. E., R. L. Given, and B. T. Miller. 2003. Binding of cationic cell-permeable peptides to plastic and glass. Peptides. 24:3–9. [DOI] [PubMed] [Google Scholar]
- 32.Heerklotz, H. H., H. Binder, and R. M. Epand. 1999. A “release” protocol for isothermal titration calorimetry. Biophys. J. 76:2606–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paula, S., A. G. Volkov, and D. W. Deamer. 1998. Permeation of halide anions through phospholipid bilayers occurs by the solubility-diffusion mechanism. Biophys. J. 74:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magzoub, M., L. E. G. Eriksson, and A. Gräslund. 2002. Conformational states of the cell-penetrating peptide penetratin when interacting with phospholipid vesicles: effects of surface charge and peptide concentration. Biochim. Biophys. Acta Biomembr. 1563:53–63. [DOI] [PubMed] [Google Scholar]
- 35.Magzoub, M., L. E. G. Eriksson, and A. Gräslund. 2003. Comparison of the interaction, positioning, structure induction and membrane perturbation of cell-penetrating peptides and non-translocating variants with phospholipid vesicles. Biophys. Chem. 103:271–288. [DOI] [PubMed] [Google Scholar]
- 36.Magzoub, M., K. Kirk, L. E. G. Eriksson, Ü. Langel, and A. Gräslund. 2001. Interaction and structure induction of cell-penetrating peptides in the presence of phospholipid vesicles. Biochim. Biophys. Acta Biomembr. 1512:77–89. [DOI] [PubMed] [Google Scholar]
- 37.Ziegler, A., X. L. Blatter, A. Seelig, and J. Seelig. 2003. Protein transduction domains of HIV-1 and SIV TAT interact with charged lipid vesicles. Binding mechanism and thermodynamic analysis. Biochemistry. 42:9185–9194. [DOI] [PubMed] [Google Scholar]
- 38.Matsuzaki, K., S. Yoneyama, and K. Miyajima. 1997. Pore formation and translocation of melittin. Biophys. J. 73:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuzaki, K., O. Murase, N. Fujii, and K. Miyajima. 1996. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. 35:11361–11368. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi, S., K. Takeshima, C. B. Park, S. C. Kim, and K. Matsuzaki. 2000. Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: proline as a translocation promoting factor. Biochemistry. 39:8648–8654. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki, T., S. Futaki, M. Niwa, S. Tanaka, K. Ueda, and Y. Sugiura. 2002. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 277:2437–2443. [DOI] [PubMed] [Google Scholar]
- 42.Ziegler, A., and J. Seelig. 2004. Interaction of the protein transduction domain of HIV-1 TAT with heparan sulfate: binding mechanism and thermodynamic parameters. Biophys. J. 86:254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonçalves, E., E. Kitas, and J. Seelig. 2005. Binding of oligoarginine to membrane lipids and heparan sulfate: structural and thermodynamic characterization of a cell-penetrating peptide. Biochemistry. 44:2692–2702. [DOI] [PubMed] [Google Scholar]