

Abstract
The K1 protein of Kaposi's sarcoma-associated herpesvirus (KSHV) has been shown to be a transforming protein capable of inducing morphological changes and focus formation in rodent fibroblasts. K1 can activate B-cell receptor (BCR) signaling and upregulate activity of the NFAT and NF-κB transcription factors. In order to understand the regulation of K1 gene expression, we have analyzed sequences upstream of the K1 gene to identify the K1 promoter element. We have performed 5′ rapid amplification of cDNA ends as well as a nuclease protection assay to map the transcriptional start site of the KSHV K1 transcript. The K1 transcriptional start site lies 75 bp upstream of the translation start site. Sequences upstream of the K1 gene were characterized for their ability to activate a luciferase reporter gene in 293 epithelial cells, KSHV-negative B cells (BJAB), KSHV-positive B cells (BCBL-1), and KS tumor-derived endothelial cells (SLK-KS−). We found that a 125-bp sequence upstream of the K1 transcript start site was sufficient to fully activate the luciferase reporter gene in all cell types tested. In addition, the viral transcription factor KSHV Orf50/Rta was capable of further activating this promoter element in 293, BJAB, and BCBL-1 cells but not in SLK-KS− cells. Promoter constructs containing additional sequences upstream of the 125-bp element did not show further augmentation of transcription in the presence or absence of KSHV Orf50.
Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8, was originally isolated from KS tissues by representational difference analysis (10). KSHV has been linked to the development of KS (16, 20, 31) as well as the lymphoproliferative disorders primary effusion lymphoma and multicentric Castleman's disease (7, 8). Based on sequence information, KSHV has been classified in the gamma subfamily of herpesviruses and is closely related to rhesus monkey rhadinovirus, herpesvirus saimiri (HVS), and murine herpesvirus 68 (1, 2, 44).
At the far left end of its genome, KSHV encodes a 46-kDa transmembrane glycoprotein known as K1 (21). Its position is equivalent to that of the saimiri transformation-associated protein (STP) of HVS (32) and the R1 gene of rhesus monkey rhadinovirus (12). Previous research has shown that HVS STP is essential for the immortalization of primary T lymphocytes and for the development of lymphomas in marmosets (13). Expression of K1 has been shown to induce focus formation and morphological changes in Rat-1 fibroblasts, indicative of its ability to transform cells (26). K1 can also functionally substitute for the STP gene in the context of HVS to immortalize common marmoset lymphocytes to interleukin-2-independent proliferation to induce lymphomas in vivo (26), promoting speculation that it performs a similar role as STP in KSHV-associated lymphoproliferative disorders.
K1 is a type 1 transmembrane glycoprotein. The cytoplasmic tail contains an immunoreceptor tyrosine-based activation motif (25). K1, as well as R1, has been shown to initiate signaling pathways in B cells which result in the tyrosine phosphorylation of cellular proteins, mobilization of intracellular calcium, and the activation of transcription factors such as NFAT and AP-1 (11, 23, 25). K1 has also been shown to downregulate B-cell receptor complex expression in the plasma membrane (24). The amino terminus of K1 interacts with μ chains of B-cell receptor complexes to retain them in the endoplasmic reticulum, and this implies a role for K1 in the survival of KSHV-infected cells (24).
Given the signaling capabilities of K1, Samaniego et al. tested it for a role in mediating paracrine activation of endothelial cells and found that cells expressing K1 showed increased NF-κB-dependent promoter activity (39). NF-κB is responsible for activating a number of inflammatory responses. K1 may therefore activate uninfected endothelial cells in a paracrine manner through the activation of NF-κB-dependent promoters and secretion of inflammatory cytokines (39).
As a herpesvirus, KSHV can maintain its genome in an episome and remain latent until reactivated. Such reactivation is dependent on the transcriptional activator KSHV Orf50/Rta, since dominant-negative Orf50 mutants inhibit reactivation in BCBL-1 (body cavity-based lymphoma 1) cells (27, 28). A cycle of lytic viral replication can also be artificially induced by the addition of the phorbol esters 12-O-tetradecanoylphorbol-13-acetate (TPA) and n-butyrate (30, 37).
Present evidence suggests that K1 is a lytic protein. Lagunoff and Ganem demonstrated the expression of a 1.3-kb transcript when BCBL-1 cells were induced with TPA (21). Samaniego et al. have shown that a 1.3- and a 3-kb K1 transcript are expressed in uninduced BC-3 cells but that both transcripts are upregulated in TPA-induced BC-3 cells (39). In addition they observe that the K1 transcript is expressed in KS tumor cells (39). Analysis of the KSHV transcriptional program during lytic reactivation in primary effusion lymphomas by Northern hybridization and microarrays (19, 34, 40) and real-time PCR analysis (14) indicated that K1 expression is upregulated during the lytic cycle. However, its pattern of expression was found to cluster with those of classically latent genes such as Orf73 (LANA), Orf72 (cyclin), and Orf71 (vFLIP) (14). Taken together, these data suggest that K1 is clearly upregulated during the lytic cycle but that there is a possibility that very low amounts of K1 transcript may also be expressed during latency.
Recent data from Lagunoff et al. suggest that K1 may augment reactivation of KSHV from its latent state and play a role in lytic gene expression (22). Expression of a dominant-negative mutant of K1 was shown to decrease lytic gene expression by as much as 80% (22). The authors concluded that K1 plays a role in lytic replication but is not absolutely required.
In order to develop a better understanding of the expression and regulation of the K1 gene, we have attempted to identify the K1 promoter. At present, nothing is known about this promoter or the K1 transcriptional start site. Using 5′ rapid amplification of cDNA ends (5′ RACE), we have identified the transcription start site and confirmed it by RNase protection assays (RPAs). We have analyzed sequences upstream of this start site for their ability to activate a luciferase reporter construct. Since KSHV can infect human endothelial cells, B cells, epithelial cells, and foreskin fibroblasts (4-6, 15, 36, 43), we have confined our experiments to endothelial cells (SLK-KS−), B cells (BJAB and BCBL-1), and epithelial cells (293). In addition, we have demonstrated the effects of the KSHV viral transcription factor Orf50/Rta (28, 41) and TPA induction on K1 promoter activity in all four cell types.
MATERIALS AND METHODS
5′ RACE.
To define the transcription start site for the K1 gene, 5′ RACE was performed with RNA isolated from KSHV-positive B cells treated with either TPA (25 ng/ml) or an equivalent volume of dimethyl sulfoxide (DMSO) (negative control) for 48 h. Total RNA was isolated by using the RNA STAT-60 kit (Tel-Test “B,” Inc.), and mRNA was isolated from total RNA by using the mRNA STAT-30 kit (Tel-Test “B,” Inc.). Reverse transcription and PCRs were carried out by using the Clontech SMART RACE kit in the absence or presence of reverse transcriptase. Amplified cDNAs were separated on a 2% agarose gel, isolated from the gel, and cloned into the pCR2.1-TOPO vector (Invitrogen TOPO cloning kit). The TOPO clones were transformed into DH5-α cells and were plated on Luria-Bertani plates containing ampicillin. Multiple TOPO colonies were picked and sequenced to identify the 5′ end of the K1 transcript.
K1 promoter cloning.
PCR primers were designed to amplify sequences upstream of the K1 gene of clade C KSHV. BCBL-1 genomic DNA was used as a source of KSHV template from which each K1 fragment was generated. A total of four K1 PCR fragments were generated and contain 25, 125, 225, and 325 nucleotides upstream of the K1 start site and includes 75 base pairs of the K1 5′ untranslated region (UTR). SacI and XhoI restriction enzyme sites were engineered into each K1 primer set to permit insertion of the PCR-amplified fragment into the pGL2-Basic luciferase vector (Promega). The 25- and 125-bp promoter fragments were amplified by PCR with Pfu polymerase under the following conditions: 94°C for 2 min; 35 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 3 min; and a final incubation at 72°C for 2 min. Due to high guanine-cytosine content in the larger-sized fragments, the remaining two promoter sequences were amplified with the Advantage-GC genomic PCR system (Clontech) and the Eppendorf Mastercycler gradient thermal cycler. Amplification conditions were as follows: 95°C for 1 min, 35 cycles of 94°C for 30 s and 68°C for 3 min, and finally 68°C for 3 min with KSHV genomic DNA from BCBL-1 cells as template.
The PCR-amplified K1 promoter DNA was cut with the restriction enzymes SacI and XhoI (New England Biolabs) and cloned into the SacI and XhoI sites of the pGL2-Basic vector. Constructs were verified by sequencing.
To clone the promoter region upstream of the K1 gene from BC-1 and BC-3 cells, the primer used to amplify the 325-nucleotide fragment was used in conjunction with a K1 internal primer to PCR amplify the fragment. The fragment was TOPO cloned and sequenced.
Cell culture.
KSHV-positive BCBL-1 cells and BJAB B cells were grown and maintained in RPMI medium 1640 (Gibco BRL) with l-glutamine and 10% fetal bovine serum (FBS) supplemented with penicillin (100 U/ml) and streptomycin (50 μg/ml). 293 epithelial cells were grown in Dulbecco modified Eagle medium (DMEM) (Gibco BRL) with 10% FBS, Glutamax I, high glucose, 110 mg of sodium pyruvate/ml, and pyridoxine-HCl and supplemented with penicillin (100 U/ml) and streptomycin (50 μg/ml). SLK-KS−, a KSHV-negative endothelial cell line, was grown in DMEM (Gibco BRL) with 10% FBS, Glutamax I, high glucose, 110 mg of sodium pyruvate/ml, and pyridoxine-HCl and supplemented with penicillin (100 U/ml) and streptomycin (50 μg/ml). All cells were grown at 37°C with 5% carbon dioxide.
Transfections.
Each of the following transfections was performed in duplicate, and each experiment was repeated at least five times.
Transfections of BCBL-1 and BJAB cells were performed with and without pcDNA3-Orf50 expression plasmid and with and without TPA induction. For each transfection, 1 × 106 to 5 × 106 cells were added to 0.4-cm-gap cuvettes in a total volume of 400 μl of RPMI 1640 (no serum or antibiotics) along with the specified amounts of DNA to be transfected. Orf50 transfections were carried out with 15 μg of K1 promoter plasmid, 15 μg of Orf50 expression plasmid, and 2 μg of a β-galactosidase reporter construct. As a negative control, 15 μg of pcDNA3 vector was transfected with 15 μg of each construct and 2 μg of the β-galactosidase reporter construct. The cells were electroporated at 300 V and 950 μF and then left at room temperature for 15 min to recover. The electroporated cells were added to separate wells on 12-well plates containing 4 ml of RPMI medium with 10% FBS, penicillin, and streptomycin and were incubated at 37°C with 5% CO2 for 48 h. At 48 h, the cells were lysed in 200 μl of Promega reporter lysis buffer and were used for luciferase and β-galactosidase assays.
To determine the effects of TPA induction on K1 promoter activity, BCBL-1 and BJAB cells were transfected with each of the constructs as described above. The electroporated cells were then treated with 25 ng of TPA/ml (or an equivalent volume of DMSO as a negative control). The cells were incubated for 48 h at 37°C with 5% CO2 and were lysed in 200 μl of Promega reporter lysis buffer for use in luciferase and β-galactosidase assays.
Transfections in 293 cells were done with Lipofectamine reagent (Gibco BRL). As with the BCBL-1 and BJAB experiments, transfections of 293 cells were designed to measure K1 promoter activity following cotransfection of an Orf50 expression plasmid and following treatment with TPA. Twenty-four hours prior to transfection, 5 × 105 cells were seeded in six-well plates in DMEM containing 10% FBS, supplemented with penicillin and streptomycin. Cells were grown overnight at 37°C with 5% CO2. For the experiment with and without Orf50, 3 μg of the appropriate K1 promoter construct, 2 μg of Orf50 expression plasmid, and 1 μg of β-galactosidase reporter construct were added to 500 μl of DMEM (no serum and no penicillin or streptomycin) for each well. Ten microliters of Lipofectamine (Gibco-BRL) was used per transfection according to the manufacturer's protocol. Cells were washed once with phosphate-buffered saline prior to addition of the transfection solution. Plates were incubated overnight at 37°C with 5% CO2. Twelve hours posttransfection, 1.5 ml of DMEM containing 20% FBS and 2× penicillin-streptomycin was added to each well. Cells were further incubated for 36 h at 37°C with 5% CO2. Forty-eight hours posttransfection, cells were harvested and lysed in 200 μl of Promega lysis buffer. For the experiment with and without TPA, a similar transfection protocol was used with either 25 ng of TPA/ml or an equivalent volume of DMSO (negative control). Cells were harvested and lysed as described above.
SLK-KS− transfections were performed with the Superfect transfection reagent (Qiagen). A total of 5 × 105 cells were plated 12 h prior to transfection and grown at 37°C with 5% CO2 in DMEM with 10% FBS, penicillin, and streptomycin. For the Orf50 cotransfection experiments, cells were transfected with 2 μg of K1 promoter construct, 3 μg of Orf50 expression plasmid or pcDNA3 vector, 1 μg of β-galactosidase, and 7.5 μl of Superfect per the manufacturer's protocol. Twelve hours posttransfection the transfection medium was replaced with fresh complete DMEM. Cells were harvested and lysed in 200 μl of Promega reporter lysis buffer 48 h posttransfection. The experiment with and without TPA was performed in a similar manner as that described above.
RPA.
BCBL-1 cells (5 × 107) were transfected with 40 μg of pcDNA3-Orf50 plasmid, using GenePorter 2 transfection reagent and diluent B (Gene Therapy Systems Inc.) according to the manufacturer's protocol. TPA was also added to a final concentration of 25 ng/ml. The cells were incubated at 37°C for 48 h, and RNA was isolated with the RNA STAT-60 kit (Tel-Test). RPAs were performed with the Ambion RPA III RPA kit. The Promega riboprobe in vitro transcription system was used to generate a K1 antisense probe for the RPA. The template used in this reaction was a pSP72-K1 plasmid in which a 557-bp fragment, spanning 400 bp of the K1 promoter and including the first 157 bp of the K1 open reading frame, was inserted into the EcoRI site of pSP72. To make antisense probe, this plasmid was linearized with SmaI, gel isolated, and transcribed in vitro with T7 polymerase to generate a 429-bp antisense probe. The full-length probe was gel isolated on an 8% denaturing acrylamide gel. For the K1 RPA, 5 × 105 cpm of gel-isolated probe per 100 μg of total RNA of DMSO-treated or TPA-treated BCBL-1 cells was used. For the positive β-actin control, the manufacturer's protocol was followed exactly. Sequencing reactions were performed with pSP72 and a 32P-end-labeled T7 primer by using the Epicentre Sequitherm cycle sequencing kit. Reactions were carried out in a thermocycler as described in the manufacturer's protocol. The φX174/HinFI-digested marker from Promega was radiolabeled with [32P]dCTP and run on the same gel.
RESULTS
5′ RACE and promoter analysis to identify the K1 transcription start site.
Nothing is currently known about the start site of K1 transcription. The KSHV K1 gene is situated at the leftmost end of the viral genome and abuts the terminal repeats. Most of the KSHV genomes sequenced to date stop before the start of the terminal repeats due to the high GC content in this region (38). Sequences entered into the National Center for Biotechnology Information GenBank database that include the K1 open reading frame also end shortly upstream of the K1 translation start site. Importantly, Lagunoff and Ganem have sequenced an approximately 800-bp fragment that includes the genomic termini of the BCBL-1 KSHV genome upstream of the K1 coding sequence and also includes the K1 open reading frame (21). This sequence laid the foundation for our promoter studies since it contained the K1 open reading frame and was colinear with sequences upstream of the K1 translation start site. Mapping of the K1 transcription start site was an important first step toward identifying the K1 promoter element required for gene expression, and we utilized three different KSHV-positive B-cell lines, BCBL-1, BC-1, and BC-3, to identify the K1 promoter.
To identify the start site, a 5′ RACE was performed. Total RNA was harvested from uninduced and TPA-induced KSHV-positive B cells. Poly(A) mRNA was isolated from total RNA, and reverse transcription was performed with an oligo(dT) primer to produce a cDNA copy of the mRNA transcripts. K1-specific primers were used in conjunction with the SMART RACE oligomer (Clontech) to PCR amplify a double-stranded DNA fragment encompassing the 5′ end of the K1 transcript. The PCR products were separated on a 2% agarose gel and cloned into the pCR2.1-TOPO vector, and 20 colonies were picked for DNA sequencing. The K1 transcription initiation site (Fig. 1A, black arrow) was found to be 75 nucleotides upstream of the K1 translation start site (Fig. 1A, white arrow), which corresponds to nucleotide position 29 of the BC-1 KSHV genome sequence (38) or nucleotide position 410 in the genomic termini of the BCBL-1 KSHV genome (21).
FIG. 1.

Characterization of the K1 promoter region in KSHV-positive BCBL-1, BC-1, and BC-3 cells. (A) A Clustal W alignment of sequences upstream of the K1 gene in BCBL-1, BC-1, and BC-3 cells is shown. Identical nucleotides are highlighted in black boxes, and divergent nucleotides are highlighted in gray boxes. Based on data from the 5′-RACE experiments, the transcription start site (black arrow) for K1 was assigned to a position 75 bp upstream of the translation start site (white arrow). The putative TATA box (asterisk) for transcription of K1 is located 25 bp upstream of the transcription start site. (B) GenBank sequences upstream of K1 from all four clades of KSHV isolates were aligned with the BCBL-1, BC-1, and BC-3 sequences by using the Clustal W alignment program. The identical nucleotides are highlighted in black boxes, and divergent nucleotides are highlighted in gray boxes. The transcription start site is depicted with a black arrow, and the translation start site is depicted with a white arrow.
As there was very little information available to us on the sequences directly upstream of the K1 open reading frame of the BC-1 and BC-3 KSHV viral genomes, we used PCR amplification to identify the putative K1 promoter element in these two viral genomes. Using primers based on upstream sequences in the genomic termini of the BCBL-1 KSHV genome (21) and primers within the K1 coding sequence, we amplified K1 promoter fragments from the BC-1 and BC-3 KSHV genomes as well. These upstream sequences from the BC-1, BC-3, and BCBL-1 genomes were aligned by using the Clustal W multiple sequence alignment program and are depicted in Fig. 1A. There is a weak TATA box located 25 bp upstream of the transcription start site which is indicated with an asterisk in Fig. 1A. The TATA elements and the transcription start sites are very well conserved in all three KSHV viral genomes. In addition, sequences 133 bp upstream of the K1 transcription start site were almost identical, and sequences farther upstream of this position were quite well conserved. Finally, we also performed an alignment of K1 coding and upstream sequences (clades A, B, C, and D) isolated from different regions of the world by Zong et al. (45) and Poole et al. (35) and our BC-1, BC-3, and BCBL-1 sequences and found that, once again, the transcription start site and TATA boxes were very well conserved in all sequences analyzed (Fig. 1B). This is remarkable in light of the fact that this is a highly variable region of the KSHV genome and that the K1 gene is one of the most highly divergent genes among different isolates of KSHV. Sequence information farther upstream of the isolates shown in Fig. 1B was not available to us.
Analysis of the K1-specific transcript was also confirmed by RPA (Fig. 2). BCBL-1 cells were induced with TPA or DMSO (negative control), and total cellular RNA was harvested 48 h later. Total RNA from TPA-treated or DMSO-treated cells was hybridized overnight to an antisense radiolabeled, gel-purified K1 riboprobe, and this was followed by an RPA performed with a mixture of RNase A and RNase T1. Lane 3 of Fig. 2 represents the undigested 429-bp K1 antisense riboprobe used in the RPA. Lanes 4 and 5 represent the 232-bp protected K1 transcript in DMSO- and TPA-treated BCBL-1 cells, respectively. A control β-actin RPA was also performed with an antisense β-actin riboprobe. Lane 1 represents the undigested 334-bp β-actin antisense riboprobe, and lane 2 represents the 250-bp protected β-actin transcript. The products of the RPA were run on an 8% denaturing acrylamide gel in parallel with a radiolabeled DNA marker (lanes marked M) and a sequencing ladder (lanes G, A, T, and C) to determine the size of the protected fragments. The arrow in Fig. 2 indicates the 232-bp protected fragment representing K1 transcripts in both DMSO-treated and TPA-treated BCBL-1 cells. The minor bands seen below the protected K1 fragment may represent RNA species with alternative transcription initiation sites. In order to determine expression of the K1 transcript in latent versus lytic KSHV-positive B cells, we also performed Northern hybridizations and reverse transcription-PCR assays with BCBL-1 cells either induced with 25 ng of TPA/ml or uninduced (DMSO) and found that cells treated with TPA showed a 1.3- and a 3-kb transcript (data not shown), similar to the findings of Samaniego et al. with BC-3 cells (39). K1 transcripts were also seen in the uninduced lanes and could represent K1 transcription from the 5% of BCBL-1 cells undergoing spontaneous reactivation (9, 37). Alternatively it is possible that there is minimal K1 expression in latent uninduced BCBL-1 cells, and further evidence is needed to address this issue.
FIG. 2.

Analysis of the K1 transcript by RPA. BCBL-1 cells were treated with DMSO or TPA, and RNA was isolated 48 h later. Total RNA from TPA-treated or DMSO-treated cells was hybridized overnight to an antisense radiolabeled, gel-purified K1 riboprobe, and this was followed by an RPA performed with a mixture of RNase A and RNase T1. Lane 3 represents the undigested 429-bp K1 antisense riboprobe used in the RPA. Lanes 4 and 5 represent the 232-bp protected K1 transcript in DMSO- and TPA-treated BCBL-1 cells, respectively. A control β-actin RPA was also performed with an antisense β-actin riboprobe. Lane 1 represents the undigested 334-bp β-actin antisense riboprobe used in the RPA, and lane 2 represents the 250-bp protected β-actin transcript. The products of the RPA were run on an 8% denaturing acrylamide gel in parallel with a radiolabeled φX174/HinFI DNA marker from Promega (lanes M) and a sequencing ladder (lanes G, A, T, and C) to determine the size of the protected fragments in the RPA. The arrow indicates the 232-bp protected fragment representing K1 transcripts in both DMSO-treated and TPA-treated BCBL-1 cells. Numbers at right represent sizes of the marker in base pairs.
Figure 3A depicts the BCBL-1 promoter fragment that we used in our transcriptional studies. The promoter sequence was analyzed with the MatInspector program from Genomatix, and transcription factor binding sites for SP-1, AP-2, and Myc-Max are indicated. Based on the location of the K1 transcription start site, sequences 25, 125, 225, and 325 nucleotides upstream of this start site from the BCBL-1 KSHV genome were cloned upstream of a luciferase reporter gene (Fig. 3B) and were tested for their ability to drive expression of the luciferase gene. The constructs were named −25K1p, −125K1p, −225K1p, and −325K1p and included 75 base pairs of the K1 5′ UTR (Fig. 3B). We transfected these four constructs into cell lines that are tropic for KSHV: KSHV-positive (Epstein-Barr virus [EBV]-negative) BCBL-1 cells, KSHV- and EBV-negative BJAB B cells, 293 kidney epithelial cells, and SLK-KS− KS tumor-derived endothelial cells. The promoter activity of each of the four promoter constructs was measured as luciferase activity in the four different cell lines. K1 promoter responsiveness to the immediate-early transactivator KSHV Orf50/Rta (28, 41) was also examined in these four cell lines.
FIG. 3.

The K1 promoter from the BCBL-1 KSHV genome. (A) Transcription factor binding sites in the K1 promoter from BCBL-1 cells are depicted. The sequence was analyzed with the MatInspector program from Genomatix. (B) Sequences upstream of the K1 start site were cloned upstream of the luciferase reporter gene in the pGL2-Basic plasmid (Promega). These constructs contain 25, 125, 225, and 325 nucleotides upstream of the K1 transcription start site. The pGL2-Basic vector itself contains no promoter of its own. Asterisks represent the 5′ end of the different K1 promoter fragments used. Bold or underlined letters represent transcription factor binding sites, which are often overlapping.
K1 promoter activity in latently infected BCBL-1 cells.
BCBL-1 is a B-cell line that is latently infected with KSHV (37). Renne et al. (37) showed a 50- to 70-fold induction of lytic transcripts upon addition of TPA, indicating that the virus was reactivated from latency. Transfections were performed in these cells to measure the activity of the K1 promoter in the context of a latent (−TPA, −Orf50) and lytic (+TPA, +Orf50) KSHV infection. Each transfection was performed in duplicate and repeated five times, and all transfections were normalized by using a β-galactosidase expression plasmid to control for transfection efficiency. Luciferase constructs containing segments of the putative K1 promoter (Fig. 3B) were cotransfected with or without an Orf50 expression plasmid or were transfected and then stimulated with TPA or DMSO as a negative control to mimic the KSHV lytic and latent states, respectively. Figure 4A represents the transcriptional activation seen with these constructs in untreated BCBL-1 cells. Promoter activity, which was measured by luciferase units for each of the plasmids, is represented as fold activation over the pGL2-Basic vector control. With the exception of the −25K1p promoter construct, the three other promoter constructs (−125K1p, −225K1p, and −325K1p) all showed significant activity (∼12-fold) over the pGL2-Basic vector, indicating that sequences as close as 125 nucleotides upstream of the K1 transcription start site are capable of functioning as a promoter element. Next we looked at the effect of Orf50, the key viral transcriptional activator of KSHV, on the K1 promoter fragments. Orf50 was expressed from a pcDNA3-Orf50 expression plasmid, and the fold activation for each promoter fragment was compared to that for a pcDNA3 vector control (Fig. 4B). We observed that Orf50 could activate the −125K1p promoter fragment about ninefold. The larger promoter fragments, −225K1p and −325K1p, showed a slightly lower fold activation in the presence of Orf50 compared to the −125K1p fragment, suggesting the involvement of a repressor in these larger promoter elements. The same trend was seen when TPA was used to induce the transfected BCBL-1 cells into the lytic cycle (Fig. 4C), the only difference being that the overall fold activations were lower than those seen with the Orf50 transactivator. This suggests that Orf50 is a much better activator of the KSHV lytic cycle and the K1 promoter than is TPA, and this is in concordance with previous findings (41). In all three experiments, −25K1p had minimal activity regardless of the presence or absence of Orf50 or TPA. The −25K1p fragment contains only a weak TATA box, thus suggesting that it functions as a minimal promoter element and that transcription factor binding sites upstream of the TATA box are required for further activation.
FIG. 4.
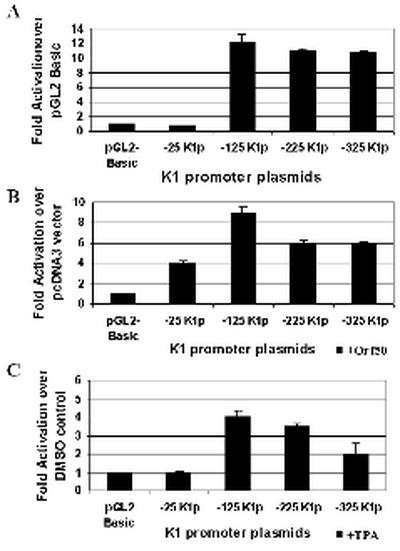
Analysis of K1 promoter activity in BCBL-1 cells. (A) K1 promoter activity in the context of latent KSHV infection was measured by transfections of the K1 promoter constructs in BCBL-1 B cells. Transfection efficiencies were normalized by using a plasmid expressing β-galactosidase. Cells were left untreated and harvested 48 h posttransfection. Luciferase activity was measured for each construct and normalized to β-galactosidase activity. Activity of each promoter construct is represented as fold activation over the pGL2-Basic vector control. (B) Each of the K1 promoter constructs was cotransfected with a pcDNA3-Orf50 expression plasmid into BCBL-1 cells. Luciferase activities were measured and normalized as described above. Activity of each promoter construct is represented as fold activation over the pcDNA3 vector control. (C) The effects of TPA induction on the K1 promoter were tested by performing the same transfections described for panel A but treating cells with TPA to induce the KSHV lytic cycle or DMSO (negative control). Activity of each promoter construct is represented as fold activation over the DMSO control.
K1 promoter activation in KSHV-negative BJAB cells.
BJAB is an EBV-negative and KSHV-negative B-cell line. BJAB cells were transfected with the same K1 promoter constructs described above (Fig. 3B) and either cotransfected with an Orf50 expression plasmid or induced with TPA to measure K1 promoter activity in a KSHV-negative B-cell line. Transfections were performed in duplicate, repeated five times, and normalized with a β-galactosidase expression plasmid to control for transfection efficiency. Figure 5A represents the transcriptional activation seen with these constructs in untreated BJAB B lymphocytes. Promoter activity, which was measured by luciferase units for each of the plasmids, is represented as fold activation over the pGL2-Basic vector control. With the exception of the −25K1p promoter construct, the three other promoter constructs (−125K1p, −225K1p, and −325K1p) all showed significant activity over the pGL2-Basic vector (∼18-fold), indicating that sequences as close as 125 nucleotides upstream of the K1 transcription start site are capable of functioning as a promoter element in KSHV-negative B cells. This finding also indicates that the K1 promoter is active in B cells in the complete absence of any KSHV Orf50/Rta, a possibility that we could not rule out in the BCBL-1 cells due to the 2 to 5% rate of spontaneous reactivation seen in these cells.
FIG. 5.
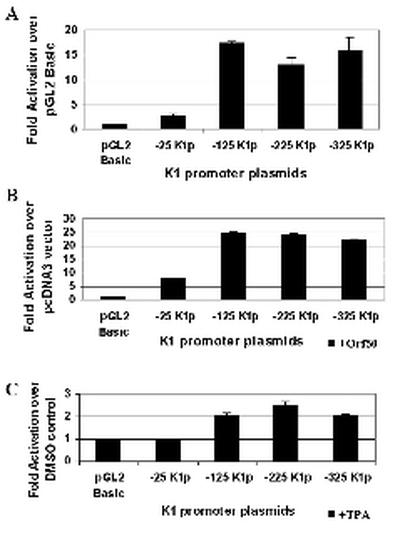
Analysis of K1 promoter activity in BJAB cells. (A) K1 promoter activity in KSHV-negative B cells was measured by transfections of the four K1 promoter constructs into BJAB B cells. Transfection efficiencies were normalized by using a plasmid expressing β-galactosidase. Cells were left untreated and harvested 48 h posttransfection. Luciferase activity was measured for each construct and normalized to β-galactosidase activity. Activity of each promoter construct is represented as fold activation over the pGL2-Basic vector control. (B) Each of the K1 promoter constructs was cotransfected with a pcDNA3-Orf50 expression plasmid into BJAB cells. Luciferase activities were measured and normalized as described above. Activity of each promoter construct is represented as fold activation over the pcDNA3 vector control. (C) The effects of TPA induction on the K1 promoter were tested by performing the same transfections described for panel A but treating cells with TPA or DMSO (negative control). Activity of each promoter construct is represented as fold activation over the DMSO control.
Next we looked at the effect of Orf50 in activating the K1 promoter fragments. Orf50 was expressed from a pcDNA3-Orf50 expression plasmid, and the fold activation for each promoter fragment was compared to that of a pcDNA3 vector control (Fig. 5B). We observed that Orf50 could activate the −25K1p promoter construct about eightfold over the pcDNA3 vector control and the −125K1p, −225K1p, and −325K1p promoter fragments about 25-fold over the pcDNA3 vector control. TPA could not significantly activate the K1 promoter in KSHV-negative BJAB cells (Fig. 5C), and this is probably due to the fact that there is no TPA-inducible endogenous Orf50 present in the BJAB cells to activate the K1 promoter. This suggests that KSHV Orf50, rather than TPA-induced cellular transcription factors, is responsible for K1 promoter activation in these cells. Interestingly, Lukac et al. have shown that KSHV Orf50 activates the delayed early promoters nut-1 and thymidine kinase (TK) or DNA binding protein (DBP) promoters 30- and 6-fold, respectively, in BJAB cells (27). Our data demonstrate that KSHV Orf50 activates the K1 promoter about 20- to 25-fold in BJAB cells, which is within this range of fold activation seen with other early-delayed-early promoters and corresponds well with published evidence indicating that K1 is an early lytic gene product.
K1 promoter activity in 293 epithelial cells.
293 epithelial cells have been previously shown to support KSHV infection in vitro (15, 36). We transfected the four K1 constructs into these cells to measure K1 promoter activity (Fig. 6). Each K1 promoter plasmid was transfected into 293 cells to test promoter activity when cotransfected with and without a pcDNA3-Orf50 expression plasmid or in the presence or absence of TPA and compared to a pcDNA3 vector control or DMSO-treated control, respectively. Each transfection was performed in duplicate repeated several times and normalized with a β-galactosidase expression plasmid to control for transfection efficiency.
FIG. 6.
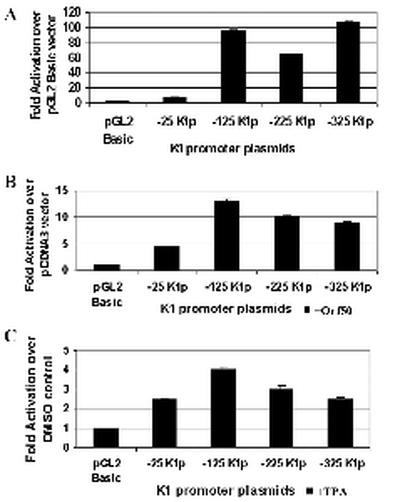
Analysis of K1 promoter activity in 293 epithelial cells. (A) K1 promoter activity in 293 epithelial cells was measured by transfections of the K1 promoter constructs into these cells. Transfection efficiencies were normalized by using a plasmid expressing β-galactosidase. Cells were left untreated and harvested 48 h posttransfection. Luciferase activity was measured for each construct and normalized to β-galactosidase activity. Activity of each promoter construct is represented as fold activation over the pGL2-Basic vector control. (B) Each of the K1 promoter constructs was cotransfected with a pcDNA3-Orf50 expression plasmid into 293 cells. Luciferase activities were measured and normalized as described above. Activity of each promoter construct is represented as fold activation over the pcDNA3 vector control. (C) The effects of TPA induction on the K1 promoter were tested by performing the same transfections described for panel A but treating cells with TPA or DMSO (negative control). Activity of each promoter construct is represented as fold activation over the DMSO control.
Figure 6A represents the transcriptional activation seen with these constructs in untreated 293 kidney epithelial cells. Promoter activity, which was measured by luciferase units for each of the plasmids, is represented as fold activation over the pGL2-Basic vector control. With the exception of the −25K1p promoter construct, the three other promoter constructs (−125K1p, −225K1p, and −325K1p) all showed significant activity, between 60- and 100-fold over the pGL2-Basic vector. This indicates that sequences 125 nucleotides upstream of the K1 transcription start site are capable of functioning as a promoter element for K1 transcription. Next we looked at the ability of Orf50 to activate the K1 promoter fragments in 293 cells. Orf50 was expressed from a pcDNA3-Orf50 expression plasmid, and the fold activation for each promoter fragment was compared to that for a pcDNA3 vector control (Fig. 6B). We observed that Orf50 could activate all three promoter fragments between 9- and 13-fold over the pcDNA3 vector control. TPA activated the K1 promoter about fourfold (Fig. 6C), which is much less than the activation seen with Orf50 and again suggests that Orf50 is a specific and significant activator of the K1 promoter.
K1 promoter activity in SLK-KS− endothelial cells.
SLK-KS− is a KS tumor-derived endothelial cell line that has since lost the virus (18). It was of particular interest to measure K1 promoter activity in these cells since KSHV can infect endothelial cells and KS lesions are comprised of a large number of infected endothelial cells. In addition, K1 has been shown to be expressed in KS lesions (39). Each K1 construct was transfected into SLK-KS− cells to test promoter activity when cotransfected with or without an Orf50 expression plasmid and with or without TPA treatment. Each transfection was performed in duplicate repeated five times, and all transfections were normalized with a β-galactosidase expression plasmid to control for transfection efficiency.
Figure 7A represents the transcriptional activation seen with these constructs in untreated SLK-KS− endothelial cells. Promoter activity, which was measured by luciferase units for each of the plasmids, is represented as fold activation over the pGL2-Basic vector control. With the exception of the −25K1p promoter construct, the three other promoter constructs (−125K1p, −225K1p, and −325K1p) all showed significant activity, between 20- and 35-fold over the pGL2-Basic vector. This indicates that sequences 125 nucleotides upstream of the K1 transcription start site are capable of functioning as a promoter element for K1 transcription. Next we looked at the ability of Orf50 to activate the K1 promoter fragments in endothelial cells. Orf50 was expressed from a pcDNA3-Orf50 expression plasmid, and the fold activation for each promoter fragment was compared to that for a pcDNA3 vector control (Fig. 7B). We observed that, although the promoter fragments alone were active in these SLK-KS− cells, KSHV Orf50 could not significantly activate any of the K1 promoter fragments over the pcDNA3 vector control. This suggests that an essential cellular factor needed for Orf50 specific transactivation of the K1 promoter is absent in these cells compared to B cells and epithelial cells. Interestingly, Lukac et al. have previously shown that other delayed-early promoters like the nut-1, DBP, and TK promoters could be activated by Orf50 in these cells but that the DNA polymerase promoter could not (27). Consistent with other cell lines that we tested, TPA only marginally activated the K1 promoter in the SLK-KS− endothelial cells. Our data suggest that the K1 promoter may be regulated differently in endothelial cells and that the activity of these promoter fragments may be regulated by cell-specific transcription factors.
FIG. 7.

K1 promoter activity in SLK-KS− endothelial cells. (A) K1 promoter activity in KSHV-negative endothelial cells was measured by transfections of the K1 promoter constructs into SLK-KS− cells. Transfection efficiencies were normalized by using a plasmid expressing β-galactosidase. Cells were left untreated and harvested 48 h posttransfection. Luciferase activity was measured for each construct and normalized to β-galactosidase activity. Activity of each promoter construct is represented as fold activation over the pGL2-Basic vector control. (B) Each of the K1 promoter constructs was cotransfected with a pcDNA3-Orf50 expression plasmid into SLK-KS− cells. Luciferase activities were measured and normalized as described above. Activity of each promoter construct is represented as fold activation over the pcDNA3 vector control. (C) The effects of TPA induction on the K1 promoter were tested by performing the same transfections described for panel A but treating cells with TPA or DMSO (negative control). Activity of each promoter construct is represented as fold activation over the DMSO control.
DISCUSSION
In this study, we have mapped the transcription start site of the K1 viral gene and have also identified the K1 promoter element necessary for K1 gene expression. Although RNA isolated from TPA-induced and uninduced KSHV-positive B cells shows the presence of the K1 transcript (39), it is possible that the K1 transcript seen in uninduced cells represents K1 lytic transcripts, due to the fact that 2 to 5% of latently infected BCBL-1 cells spontaneously undergo reactivation (36, 37). Alternatively, it is also feasible that there is a small amount of K1 transcription occurring during latent infection. Although K1 is undoubtedly expressed and upregulated during the KSHV lytic cycle, there is a possibility that it may be expressed at low levels during latency similar to LMP1, its functional homolog in EBV (29, 42). However, much more evidence will be needed to address this question.
Before detailed analysis of the K1 promoter could be performed, it was important to identify the start site of K1 transcription. 5′ RACE was performed with K1 internal primers and revealed a major transcription start site 75 bp upstream of the translation start site (Fig. 1A). These sequences upstream of the K1 gene were conserved quite well among three different isolates of KSHV from the BCBL-1, BC-1, and BC-3 cell lines. This is in marked contrast to the K1 gene, which is the most highly divergent gene in the viral genome among KSHV isolates around the world. This would suggest that the conserved sequences represent important cellular or viral transcription factor binding sites that are needed to regulate the expression of the K1 gene. The start site for K1 transcription was further confirmed by RNase protection since reverse transcriptase can be sensitive to RNA structure. Based on the transcriptional start site, we have identified a putative weak TATA box 25 bp upstream of the transcription start site. This TAATT motif has been seen upstream of cellular genes such as the UDP-glucuronosyltransferase gene (17). In addition, available sequences from different K1 genes from the A, B, C, and D clades indicate that sequences from the weak TATA box to the K1 translation start site are well conserved, indicating that these sequences may play a key role in regulation of K1 gene expression. We confirmed our 5′-RACE results with an RPA. This assay also mapped the K1 transcriptional start site at a position 75 nucleotides upstream of the K1 translation start site. With these data we were then able to construct deletion mutants that contained segments of sequences upstream of the K1 gene to assay for K1 promoter activity.
The first construct (−25K1p) contained 25 nucleotides of the putative K1 promoter sequence and included the 5′ UTR. In each cell line tested, −25K1p lacked significant promoter activity. In addition, this construct was unresponsive to KSHV Orf50 expression and TPA induction. The reasons for this inactivity may be due to the fact that this fragment contains only the putative TATA element with no upstream transcription factor binding sites. The element may be sufficient only for binding of the basal complex, TFIID, and may be inactive because it is isolated from its upstream sequence and is therefore out of its normal context. Further, studies of TFIID-dependent transcription have suggested that TFIID binding may vary from promoter to promoter. For example, experiments with DNase I footprinting demonstrated that the size of protection by TFIID differed between the adenovirus major late promoter and human hsp-70 promoter, 75 and 54 bp, respectively (33). Hence, it may be possible that the −25K1p promoter fragment is too small for TFIID to bind and initiate transcription.
The −125K1p construct typically had the highest activity of the K1 constructs in all of the cell lines tested. This construct is 100 base pairs larger than the −25K1p fragment and would likely be large enough for the basal TFIID and RNA polymerase to form a transcription initiation complex on it. Also, the putative TATA box would be in its natural context in this construct. Sequence analysis showed that this fragment contains transcription factor binding sites such as SP-1 and NF-1 that are not present in the −25K1p fragment. It is important to note that the −125K1p promoter region is very well conserved in the three different KSHV viral genomes isolated from BC-1, BC-3, and BCBL-1 cells. This suggests that certain key cellular or viral transcription factors required for regulation and activation of the K1 promoter may interact with this region of the promoter. There was also significant activity observed with the −225K1p and −325K1p promoter fragments in all four cell lines tested but not more than that seen with the −125K1p fragment. Both of these constructs included the −125K1p promoter fragment, and thus this constitutes the minimal cis element necessary for K1 expression in untreated cells. Also, since the 293, BJAB, and SLK-KS− cells do not contain the KSHV genome or express KSHV Orf50, our data show that the K1 promoter is active in all these cell types in the absence of Orf50, a possibility that we could not rule out in the BCBL-1 cells due to the 2 to 5% rate of spontaneous reactivation seen in these cells.
Cotransfection of the KSHV Orf50/Rta expression plasmid increased promoter activity of the K1 constructs in both B-cell lines and 293 epithelial cells. Since K1 is expressed as an early gene during the KSHV lytic cycle (19, 34), it is entirely feasible that the immediate-early transactivator Orf50 can activate the K1 promoter. Activation of the K1 promoter by KSHV Orf50 was 20- to 25-fold in BJAB cells, and this falls well within the range of Orf50's ability to activate other early-delayed-early promoters such as nut-1, TK, and DBP (27). Interestingly, K1 promoter activation in BCBL-1 cells was generally lower than that seen in BJAB cells. Perhaps there is a factor or factors expressed during KSHV latency in the BCBL-1 cells that can bind the K1 promoter to repress or substantially reduce K1 promoter activity in these cells. TPA induction results in the reactivation of virus in BCBL-1 cells, causing it to undergo lytic replication through expression of the key lytic transactivator protein KSHV Orf50. Although the K1 constructs were activated by the addition of TPA in BCBL-1 cells, the fold activations are much lower than those seen with Orf50, indicating that KSHV Orf50 is a more potent transactivator of the KSHV lytic cycle than is TPA. TPA has been shown to be a general activator of gene transcription through the protein kinase C and AP-1 pathways (3, 46).
Although the K1 promoter constructs were active in untreated SLK-KS− endothelial cells, addition of an Orf50 expression vector and TPA induction did not significantly increase promoter activity. These data are intriguing because they suggest that there are cell-specific transcription factors that augment Orf50's ability to transactivate the K1 promoter in B cells but not endothelial cells. Hence, the expression of K1 in endothelial cells may be regulated differently than in other cell types. Since the K1 transcript has been shown to be present in KS lesions (39), it is possible that the K1 promoter is active in endothelial cells in the absence of Orf50 and that the presence of the viral transactivator does not significantly enhance or activate its expression over basal levels.
In this paper we have examined the regulation of K1 gene expression in four relevant cell types. The identification of a major transcription start site, a TATA box, and putative transcription factor binding sites is important because they represent fundamental elements needed for transcription. Furthermore, promoter-reporter gene constructs have been used to delineate the upstream promoter region of K1 and provide a framework on which subsequent studies may be based. There is likely a significant sequence contained in the −125K1p construct that is lacking in the −25K1p construct. This sequence may be important for the binding of viral and/or cellular transcription factors. Finally, we have also shown that the K1 promoter element is very responsive to the KSHV Orf50 transactivator protein in B cells and epithelial cells. The construction of additional deletion and substitution mutations in the K1 promoter will provide some insight into the nature of the Orf50-responsive sequence and will also be useful for determining specific cellular transcription factor binding sites in the K1 promoter.
Acknowledgments
We thank Stuart Krall for assistance with the cell lines. We thank D. Dittmer for invaluable discussions and manuscript reading.
S. M. DeWire is supported, in part, by NIH training grant 5T32GM07092-26, and B. S. Bowser is supported, in part, by NIH training grant 5T32AI007419-10. This work was supported in part by grants from the V Foundation and the American Heart Association and by NIH/NCI grant CA096500 (to B. Damania).
REFERENCES
- 1.Albrecht, J. C., J. Nicholas, D. Biller, K. R. Cameron, B. Biesinger, C. Newman, S. Wittmann, M. A. Craxton, H. Coleman, B. Fleckenstein, and R. W. Honess. 1992. Primary structure of the herpesvirus saimiri genome. J. Virol. 66:5047-5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander, L., L. Denenkamp, A. Knapp, M. Auerbach, B. Damania, and R. C. Desrosiers. 2000. The primary sequence of rhesus monkey rhadinovirus isolate 26-95: sequence similarities to Kaposi's sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 74:3388-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angel, P., M. Imagawa, R. Chiu, B. Stein, R. J. Imbra, H. J. Rahmsdorf, C. Jonat, P. Herrlich, and M. Karin. 1987. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 49:729-739. [DOI] [PubMed] [Google Scholar]
- 4.Antman, K., and Y. Chang. 2000. Kaposi's sarcoma. N. Engl. J. Med. 342:1027-1038. [DOI] [PubMed] [Google Scholar]
- 5.Boshoff, C., T. F. Schulz, M. M. Kennedy, A. K. Graham, C. Fisher, A. Thomas, J. O. McGee, R. A. Weiss, and J. J. O'Leary. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274-1278. [DOI] [PubMed] [Google Scholar]
- 6.Cerimele, F., F. Curreli, S. Ely, A. E. Friedman-Kien, E. Cesarman, and O. Flore. 2001. Kaposi's sarcoma-associated herpesvirus can productively infect primary human keratinocytes and alter their growth properties. J. Virol. 75:2435-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 8.Cesarman, E., and D. M. Knowles. 1997. Kaposi's sarcoma-associated herpesvirus: a lymphotropic human herpesvirus associated with Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. Semin. Diagn. Pathol. 14:54-66. [PubMed] [Google Scholar]
- 9.Chandran, B., C. Bloomer, S. R. Chan, L. Zhu, E. Goldstein, and R. Horvat. 1998. Human herpesvirus-8 ORF K8.1 gene encodes immunogenic glycoproteins generated by spliced transcripts. Virology 249:140-149. [DOI] [PubMed] [Google Scholar]
- 10.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 11.Damania, B., M. DeMaria, J. U. Jung, and R. C. Desrosiers. 2000. Activation of lymphocyte signaling by the R1 protein of rhesus monkey rhadinovirus. J. Virol. 74:2721-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damania, B., M. Li, J. K. Choi, L. Alexander, J. U. Jung, and R. C. Desrosiers. 1999. Identification of the R1 oncogene and its protein product from the rhadinovirus of rhesus monkeys. J. Virol. 73:5123-5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duboise, S. M., J. Guo, S. Czajak, R. C. Desrosiers, and J. U. Jung. 1998. STP and Tip are essential for herpesvirus saimiri oncogenicity. J. Virol. 72:1308-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fakhari, F. D., and D. P. Dittmer. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213-6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foreman, K. E., J. Friborg, W. P. Kong, C. Woffendin, P. J. Polverini, B. J. Nickoloff, and G. J. Nabel. 1997. Propagation of a human herpesvirus from AIDS-associated Kaposi's sarcoma. N. Engl. J. Med. 336:163-171. [DOI] [PubMed] [Google Scholar]
- 16.Ganem, D. 1998. Human herpesvirus 8 and its role in the genesis of Kaposi's sarcoma. Curr. Clin. Top. Infect. Dis. 18:237-251. [PubMed] [Google Scholar]
- 17.Gong, Q. H., J. W. Cho, T. Huang, C. Potter, N. Gholami, N. K. Basu, S. Kubota, S. Carvalho, M. W. Pennington, I. S. Owens, and N. C. Popescu. 2001. Thirteen UDPglucuronosyltransferase genes are encoded at the human UGT1 gene complex locus. Pharmacogenetics 11:357-368. [DOI] [PubMed] [Google Scholar]
- 18.Herndier, B. G., L. D. Kaplan, and M. S. McGrath. 1994. Pathogenesis of AIDS lymphomas. AIDS 8:1025-1049. [DOI] [PubMed] [Google Scholar]
- 19.Jenner, R. G., M. M. Alba, C. Boshoff, and P. Kellam. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kedes, D. H., E. Operskalski, M. Busch, R. Kohn, J. Flood, and D. Ganem. 1996. The seroepidemiology of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 2:918-924. [DOI] [PubMed] [Google Scholar]
- 21.Lagunoff, M., and D. Ganem. 1997. The structure and coding organization of the genomic termini of Kaposi's sarcoma-associated herpesvirus. Virology 236:147-154. [DOI] [PubMed] [Google Scholar]
- 22.Lagunoff, M., D. M. Lukac, and D. Ganem. 2001. Immunoreceptor tyrosine-based activation motif-dependent signaling by Kaposi's sarcoma-associated herpesvirus K1 protein: effects on lytic viral replication. J. Virol. 75:5891-5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagunoff, M., R. Majeti, A. Weiss, and D. Ganem. 1999. Deregulated signal transduction by the K1 gene product of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 96:5704-5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee, B. S., X. Alvarez, S. Ishido, A. A. Lackner, and J. U. Jung. 2000. Inhibition of intracellular transport of B cell antigen receptor complexes by Kaposi's sarcoma-associated herpesvirus K1. J. Exp. Med. 192:11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, H., J. Guo, M. Li, J. K. Choi, M. DeMaria, M. Rosenzweig, and J. U. Jung. 1998. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi's sarcoma-associated herpesvirus. Mol. Cell. Biol. 18:5219-5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, H., R. Veazey, K. Williams, M. Li, J. Guo, F. Neipel, B. Fleckenstein, A. Lackner, R. C. Desrosiers, and J. U. Jung. 1998. Deregulation of cell growth by the K1 gene of Kaposi's sarcoma-associated herpesvirus. Nat. Med. 4:435-440. [DOI] [PubMed] [Google Scholar]
- 27.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 29.Martel-Renoir, D., V. Grunewald, R. Touitou, G. Schwaab, and I. Joab. 1995. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gen. Virol. 76:1401-1408. [DOI] [PubMed] [Google Scholar]
- 30.Miller, G., L. Heston, E. Grogan, L. Gradoville, M. Rigsby, R. Sun, D. Shedd, V. M. Kushnaryov, S. Grossberg, and Y. Chang. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore, P. S., and Y. Chang. 1995. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N. Engl. J. Med. 332:1181-1185. [DOI] [PubMed] [Google Scholar]
- 32.Murthy, S. C., J. J. Trimble, and R. C. Desrosiers. 1989. Deletion mutants of herpesvirus saimiri define an open reading frame necessary for transformation. J. Virol. 63:3307-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakajima, N., M. Horikoshi, and R. G. Roeder. 1988. Factors involved in specific transcription by mammalian RNA polymerase II: purification, genetic specificity, and TATA box-promoter interactions of TFIID. Mol. Cell. Biol. 8:4028-4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulose-Murphy, M., N. K. Ha, C. Xiang, Y. Chen, L. Gillim, R. Yarchoan, P. Meltzer, M. Bittner, J. Trent, and S. Zeichner. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 75:4843-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poole, L. J., J. C. Zong, D. M. Ciufo, D. J. Alcendor, J. S. Cannon, R. Ambinder, J. M. Orenstein, M. S. Reitz, and G. S. Hayward. 1999. Comparison of genetic variability at multiple loci across the genomes of the major subtypes of Kaposi's sarcoma-associated herpesvirus reveals evidence for recombination and for two distinct types of open reading frame K15 alleles at the right-hand end. J. Virol. 73:6646-6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renne, R., D. Blackbourn, D. Whitby, J. Levy, and D. Ganem. 1998. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 72:5182-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renne, R., W. Zhong, B. Herndier, M. McGrath, N. Abbey, D. Kedes, and D. Ganem. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342-346. [DOI] [PubMed] [Google Scholar]
- 38.Russo, J. J., R. A. Bohenzky, M. C. Chien, J. Chen, M. Yan, D. Maddalena, J. P. Parry, D. Peruzzi, I. S. Edelman, Y. Chang, and P. S. Moore. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 93:14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samaniego, F., S. Pati, J. Karp, O. Prakash, and D. Bose. 2001. Human herpesvirus 8 K1-associated nuclear factor-kappa B-dependent promoter activity: role in Kaposi's sarcoma inflammation? J. Natl. Cancer Inst. Monogr. 28:15-23. [DOI] [PubMed] [Google Scholar]
- 40.Sarid, R., J. S. Wiezorek, P. S. Moore, and Y. Chang. 1999. Characterization and cell cycle regulation of the major Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J. Virol. 73:1438-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tierney, R. J., N. Steven, L. S. Young, and A. B. Rickinson. 1994. Epstein-Barr virus latency in blood mononuclear cells: analysis of viral gene transcription during primary infection and in the carrier state. J. Virol. 68:7374-7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vieira, J., P. O'Hearn, L. Kimball, B. Chandran, and L. Corey. 2001. Activation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J. Virol. 75:1378-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Virgin, H. W., IV, P. Latreille, P. Wamsley, K. Hallsworth, K. E. Weck, A. J. Dal Canto, and S. H. Speck. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894-5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zong, J. C., D. M. Ciufo, D. J. Alcendor, X. Wan, J. Nicholas, P. J. Browning, P. L. Rady, S. K. Tyring, J. M. Orenstein, C. S. Rabkin, I. J. Su, K. F. Powell, M. Croxson, K. E. Foreman, B. J. Nickoloff, S. Alkan, and G. S. Hayward. 1999. High-level variability in the ORF-K1 membrane protein gene at the left end of the Kaposi's sarcoma-associated herpesvirus genome defines four major virus subtypes and multiple variants or clades in different human populations. J. Virol. 73:4156-4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.zur Hausen, H., F. J. O'Neill, U. K. Freese, and E. Hecker. 1978. Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature 272:373-375. [DOI] [PubMed] [Google Scholar]