

Abstract
The nature of chiral interactions among chiral biopolymers, such as DNA, protein α-helices, and rodlike virus particles, remains elusive. In particular, a satisfactory model connecting molecular chiral interactions and the pitch of the resulting chiral mesophases is lacking. We report the measurement of short-fragment (146-bp) DNA cholesteric spherulite pitch as a function of osmotic pressure, average DNA interaxial spacing, and salt concentration. We determined cholesteric pitch and interaxial spacing by polarizing optical microscopy and x-ray scattering, respectively, from which the twist-angle between DNA molecules can be calculated. Surprisingly, we found that decreasing ionic strength resulted in weaker chiral interactions between DNA chains, as evidenced by the decrease in the twist-angle, and consequent increase in the cholesteric pitch, for a fixed interaxial spacing. We propose that this behavior can be explained by increased smearing-out of the helical charge pattern along DNA as the Debye screening length is increased.
INTRODUCTION
Any object that is not superimposable onto its mirror image is chiral. Nearly all molecules synthesized by living organisms are chiral. For example, L-amino acids and D-sugars are prevalent over their mirror-image isomers. In biomolecular liquid crystals formed by DNA (1–4), the α-helical polypeptide poly-γ-benzyl-L-glutamate (5), and fd virus suspensions (6,7), chirality leads to the formation of such twisted liquid crystalline structures as cholesterics, blue phases, and twisted smectics (8,9). There is even evidence that DNA can form a cholesteric phase in vivo, as observed for dinoflagellate chromosomes (10–13). Ironically, despite the vast number of examples of chirality in nature, some important details of chiral interactions are still poorly understood. Although chirality has a strong structural impact on its liquid crystalline phases, the strength of chiral interactions is surprisingly small. In a typical cholesteric, the average twist-angle ψ between neighboring molecules (Fig. 1) amounts to a fraction of a degree. The resulting cholesteric pitch P, defined as the length along the twist axis corresponding to a full turn of the nematic director, is in the few-hundred-Ångstroms-to-micrometer range. Recently, this problem has been addressed theoretically (14,15), and experiments are now urgently needed to test these novel ideas.
FIGURE 1.

Illustration of the twist angle ψ between two DNA molecules. This angle, which is <1°, is directly related to the chiral interaction energy.
To study chiral interactions in detail, we chose to investigate DNA cholesterics. This is because the DNA molecular structure is very well characterized, and by changing the liquid crystalline density and salt concentration, we can control the microscopic interactions. The ultimate goal for this project is to make a connection between the microscopic interactions and the mesoscopic behavior (e.g., cholesteric pitch). Recently, a theory of interactions between helical molecules has been put forward that especially focused on interactions between DNA helices (16–18). We will review the relevant facts of these efforts for comparison with our experimental observations.
For cholesterics, the liquid crystalline free energy density F is given by
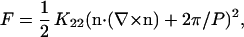 |
(1) |
where K22 is the twist elastic constant, n is the director field, and P is the cholesteric pitch (9). From the microscopic screened electrostatic interactions between two DNA double helices, Kornyshev and Leikin (18) estimate K22 and the chiral torque Kt from the chiral interaction. The cholesteric pitch is then given by P = 2πK22/Kt. However, the magnitude of P depends sensitively on the assumed distribution of condensed counterions on the DNA molecules. The theory predicts cholesteric unwinding at the cholesteric to isotropic and cholesteric to hexagonal transitions due to a loss of biaxial correlations between DNA molecules. At intermediate interaxial spacings di, a minimum in the cholesteric pitch Pmin is found at ∼di = 3.5–3.7 nm, which is a reflection of the pitch of the DNA double helix H (= 3.4 nm). This originates from a maximum in the torque-to-twist elasticity ratio found at a distance approximately equal to H.
In our study, we are observing the structure and pitch of short-fragment (146-bp) DNA cholesteric spherulites as a function of osmotic pressure, interaxial spacing, and salt concentration. This is an extension of our previous research on this system (4), where we are now simultaneously measuring di and P to calculate ψ between DNA helices. We are then following changes in P and ψ for varying ionic strength to gain an understanding of the electrostatic contribution to DNA chiral interactions. We chose DNA for this study because of its well-known structure and charge placement (19). DNA in monovalent salt solutions exhibits the following liquid crystalline phases with decreasing DNA concentration: hexagonal, line hexatic, cholesteric, and blue phases before reaching the isotropic phase (3). From previous osmotic stress studies there emerged a picture that the interactions between DNA molecules in a liquid crystal can be understood in terms of screened electrostatic interactions with additional contributions from bending fluctuations (20,21).
MATERIALS AND METHODS
DNA droplets
Short-fragment (146-bp) DNA prepared from chicken erythrocytes was dissolved in 10:1 TE (10 mM Tris-HCl, 1 mM EDTA) at pH 7.0 with 300 mM sodium acetate at a DNA concentration of 5 mg/ml. DNA was precipitated and washed with an ethanol/water mixture (3:1). Pellets (1 mg each) of short-fragment DNA were dried in a Speedvac (Savant Instruments, Farmingdale, NY) and resuspended in 5 mL of 10:1 TE (pH 7.8) buffer solution containing 10–19 wt % poly(ethylene glycol) of 35,000-mol wt (PEG 35,000, Fluka, MicroSelect for Molecular Biology, St. Louis, MO) and 0.2–1.0 M NaCl. The PEG 35,000 concentration controls the DNA density through osmotic stress (22–24). Cholesteric droplets were then generated by gently shaking the samples after an equilibration time of five days.
Polarizing optical microscopy
Polarizing optical microscopy was performed with a Zeiss Axiovert S100 TV inverted polarizing microscope (objective: Zeiss Plan-Neofluar 40×/0.85 pol; Carl Zeiss, Jena, Germany) equipped with an LC Pol-Scope retardance imaging system (Cambridge Research and Instrumentation, Boston, MA), which simultaneously measures the magnitude and direction of birefringence. The DNA droplets and equilibrating PEG solution were prepared on glass slides using vacuum grease to seal the samples under a coverslip. Birefringence images I(x,y) of individual cholesteric droplets were Fourier-transformed I(qx,qy) and the pitch was measured using the location of the first maximum in the angular averaged power spectrum I(q)I*(q), with 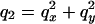 .
.
X-ray scattering
Small-angle x-ray scattering measurements were performed on a Rigaku RU-H3R rotating anode x-ray diffractometer (Rigaku, Tokyo, Japan) equipped with an Osmic multilayer focusing optic ((100 μm)2 point focus) and an evacuated Statton-type scattering camera. The sample-to-detector distance was 460 mm, which corresponds to a q range of 0.698 nm−1 ≤ q ≤ 6.25 nm−1 with q = (4π/λ)sin(θ/2), where θ is twice the Bragg angle. The incident beam wavelength was 0.154 nm, corresponding to 8 keV Cu Kα radiation. Samples were sealed in glass capillaries to isolate them from vacuum. Scattering patterns were acquired with 10 cm × 15 cm Fuji ST-VA image plates in conjunction with a Fuji BAS-2500 image plate scanner (Fuji, Elmsford, NY), and intensity profiles were obtained from radial averages of the scattering pattern intensities. The scattering intensity profiles I(q) = |F(q)|2 S(q) were fitted with a first-order Bessel function of the first kind for the cylindrical form factor of DNA F(q) (radius = 1 nm) and a Lorentzian peak for the structure factor S(q). The interaxial spacing was calculated from the Bragg spacing dBragg as 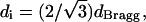 assuming local hexagonal packing. The DNA concentration cDNA also was calculated from dBragg by assuming local hexagonal packing and using 50 nm for the length of the DNA fragments and 660 g/mol as the basepair molecular weight.
assuming local hexagonal packing. The DNA concentration cDNA also was calculated from dBragg by assuming local hexagonal packing and using 50 nm for the length of the DNA fragments and 660 g/mol as the basepair molecular weight.
RESULTS
Cholesteric spherulites (25) of short-fragment DNA (Fig. 2) are stabilized by surface tension between the liquid crystal and the PEG solution (4). Both diametrical and radial defect lines (26) are found within the DNA cholesteric spherulites studied. Fig. 2 a shows a double spiral structure having a diametrical χ disclination line of strength = 1 located in the center of the spherulite. This defect line typically is seen at lower DNA densities and penetrates through the full diameter of the spherulite perpendicular to the viewing plane. A radial χ disclination line of strength = 2 (Fig. 2 b) running parallel to the viewing plane typically is observed at higher DNA densities. Also, as the cholesteric to hexagonal transition is approached at the higher DNA densities, birefringence in the center of the spherulites is lost. These results are the same as those observed previously (4). Since the center of the spherulites should maintain liquid crystallinity, they suggest that the center is a nematic phase, oriented perpendicular to the viewing plane where an enhancement of the cholesteric liquid crystalline bending stiffness close to the transition is responsible for expelling cholesteric twist.
FIGURE 2.

DNA cholesteric spherulites bathed in (a) 11 and (b) 17 wt % PEG 35,000 solutions (10:1 TE at pH 7.8, 0.5 M NaCl; scale bar = 5 μm). The distance between two striations represents the cholesteric pitch, which is ∼2.7 μm for each of the spherulites shown. Both diametrical (a) and radial χ disclination (b) lines are observed in the spherulites.
Fig. 3 shows P of the DNA cholesteric spherulites as a function of osmotic pressure Π for 0.2, 0.5, and 1.0 M NaCl. Osmotic pressure measurements of PEG 35,000 with varying NaCl concentration were performed using sedimentation equilibrium ultracentrifugation as described previously (27). Raising Π increases the DNA density due to removal of water from within the liquid crystalline array. For these salt concentrations, P is found to vary between 2 and 5 μm within di = 3.3 to 4.9 nm (Fig. 4). Increasing ionic strength reduces the electrostatic repulsion force between DNA molecules exponentially through a reduction in the Debye screening length 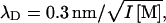 where I = ionic strength. This results in a decrease in di and thus, closer DNA packing.
where I = ionic strength. This results in a decrease in di and thus, closer DNA packing.
FIGURE 3.

Cholesteric pitch of short fragment DNA as a function of osmotic pressure for 0.2, 0.5, and 1.0 M NaCl. Increasing osmotic pressure corresponds to decreasing DNA density via removal of water from within the liquid crystalline array. The nonmonotonic nature of P with varying DNA density is apparent at all salt concentrations.
FIGURE 4.

Force curves for short fragment DNA at 0.2, 0.5, and 1.0 M NaCl obtained by x-ray scattering. Higher ionic strength reduces electrostatic repulsion, thus allowing DNA chains to come into closer proximity. For the range of DNA interaxial spacings observed in the cholesteric regime, the interaxial force decays exponentially with approximately four times the Debye screening length, indicating fluctuation-enhanced electrostatic interactions dominate (20,21).
Fig. 5 a shows P of the spherulites as a function of di for 0.2, 0.5, and 1.0 M NaCl. The corresponding cDNA calculated from di is also given. The twist-angle ψ between neighboring DNA molecules (Fig. 1) can be calculated from the experimentally determined P and di as ψ = 360°(di/P) and is found to be within 0.28–0.67° in this di range (Fig. 5 b). For all salt concentrations studied, P increases as the cholesteric to isotropic and cholesteric to hexagonal phase transitions are approached. Above ∼19 wt % PEG (P = 4.2 μm), the cholesteric phase ceases to exist, indicating a discontinuous jump to P = ∞ (and ψ = 0°) as the transition to the hexagonal phase occurs. However, because we use PEG to control DNA density, it is difficult to determine the location of the cholesteric to isotropic phase transition since PEG enters the DNA phase for densities just below the range of our measurements. This disrupts the cholesteric phase we are observing. Strzelecka and Rill (28) have used 31P NMR to measure phase transitions for 50-nm-length DNA with varying ionic strength. They showed that the first appearance of an anisotropic phase occurs at cDNA = 130–170 mg/ml for 0.01 to 1 M NaCl, respectively, and a transition from biphasic to fully liquid crystalline occurs at ∼cDNA = 260–270 mg/ml for the same respective salt-concentration range.
FIGURE 5.

(a) The DNA cholesteric pitch as a function of interaxial spacing for 0.2, 0.5, and 1.0 M NaCl. The top axis gives the DNA concentration calculated from the interaxial spacing. The pitch unwinds at low and high DNA densities, where the cholesteric to isotropic and cholesteric to hexagonal phase transitions are approached, respectively. The pitch minimum, occurring between di = 3.6 and 4.2 nm, is found to shift toward smaller interaxial spacings with increasing ionic strength. (b) The calculated twist-angle shows similar trends to the cholesteric pitch.
Changes in chiral interactions with di also can be determined by monitoring ψ (Fig. 5 b), since the energy of chiral interactions is directly proportional to this molecular parameter (16–18). For 0.2–1.0 M NaCl, ψ shows a maximum in chiral interaction energy between di = 3.8 and 4.3 nm and a divergence toward zero at DNA densities approaching the cholesteric to isotropic and cholesteric to hexagonal phase transitions. In addition, Pmin and di at which Pmin occurs decrease, while cDNA increases, with increasing ionic strength (Table 1). The values of ψmax, and the corresponding di and cDNA, are also given in Table 1. Their dependence on ionic strength is in agreement with the trend observed for Pmin. In principle, K22 and hence, Kt, also can be calculated for comparison with theory by measuring the critical magnetic-field strength required to induce the cholesteric to nematic phase transition (9). Dogic and Fraden (6) performed such measurements for fd virus. Unfortunately, DNA displays a negative anisotropy in diamagnetic susceptibility that tends to align the DNA axis perpendicular to the magnetic field. This makes similar pitch-unwinding experiments quite difficult.
TABLE 1.
Ionic-strength dependence of the pitch minimum and twist-angle maximum
| I (M NaCl) | Pmin (μm) | di (nm) | cDNA* (mg/mL) | ψmax (deg) | di (nm) | cDNA* (mg/mL) |
|---|---|---|---|---|---|---|
| 0.2 | 2.81 ± 0.05 | 4.14 | 249 | 0.54 ± 0.02 | 4.26 | 235 |
| 0.5 | 2.29 ± 0.02 | 3.86 | 286 | 0.61 ± 0.01 | 3.86 | 286 |
| 1.0 | 2.03 ± 0.03 | 3.68 | 315 | 0.67 ± 0.02 | 3.79 | 297 |
DNA concentration calculated from di as described in Materials and Methods.
DISCUSSION
The theory of chiral interactions in cholesteric liquid crystals of DNA developed by Kornyshev and Leikin (18) predicts the nonmonotonic dependence of P on di, where cholesteric unwinding occurs at the cholesteric to isotropic and cholesteric to hexagonal transitions at ∼di = 5 and 3 nm, respectively. This resembles our experimental findings. The theory also predicts Pmin at ∼di = 3.5–3.7 nm, which is a reflection of the pitch of the double helix. Again, the theory is in close proximity to the range (di = 3.7–4.1 nm) that we find, but best agreement is found for DNA cholesterics at 1.0 M NaCl (where Pmin occurs at di = 3.68 nm). However, it was predicted that despite the electrostatic origin of these chiral interactions, the salt dependence of P should be insignificant for 1:1 electrolytes at concentrations of 0.01 to 1.0 M. We find experimentally, however, that both P and Pmin do have a significant dependence on salt concentration for 0.2–1.0 M NaCl. As ionic strength is decreased from 1.0 to 0.2 M NaCl, P of the DNA cholesteric spherulites increases for a fixed di (Fig. 5 a), indicating chiral interactions between DNA molecules are reduced.
Since the dominant contribution to chiral interactions comes from electrostatic forces, with the contributions from steric and van der Waals forces being negligible within this range for DNA cholesterics (18), there is a question as to how chiral interactions can be reduced by decreasing ionic strength. The naïve prediction, as mentioned by Van Winkle and co-workers (1), would be to assume that as ionic strength decreases, the interaxial force at constant interaxial spacing increases and therefore, the pitch should decrease. However, we observe the opposite trend.
Despite the increase in DNA intermolecular interactions with decreasing ionic strength, we attribute the resulting reduction in chiral interactions to the consequent increase in intramolecular interactions along the DNA molecules, which effectively smears out the helical charge pattern of the DNA. Decreasing the ionic strength increases the Debye screening length while augmenting the strength of the interhelical electrostatic repulsion. At the same time, a larger screening length means that fewer details of the helix are visible to the adjacent helix, especially since the DNA helical pitch is in the range of accessible Debye screening lengths. Since we observe a decrease in chiral interactions while decreasing ionic strength, the latter effect dominates our system. In the fd virus cholesteric system studies by Grelet and Fraden (7), an increase in chiral interactions with decreased ionic strength is observed. The naïve prediction applies here because the molecular pitch of the fd virus coat is not believed to transmit chiral interactions but rather, the micron-scale helical shape of the virus molecule, which is much larger than λD, is responsible for manifesting the cholesteric mesophase. It also should be pointed out that in the range of interaxial spacings in the DNA cholesteric regime, the interaxial force decays exponentially with ∼4λD, which indicates that bending fluctuation-enhanced repulsion dominates (20,21). With surface-to-surface separations between DNA molecules being 3–7 times larger than λD, it is believed that such fluctuation-enhanced electrostatic interactions are responsible for transmitting these intermolecular chiral interactions. The observed decrease in di at which Pmin and ψmax occur with increasing ionic strength (Table 1) is attributed to the reduced amplitude of intermolecular electrostatic interactions, thus requiring the DNA molecules to come into closer proximity. Theoretical calculations incorporating the ionic-strength dependence of chiral interactions would be necessary to describe this phenomenon quantitatively and in more detail.
CONCLUSIONS
From our experimental data we conclude that decreasing ionic strength from 1.0 to 0.2 M NaCl diminishes chiral interactions between DNA chains in a cholesteric liquid crystal. Although the increase in Debye screening length enhances intermolecular interactions between DNA, the consequent increase in intramolecular interactions smears out the helical charge pattern along DNA molecules. We attribute this effect to the observed weakening of chiral interactions between DNA molecules with decreasing ionic strength.
With DNA surface-to-surface separations up to seven times larger than λD at the salt concentrations studied, fluctuation-enhanced electrostatic interactions (20,21) are most likely responsible for the resulting intermolecular chiral interactions. Although the theory by Kornyshev and Leikin (18) accounts for the nonmonotonic dependence of P on DNA density and accurately predicts the range of the DNA cholesteric phase that we observe experimentally, their prediction for Pmin only matches our data for 1.0 M NaCl and no ionic-strength dependence for chiral interactions is predicted. We suggest that incorporating fluctuation-enhanced electrostatic interactions within a suitable theoretical description for DNA chiral interactions may prove useful in capturing the ionic-strength dependence of P and ψ we observe experimentally.
Acknowledgments
We acknowledge preliminary work by Lawrence Chiang and discussions with V. Adrian Parsegian and Rudi Podgornik.
This research was supported by the National Science Foundation through the University of Massachusetts, Amherst Materials Research Science and Engineering Center (grant No. DMR-0213695) and a National Science Foundation Career Award (Chiral Biopolymer Liquid Crystals, award No. DMR-9984427).
Christopher Stanley's present address is Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD 20899.
References
- 1.Van Winkle, D. H., M. W. Davidson, W. X. Chen, and R. L. Rill. 1990. Cholesteric helical pitch of near persistence length DNA. Macromolecules. 23:4140–4148. [Google Scholar]
- 2.Livolant, F. 1991. Ordered phases of DNA in vivo and in vitro. Physica A (Amsterdam). 176:117–137. [Google Scholar]
- 3.Livolant, F., and A. Leforestier. 1996. Condensed phases of DNA: structures and phase transitions. Prog. Polym. Sci. 21:1115–1164. [Google Scholar]
- 4.Leonard, M., H. Hong, N. Easwar, and H. H. Strey. 2001. Soft matter under osmotic stress. Polym. 42:5823–5827. [Google Scholar]
- 5.DuPre, D. B., and R. W. Duke. 1975. Temperature, concentration, and molecular weight dependence of the twist elastic constant of cholesteric poly-γ-benzyl-L-glutamate. J. Chem. Phys. 63:143–148. [Google Scholar]
- 6.Dogic, Z., and S. Fraden. 2000. Cholesteric phase in virus suspensions. Langmuir. 16:7820–7824. [Google Scholar]
- 7.Grelet, E., and S. Fraden. 2003. What is the origin of chirality in the cholesteric phase of virus suspensions? Phys. Rev. Lett. 90:198302. [DOI] [PubMed] [Google Scholar]
- 8.Chandrasekhar, S. 1992. Liquid Crystals. Cambridge University Press, New York.
- 9.de Gennes, P. G., and J. Prost. 1993. The Physics of Liquid Crystals. Oxford University Press, New York.
- 10.Livolant, F., and Y. Bouligand. 1978. New observations on the twisted arrangement of dinoflagellate chromosomes. Chromosoma. 68:21–44. [Google Scholar]
- 11.Livolant, F. 1984. Cholesteric organization of DNA in vivo and in vitro. Eur. J. Cell Biol. 33:300–311. [PubMed] [Google Scholar]
- 12.Livolant, F., and M. F. Maestre. 1988. Circular dichroism microscopy of compact forms of DNA and chromatin in vivo and in vitro: cholesteric liquid-crystalline phases of DNA and single dinoflagellate nuclei. Biochemistry. 27:3056–3068. [DOI] [PubMed] [Google Scholar]
- 13.Rill, R. L., F. Livolant, H. C. Aldrich, and M. W. Davidson. 1989. Electron microscopy of liquid crystalline DNA: direct evidence for cholesteric-like organization of DNA in dinoflagellate chromosomes. Chromosoma. 98:280–286. [DOI] [PubMed] [Google Scholar]
- 14.Harris, A. B., R. D. Kamien, and T. C. Lubensky. 1997. Microscopic origin of cholesteric pitch. Phys. Rev. Lett. 78:1476–1479. [Google Scholar]
- 15.Harris, A. B., R. D. Kamien, and T. C. Lubensky. 1999. Molecular chirality and chiral parameters. Rev. Mod. Phys. 71:1745–1757. [Google Scholar]
- 16.Kornyshev, A. A., and S. Leikin. 2000. Twist in chiral interaction between biological helices. Phys. Rev. Lett. 84:2537–2540. [DOI] [PubMed] [Google Scholar]
- 17.Kornyshev, A. A., and S. Leikin. 2000. Electrostatic interaction between long, rigid helical macromolecules at all interaxial angles. Phys. Rev. E. 62:2576–2596. [DOI] [PubMed] [Google Scholar]
- 18.Kornyshev, A. A., S. Leikin, and S. V. Malinin. 2002. Chiral electrostatic interaction and cholesteric liquid crystals of DNA. Eur. Phys. J. E. 7:83–93. [Google Scholar]
- 19.Bloomfield, V. A., D. M. Crothers, and I. Tinoco. 2000. Nucleic Acids: Structures, Properties, and Functions. University Science Books, Sausalito, CA.
- 20.Strey, H. H., V. A. Parsegian, and R. Podgornik. 1997. Equation of state for DNA liquid crystals: fluctuation enhanced electrostatic double layer repulsion. Phys. Rev. Lett. 78:895–898. [Google Scholar]
- 21.Strey, H. H., V. A. Parsegian, and R. Podgornik. 1999. Equation of state for polymer liquid crystals: theory and experiment. Phys. Rev. E. 59:999–1008. [Google Scholar]
- 22.Parsegian, V. A., R. P. Rand, N. L. Fuller, and D. C. Rau. 1986. Osmotic stress for the direct measurement of intermolecular forces. Methods Enzymol. 127:400–416. [DOI] [PubMed] [Google Scholar]
- 23.Parsegian, V. A., R. P. Rand, and D. C. Rau. 1995. Macromolecules and water: probing with osmotic stress. Methods Enzymol. 259:43–94. [DOI] [PubMed] [Google Scholar]
- 24.Parsegian, V. A., R. P. Rand, and D. C. Rau. 2000. Osmotic stress, crowding, preferential hydration, and binding: a comparison of perspectives. Proc. Natl. Acad. Sci. USA. 97:3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouligand, Y., and F. Livolant. 1984. The organization of cholesteric spherulites. J. Phys. (Fr). 45:1899–1923. [Google Scholar]
- 26.Bezic, J., and S. Zumer. 1992. Structures of the cholesteric liquid crystal droplets with parallel surface anchoring. Liquid Cryst. 11:593–619. [Google Scholar]
- 27.Stanley, C. B., and H. H. Strey. 2003. Measuring osmotic pressure of poly(ethylene glycol) solutions by sedimentation equilibrium ultracentrifugation. Macromolecules. 36:6888–6893. [Google Scholar]
- 28.Strzelecka, T. E., and R. L. Rill. 1991. Phase transitions in concentrated DNA solutions, ionic strength dependence. Macromolecules. 28:5124–5133. [Google Scholar]