

Abstract
Incoherent elastic neutron scattering experiments on members of the cholinesterase family were carried out to investigate how molecular dynamics is affected by covalent inhibitor binding and by differences in primary and quaternary structure. Tetrameric native and soman-inhibited human butyrylcholinesterase (HuBChE) as well as native dimeric Drosophila melanogaster acetylcholinesterase (DmAChE) hydrated protein powders were examined. Atomic mean-square displacements (MSDs) were found to be identical for native HuBChE and for DmAChE in the whole temperature range examined, leading to the conclusion that differences in activity and substrate specificity are not reflected by a global modification of subnanosecond molecular dynamics. MSDs of native and soman-inhibited HuBChE were identical below the thermal denaturation temperature of the native enzyme, indicating a common mean free-energy surface. Denaturation of the native enzyme is reflected by a relative increase of MSDs consistent with entropic stabilization of the unfolded state. The results suggest that the stabilization of HuBChE phosphorylated by soman is due to an increase in free energy of the unfolded state due to a decrease in entropy.
INTRODUCTION
Protein dynamics and function occur on timescales spanning several decades, from femtosecond electronic rearrangements (e.g., in photosynthesis) to folding processes that can take minutes or longer (1). An important time window in the picosecond-to-nanosecond time range is occupied by atomic thermal fluctuations, which have been extensively investigated by molecular dynamics (MD) simulations (2), and by various experimental approaches including neutron scattering (3,4) and nuclear magnetic resonance (NMR) (5). It has been suggested that thermal fluctuations are correlated with and act as a lubricant for slower, functional motions taking place on the microsecond or longer timescales (6). Large-amplitude thermal fluctuations, requiring adequate protein hydration, have been shown to be mandatory for the function of bacteriorhodopsin, the light-driven proton pump in the purple membrane of Halobacterium salinarum (7). However, a direct correlation of the dynamical transition of fast, thermal fluctuations with the onset of enzymatic activity has been questioned (8). Establishing quantitative relationships between thermal fluctuations and functional motions is challenging, taking into consideration the vast variety of protein functions (synthesis, catalysis, electron- and proton transfer, etc.) that are likely to require completely different dynamical behavior on an extended timescale. Comparing enzymes of the same family would certainly be of value in this context.
Several NMR studies have been carried out on protein-ligand complexes (reviewed in Palmer (5)), suggesting that both increases and decreases in protein entropy may occur upon ligand-binding. These findings are supported by MD simulations that suggest that the flexibility of protein residues may either decrease or increase upon ligand-binding (9). Incoherent inelastic neutron scattering experiments probing the vibrational density of states are scarce but seem to favor an increase of low-frequency, collective motions resulting in increased protein flexibility upon ligand binding (10). In considering data obtained by x-ray crystallography, Frauenfelder and Petsko (11) found that binding of oxygen was accompanied by a reduction in B-factors of residues close to the heme pocket in myoglobin. James et al. (12) reported localized B-factor reductions within the substrate binding site in serine protease A upon binding of an inhibitor. In contrast, binding of xenon in the four internal cavities of myoglobin was shown to reduce the overall protein B-factors by ∼15% (13). It should be noted, however, that B-factors from x-ray crystallography, in general, include contributions from static disorder, and care must be taken in comparing them to thermal atomic fluctuations (14).
Denaturation of proteins, whether to a totally unfolded conformation or to a partially unfolded molten globule, has been shown to be reflected in increased atomic thermal motions relative to the native state by several techniques, including NMR (Idiyatullin et al. (15) and references therein), MD simulations (16), and incoherent neutron scattering (17).
To contribute to the ongoing discussion and to investigate the relationship among molecular dynamics, activity, inhibitor-binding, and unfolding, we carried out incoherent elastic neutron scattering experiments on two members of the cholinesterase (ChE) family: Tetrameric human butyrylcholinesterase (HuBChE), both native and covalently inhibited by the organophosphate nerve agent, soman (18), and dimeric native Drosophila melanogaster acetylcholinesterase (DmAChE).
Cholinesterases belong to the α/β hydrolase fold family (19). The principal function of acetylcholinesterase (AChE, EC. 3.1.1.7) is to terminate impulse transmission at cholinergic synapses by rapid hydrolysis of the neurotransmitter acetylcholine (ACh). Due to its crucial physiological role, AChE is the target of nerve agents and insecticides, as well as of the first generation of anti-Alzheimer medications (for literature, see (20)). Butyrylcholinesterase (BChE, EC. 3.1.1.8) is structurally and catalytically very similar to AChE (21). Although its physiological role is still unknown (22), it has found use as a stoichiometric bioscavenger for toxic organophosphate nerve agents (23). Indeed, administration of high doses of HuBChE confers efficient protection against otherwise lethal doses of nerve agents (24).
ChEs provide a particularly suitable system for studying relationships between activity, ligand binding, unfolding, and thermal molecular fluctuations due to the following properties:
The catalytic turnover number of ChEs is among the highest known, approaching the diffusion limit (25). This is particularly intriguing, since its three-dimensional structure reveals that the active site of AChE, which, like other serine hydrolases, features a Ser-His-Glu catalytic triad, is buried at the bottom of a deep and narrow gorge lined by aromatic residues (26). The three-dimensional structure of BChE is very similar (21). Since the diameter of this gorge, at its narrowest point, is smaller than the cross-section of the quaternary group of ACh, substantial breathing motions of the whole AChE catalytic subunit are necessary to allow the substrate to penetrate to the active site. A number of MD simulations have been carried out to shed light on this topic. Shen et al. (27) conducted a 500-ps MD simulation focusing on the gorge radius. Most interestingly, they found that a substantial part of the AChE molecule is involved in the gorge opening and not just a few gorge residues. In another MD simulation, Tai et al. (28) found by principal component analysis that gorge-opening behavior is not dominated by a few principal dynamical components but rather by a hierarchy of motions on different timescales acting together. Other MD studies have demonstrated that transient openings in the wall of the gorge, so-called back-doors or side-doors, provide putative alternate routes for traffic of substrate, products, and/or solvent (29,30).
Members of the ChE family differ from each other in turnover number. DmAChE, for example, hydrolyses its natural substrate (turnover number ∼1000 s−1) 50% faster than HuBChE ((31) and references therein). This factor, however, depends on the substrate. For example, the calculated ratio of kcat (DmAChE/HuBChE) for hydrolysis of various substrates at 25°C is 1.8 for acetylthiocholine, 1.4 for propionylthiocholine, and 0.6 for butyrylthiocholine (32,33). Are these activity differences on the millisecond timescale reflected in differences in atomic thermal fluctuations on the pico- to nanosecond timescale?
It has been proposed that binding of an inhibitor can modify ChE molecular dynamics. Tara et al. (9) compared root-mean-square atomic fluctuations of free AChE and of the AChE-huperzine A complex by a 1-ns MD simulation. Although differences between the two simulations were small, their results suggest that binding of the inhibitor results in a decrease in global molecular flexibility. Moreover, irreversible inhibition by the organophosphate, soman, produces a so-called aged conjugate in which a salt bridge between the organophosphate moiety and the active-site histidine contributes to inhibitor stabilization (34). This has been shown to alter the conformational stability of HuBChE (35), and to raise stability against thermal denaturation by 10°C from 63 to 73°C (18). Are the biochemical modifications produced upon binding of soman reflected by changes in molecular dynamics?
In the following, we use incoherent elastic neutron scattering (IENS) to investigate ChE molecular dynamics and to address the above issues experimentally. IENS is a technique sensitive to global hydrogen molecular dynamics on an Ångström-nanosecond scale (depending on the instrumental energy resolution and angular range). In the usual space-time windows of IENS experiments, this represents dynamics of larger atomic groups such as protein side chains (3). At physiological temperatures, atomic mean-square displacements (MSDs) measured by IENS reflect sampling of conformational substates (entropy) as well as vibrational amplitudes. IENS is, therefore, well suited for experimental investigations of thermal fluctuations within biological macromolecules under different biochemical conditions on timescales that match those of MD theoretical calculations.
In a first experiment, we compared MSDs of native HuBChE and soman-inhibited HuBChE over a large temperature range (20 to ∼349 K), passing the denaturation temperature of native HuBChE. In a second experiment, we compared dimeric DmAChE and tetrameric HuBChE. These two enzymes also differ in the extent of their glycosylation; sugar residues account for only 10% of the mass of DmAChE, but for 25% of that of HuBChE.
MATERIALS AND METHODS
Enzyme purification and characterization
HuBChE was purified to homogeneity from outdated human plasma according to an improved protocol based on that of Grunwald et al. (23). The tetrameric enzyme has nine glycosylation sites per monomer, and carbohydrates account for ∼25% of its dry weight. The highly purified HuBChE was stored at 4°C after lyophilization from 10 mM sodium phosphate, pH 8.0. The HuBChE samples were assayed before and after neutron scattering experiments by the Ellman procedure (36), with butyrylthiocholine iodide (1 mM) as the substrate. Activity of the native batch (258 U/mg) was not significantly reduced by the neutron scattering experiments. The activity of the inhibited batch was <2 U/mg. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% acrylamide) in the presence of β-mercaptoethanol (37) revealed a dominant 85-kDa band, assigned to the HuBChE monomer, and a weaker band at ∼170 kDa assigned to residual, nonreducible, covalent HuBChE dimer. Contamination by other proteins was negligible.
Wild-type DmAChE was expressed in the baculovirus system (38). The C-terminal hydrophobic peptide was deleted by insertion of a stop codon to obtain a soluble protein, and the loop 103–136 was replaced by three histidines to facilitate purification. Secreted DmAChE was purified to homogeneity using the following steps: 70% ammonium sulfate precipitation, dialysis, affinity chromatographies on a procainamide-Sepharose column and on nitrilotriacetic acid-nickel agarose (39). The overexpressed dimeric enzyme has four glycosylation sites per monomer, with carbohydrates accounting for ∼10% of the dry weight. Protein concentration was determined from the absorption at 280 nm and enzymatic activity was tested before and after neutron scattering experiments (36) at 25°C in a 25-mM phosphate buffer (pH 7.0) with 1-mM acetylthiocholine iodide as substrate. A 45% decrease in activity was recorded after the neutron scattering experiment. Circular dichroism experiments were carried out on the DmAChE sample before and after the neutron scattering experiment, to see if any conformational change had occurred. The optical pathlength was 1 mm and the protein concentration (determined from the absorption at 280 nm) varied from 0.08 to 0.25 mg/ml. No significant difference in the circular dichroism spectrum was seen in either the far UV (190–230 nm) or the near UV (260–300 nm) before and after the neutron scattering measurement.
Sample preparation for neutron scattering
A batch of ∼250 mg of HuBChE was dialyzed against 25 mM ammonium acetate, pH 7.0. Since this buffer is completely volatile, lyophilization resulted in salt-free protein powder. Approximately half of the HuBChE was inhibited with soman, as described by Masson and Goasdoue (35), before dialysis and lyophilization. Both lyophilized powders were placed in aluminum sample holders (30 × 40 × 0.2 mm3), dried for two days at atmospheric pressure over silica gel, and weighed. The measured weights were taken as their dry weights (h = 0 g water/g dry powder, denoted by g/g in the following). For the neutron scattering experiments in which native and soman-inhibited HuBChE were compared, hydration of the dry samples was carried out by D2O vapor exchange during 2–3 days over pure water (= 100% relative humidity) at 25°C. The final water content was measured by weighing shortly before closing the sample holders and was 0.45 g/g for both samples. The samples are denoted HuBChE-D2O and Soman/HuBChE-D2O, respectively.
A sample of ∼200 mg DmAChE was dialyzed against, and then lyophilized from 0.1 M ammonium acetate, pH 7.0. After lyophilization, the powder was dried over silica gel for two days and hydrated by H2O vapor exchange for two days over a saturated KNO3 salt solution (93% relative humidity) at 25°C. Its water content (0.48 g/g) was measured by weighing shortly before closing the sample holder. In the following, this sample is denoted as DmAChE-H2O. It was compared to a native HuBChE sample, denoted HuBChE-H2O (0.44 g/g), prepared as described above, but using H2O as the hydration medium rather than D2O.
To verify that no loss of material had occurred, all samples were weighed before and after the neutron scattering experiments. No losses were detected for any sample.
Incoherent elastic neutron scattering
Considering only incoherent scattering from the sample hydrogen atoms, the theoretical scattering law can be written in the form
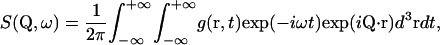 |
(1) |
with the intermediate scattering function
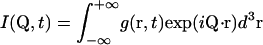 |
(2a) |
and the hydrogen atom autocorrelation function
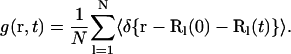 |
(2b) |
Q is the wave vector transfer in a scattering event, i.e., the neutron momentum exchange in units of h bar. For confined atomic motions with no preferential orientation, exhibiting no time-dependence on the timescale of the instrument (40), and complying with the condition
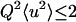 |
(3) |
(where 〈u2〉 are the MSDs), the elastic intensity S(Q,0) can be approximated by
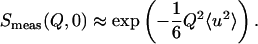 |
(4) |
This is the so-called Gaussian approximation, which is in mathematical analogy to the Guinier approximation (41) in small-angle scattering (SAS). The MSDs 〈u2〉, defined by Eq. 4, correspond to twice the radius of gyration Rg in SAS and represent the full amplitudes of the thermal fluctuations (3).
Instrumental aspects and data analysis
Neutron scattering experiments were carried out on the backscattering spectrometer IN16 (energy resolution ΔE = 0.9 μeV, corresponding to a time window of ∼730 ps; accessible Q-range: 0.19–1.93 Å−1) at the Institute Laue Langevin, Grenoble, France.
Transmission values varied from 0.9 to 0.95, according to the sample. The scattered signals were corrected for container scattering, absorption, and instrumental resolution (determined by separate runs of the empty container and of a vanadium sample) by a standard FORTRAN 77 program Iq0 based on the correction formula of Paalman-Pings coefficients (42). Due to high transmission values, data were not corrected for multiple scattering.
To assure the absence of (salt or water) crystalline structures in the samples, we used the IN16 option to record a diffraction pattern in parallel to the elastic spectra (43). For all samples and all measured temperature points, we observed no Bragg peaks corresponding to water or salt crystalline structures.
Determination of atomic mean-square displacements
The elastic intensity (Eq. 4) of each sample at a given temperature T was normalized to its elastic intensity at the lowest temperature (∼20 K) for each detector, and the values so obtained were plotted in a semilogarithmic plot versus Q2. We refer to these plots in the following as Guinier plots, due to their analogy to the Guinier plots in SAS. MSDs were extracted from the slopes of linear regimes.
The plots can be interpreted in analogy to Guinier plots of polydisperse dilute solutions: In SAS experiments, linear regimes can be assigned to populations of distinct radii of gyration Rg, provided Q*Rg < 1 (41). In analogy, if several linear regimes are present in IENS Guinier plots, they can be associated to dynamically different atomic populations with distinct MSDs. Nonlinear Guinier-plots (lnI versus Q2) will arise, in general, from large amplitude motions for which the condition Q*Rg < 1 breaks down in the Q time domain. In the monodisperse non-Gaussian case, the intensity function is expected to vary smoothly; a pronounced kink is more likely to occur in the polydisperse case of motion populations with distinct MSD values (40).
Whereas at low temperatures, intensities decreased linearly over the whole Q-range measured for all samples, a more or less pronounced kink appeared in the Guinier plots above ∼200 K (Fig. 1), permitting separation into two linear Q-ranges. We extracted MSDs only from the smaller Q-range, 0.19 Å−2 < Q2 < 1.13 Å−2. The separation into two Q-ranges and its implication for populations and dynamical heterogeneity has been described elsewhere (44). The criterion (Eq. 3) for the validity of the Gaussian approximation was checked a posteriori: The maximum value found for the product 〈u2〉Q2 was 2.7.
FIGURE 1.

Guinier plot of HuBChE-D2O at 25 and 344 K with error bars. The lines define the linear fits used to extract MSDs.
RESULTS
Mean-square displacements
DmAChE-H2O versus HuBChE-H2O
Fig. 2 shows that the MSDs for the two enzyme samples are identical, within experimental error, over the whole temperature range measured (20–285 K). MSDs increase continuously, but display a dynamical transition at ∼200 K. Linear regimes are hard to identify. In both cases, the rate of increase of the MSDs decreases drastically at the highest measured temperatures (T > 270 K), as observed previously (44). The effect is due to the limitation of the instrumental resolution regarding the measurement of hydration water large-scale motions (40).
FIGURE 2.

MSDs of DmAChE-H2O (open circles) and HuBChE-H2O (solid circles) obtained from linear fits in the Guinier plots. Error bars are shown for one data point for each data set. At the lowest measured temperatures, the errors are ∼20% smaller.
HuBChE-D2O versus Soman/HuBChE-D2O
MSDs for the native and the inhibited enzyme are identical in the temperature range below 334 K (61°C) and increase smoothly (Fig. 3). Above that temperature, the MSDs for HuBChE-D2O are larger than those for Soman/HuBChE-D2O, which remain in a quasiharmonic regime, as shown by their linear dependence on temperature (Fig. 4). These data reveal an increased flexibility of native HuBChE relative to its soman-inhibited counterpart above 334 K (61°C).
FIGURE 3.

MSDs of HuBChE-D2O (open circles) and Soman/HuBChE-D2O (solid circles) obtained from linear fits in the Guinier plots. Error bars are shown for one data point for each data set. At the lowest measured temperatures, the errors are ∼20% smaller.
FIGURE 4.

MSDs of HuBChE-D2O (open circles) and Soman/HuBChE-D2O (solid circles) in the temperature range of 300–349 K (27–76°C). The denaturation midpoint temperatures of native HuBChE 337.5 K (64.5°C) and soman-inhibited HuBChE 347.5 K (74.5°C) in D2O solution (18) are depicted by vertical lines. Error bars are shown for both data sets.
A control comparison of HuBChE-H2O and HuBChE-D2O (not shown) revealed identical MSDs below 260 K, but higher MSDs of the H2O-hydrated sample at higher temperatures. This latter observation has been ascribed elsewhere to contributions to the scattering signal from the hydration water protons, which are more flexible than protein protons in the temperature range above 260 K (44).
DISCUSSION
The effect of covalent inhibition by soman on the molecular dynamics of HuBChE
The structure and stability of the aged organophosphate conjugate of HuBChE produced by inhibition with soman are known to be modified compared to native HuBChE. Studies involving thermal, urea or pressure denaturation all show that the conformational stability of the enzyme is increased in the presence of soman (18,35,45). The inhibited enzyme has a denaturation midpoint temperature in D2O-solution which is shifted by 10°C from 337.5 K (64.5°C) to 347.5 K (74.5°C) with respect to its native counterpart. MD simulations of the binding of both fasciculin (46) and huperzine A (9) to AChE suggest that enzyme molecular dynamics are modified upon binding of inhibitor. How can our results, i.e., no significant differences in global molecular dynamics on an Ångström-nanosecond scale between native and soman-inhibited HuBChE below 334 K (61°C), but increased flexibility of the native sample with respect to its inhibited counterpart above that temperature—be reconciled with the cited studies?
Results below 61°C
Identical MSDs of two samples over a temperature range as extended as 20–334 K means that the atoms of both samples are placed in common mean free-energy surfaces (47). Since the dealkylated soman moiety bound to the HuBChE monomer subunit contributes only three hydrogen atoms (P-CH3) to the enzyme-inhibitor conjugate, while the deglycosylated native enzyme monomer itself accounts for >3500 nonexchangeable hydrogen atoms, the contribution of the inhibitor is negligibly small. Thus, in both cases the measured signal represents HuBChE molecular dynamics. Consequently, our experimental data are at variance with the MD simulations of Tara et al. (9), mentioned above, who found that binding of huperzine A to AChE resulted in decreased MSDs. The binding of soman to HuBChE via a single covalent bond (plus a salt bridge between a negatively charged oxygen on the phosphoryl moiety, and the protonated imidazole ring of His-438) may explain why it does not noticeably affect global HuBChE molecular dynamics. However, Tara et al. (9) recorded MSDs for the huperzine A/AChE complex that were also larger than the MSDs for the free enzyme in a number of picosecond simulation periods during a total simulation time of 1 ns. Since IENS averages 730 ps in our experiments over the total instrumental resolution time, it is possible that putative dynamical differences of opposite sign, on shorter timescales of ∼10 ps, might simply have been cancelled out. We do not, therefore, see a fundamental contradiction between our experimental results and the MD simulations.
In this context we wish to stress that, since IENS measures global mean atomic fluctuations (i.e., it weighs contributions from all atoms), it cannot be ruled out a priori that soman-inhibition decreases fluctuations in some residues (e.g., close to the active-site gorge), while at the same time increasing fluctuations in some other part of the protein (see, e.g., (48)), thus compensating for the reduced flexibility adjacent to the binding site. It would, however, be surprising if such a dynamical modification were to give rise to exactly the same global MSDs as those for native HuBChE over a temperature range as large as 300 K.
Low-frequency modes have been shown to be affected by complex formation by MD simulations (49,50), and inelastic neutron scattering experiments (10). Our results, however, can neither confirm nor exclude a modification in collective, low-frequency modes of HuBChE upon soman binding, which would give rise to changes in MSDs below the detection threshold of our technique (∼5%).
MSDs above 61°C
The finding that MSDs of native HuBChE tend to be larger than those of soman-inhibited HuBChE starting ∼3° below the denaturation temperature of native HuBChE (∼64°C) is in line with results from several techniques indicating that denaturation increases atomic flexibility of proteins (17,51,52). For HuBChE, it supports the notion that its denaturation is a concerted process, involving a large number of amino acid residues. Indeed, several lines of evidence indicate that both pressure-induced and thermal denaturation of HuBChE are multistep processes that are initiated by penetration of water into the active-site gorge, leading to protein swelling and, eventually, unfolding (18,53,54).
The stability of a particular configuration is given by the free energy difference between the folded and unfolded states. The unfolded state may be either completely unfolded or a stable intermediate, such as the molten globule. The observation of identical MSDs and identical MSDs changes with temperature (and thus force constants, (55)) for native and soman-inhibited HuBChE below 334 K (61°C) suggest that wells A and B (Fig. 5) are identical for the folded native and soman-inhibited enzymes. Since soman inhibition is known to stabilize the folded state, this implies that the soman-inhibited unfolded state, D (Fig. 5), must be at a higher free energy than the native unfolded state, C (Fig. 5). The stabilization of state C is probably due to an increase of its entropy, related to larger MSDs (in line with neutron scattering experiments on denatured α-amylase, (51)). In soman-aged HuBChE, the organophosphoryl moiety remains covalently attached to the unfolded protein, and the salt-bridge between the protonated imidazole ring of His-438 and the negatively charged phosphoryl oxygens may contribute to the observed shift in the onset of unfolding to a higher temperature compared to the native enzyme. The destabilization of the soman-bound unfolded state is, therefore, likely to arise from a decrease in its entropy, as a consequence of certain configurations no longer being available to the unfolded structure due to the presence of the covalently bound inhibitor.
FIGURE 5.

Schematic free-energy diagram of the folded and unfolded states of native and soman-inhibited HuBChE. The width of each well is a qualitative indication of the root-mean-square fluctuation, √〈u2〉, and of the entropy of the corresponding state, whereas the steepness of its sides is an indication of the effective force constant, k', or resilience.
The native state of HuBChE is also stabilized by the kosmotropic sodium phosphate buffer; its flexibility is reduced and its resilience increased over a large temperature range as established by neutron scattering (44). In this case, however, stabilization of the native state is most probably due to a decrease in free energy of the native state; phosphate, like sulfate, is a salting-out ion that stabilizes folded protein structures and favors aggregation and precipitation. In contrast to the single covalently bound organophosphoryl moiety, the salt ions interact with essentially the entire surface of the enzyme and with the hydration water molecules, thus affecting a much larger number of protein residues. The relationship observed between reduced protein flexibility and increased thermal stability is in line with other experiments on trehalose-coated myoglobin (56) and on lysozyme in glycerol (57,58). We conclude that stabilization, and corresponding effects on the MSDs of HuBChE, by solvent/buffer and by the covalently bound inhibitor involve two different mechanisms. Solvent stabilization by salting-out ions is a consequence of a decrease in free energy of the native state, due to both entropic and enthalpic contributions; in particular, the effect on hydration water is expected to be mainly entropic, whereas the observed decrease in protein MSDs (44) indicates a tighter packing of the apolar residues in the core of the protein as suggested for hydrophobic stabilization by calorimetry experiments (59). Our experiments are consistent with soman-induced stabilization resulting from an elevation of the free energy of the unfolded state, which arises mainly from a decrease in its entropy due to the covalent modification by soman.
The effects of glycosylation, oligomeric state and differences in catalytic activity on molecular dynamics: AChE versus BChE
Glycosylation appears to play a role in prolonging the circulatory lifetime of BChE and AChE (60,61), although it is not required for enzymatic activity. Indeed, engineered HuBChE with a reduced degree of glycosylation displays catalytic properties similar to those of the wild-type enzyme (62). Similarly, there does not appear to be any effect of the extent of glycosylation on the kinetic characteristics of either Drosophila (63) or human (64) AChE. There is, however, evidence that glycosylation may influence the molecular dynamics of some proteins. NMR experiments in solution on RNase B and the proteinase inhibitor PMP-C (65,66) have shown that amide hydrogen exchange rates are correlated with the degree of glycosylation. Although glycosylation does not affect the structure, protein dynamic fluctuations decrease globally, decreasing amide hydrogen exchange rates by up to sixfold throughout the entire protein. This decrease in protein fluctuations was ascribed to a reduction in the frequency of breakage of hydrogen bonds due to steric hindrance by the glycan chains. A dependence on the number of the glycosylation sites was found for only a small percentage of the exchangeable amide H-atoms (65), the exchange rate decreasing with increasing glycosylation. Notwithstanding these experimental results, we were not able to detect any differences in global MSDs between DmAChE and HuBChE, despite the former having a much lower degree of glycosylation than the latter. The fact that the HuBChE preparation is a tetramer, whereas the DmAChE preparation is a dimer, does not find expression in a difference in their global MSDs. This is in agreement with MD simulation data: Modifications in the dynamics of large protein complexes relative to their component subunit are dominated by low-frequency collective modes which take place on the 10-ps range upwards (2,49), and may be invisible in the experimental space-time window chosen. Modifications induced upon oligomerization in high-frequency, small amplitude/localized modes, which do lie within the sensitivity of our experimental window, might be rather small.
It is widely accepted that pico- to nanosecond thermal fluctuations serve as a lubricant for functionally relevant motions occurring on a micro- to millisecond timescale (6). It is interesting that no difference in thermal fluctuations between HuBChE and DmAChE is observed, despite their different catalytic turnover rates. Our results suggest, therefore, that dynamically identical thermal fluctuations are the lubricant for functional motions involved in catalytic rates that differ by a factor of ∼2.
Thus, the relationship between thermal atomic fluctuations and functionally important motions is not straightforward. A more likely relationship may be of a kinetic nature, comprising barrier-crossing and activation processes that are of a statistical nature.
CONCLUSIONS
A series of incoherent elastic neutron scattering experiments were carried out on native and soman-inhibited human butyrylcholinesterase, as well as on Drosophila melanogaster acetylcholinesterase, to investigate the influence of irreversible chemical modifications (active-site phosphorylation) and structural differences (glycosylation, oligomeric state) on protein molecular dynamics. Mean-square displacements were extracted within the Gaussian approximation, and compared in a temperature range of 20–349 K.
The influence of inhibitor-binding on global enzyme molecular dynamics
Although several studies using different techniques have shown a modification of global protein molecular dynamics on an Ångström-nanosecond scale upon inhibitor-binding, we were not able to detect any significant differences between the native and the inhibited HuBChE, at comparable degrees of hydration, up to ∼334 K (61°C). We conclude that the modifications of HuBChE molecular dynamics on an Ångström-nanosecond scale upon covalent binding of the small inhibitor soman are, if present, either local (e.g., being limited to a small number of amino acid residues in the active-site pocket) or collective (e.g., changes in low-frequency modes leading to variations in MSDs below the sensitivity of our technique). However, above the denaturation temperature of the native enzyme, 337.5 K (64.5°C), mean-square displacements of native HuBChE were larger than those of its inhibited counterpart, which denatures only at 347.5 K (74.5°C). We conclude that HuBChE stabilization by covalent modification with soman is due to an increase in the free energy of the unfolded state. This is in contrast with stabilization by kosmotropic buffers, which decrease the free energy of the native state.
The influence of small differences in primary and quaternary structure and in degree of glycosylation
Differences in primary and quaternary structure, degree of glycosylation, and oligomeric state, between DmAChE and HuBChE, although they affect various structural and functional parameters, are not reflected in changes in global dynamics on an Ångström-nanosecond scale in the temperature range investigated. We conclude that functional differences arising from these structural differences are either of a local nature (e.g., limited to the active site), concern low-frequency collective modes below the sensitivity of the technique used and/or present only on a longer timescale (>1 ns), and not directly related to fast thermal fluctuations.
Acknowledgments
The authors thank Jacques Colletier for useful discussions on cholinesterase biochemistry as well as the IN16 instrumental team (Bernhard Frick, Thilo Seydel, and Matthias Elender) and the IN13-CRG team (Marc Bée, Francesca Natali, and Sebastien Vial) at the Institute Laue-Langevin, Grenoble, France.
I.S. was a visiting professor at the Université Joseph-Fourier in Grenoble, France.
The study was supported by the European Union program under contracts No. HPRI-CT-2001-50035 and RII3-CT-2003-505925.
F. Gabel's present address is European Molecular Biology Laboratory, Heidelberg, Germany.
References
- 1.Creighton, T. 1993. Proteins: Structures and Molecular Properties. W.H. Freeman and Company, New York.
- 2.McCammon, J. A., and S. C. Harvey. 1987. Dynamics of Proteins and Nucleic Acids. Cambridge University Press, Cambridge, UK.
- 3.Smith, J. C. 1991. Protein dynamics: comparison of simulations with inelastic neutron scattering experiments. Q. Rev. Biophys. 24:227–291. [DOI] [PubMed] [Google Scholar]
- 4.Gabel, F., D. Bicout, U. Lehnert, M. Tehei, M. Weik, and G. Zaccai. 2002. Protein dynamics studied by neutron scattering. Q. Rev. Biophys. 35:327–367. [DOI] [PubMed] [Google Scholar]
- 5.Palmer, A. G. 2004. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 104:3623–3640. [DOI] [PubMed] [Google Scholar]
- 6.Brooks III, C. L., M. Karplus, and B. M. Pettitt. 1988. Proteins: a theoretical perspective of dynamics, structure, and thermodynamics. In Advances in Chemical Physics, Vol. LXXI. I. Prigogine and S.A. Rice, editors. John Wiley and Sons, New York. 94–95.
- 7.Lehnert, U., V. Reat, M. Weik, G. Zaccai, and C. Pfister. 1998. Thermal motions in bacteriorhodopsin at different hydration levels studied by neutron scattering: correlation with kinetics and light-induced conformational changes. Biophys. J. 75:1945–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daniel, R. M., R. V. Dunn, J. L. Finney, and J. C. Smith. 2003. The role of dynamics in enzyme activity. Annu. Rev. Biophys. Biomol. Struct. 32:69–92. [DOI] [PubMed] [Google Scholar]
- 9.Tara, S., T. P. Straatsma, and J. A. McCammon. 1999. Mouse acetylcholinesterase unliganded and in complex with huperzine A: a comparison of molecular dynamics simulations. Biopolymers. 50:35–43. [DOI] [PubMed] [Google Scholar]
- 10.Balog, E., T. Becker, M. Oettl, R. Lechner, R. Daniel, J. Finney, and J. C. Smith. 2004. Direct determination of vibrational density of states change on ligand binding to a protein. Phys. Rev. Lett. 93:028103. [DOI] [PubMed] [Google Scholar]
- 11.Frauenfelder, H., and G. A. Petsko. 1980. Structural dynamics of liganded myoglobin. Biophys. J. 32:465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.James, M. N., A. R. Sielecki, G. D. Brayer, L. T. Delbaere, and C. A. Bauer. 1980. Structures of product and inhibitor complexes of Streptomyces griseus protease A at 1.8 Å resolution. A model for serine protease catalysis. J. Mol. Biol. 144:43–88. [DOI] [PubMed] [Google Scholar]
- 13.Tilton, R. F. J., I. D. Kuntz, Jr., and G. A. Petsko. 1984. Cavities in proteins: structure of a metmyoglobin-xenon complex solved to 1.9 Å. Biochemistry. 23:2849–2857. [DOI] [PubMed] [Google Scholar]
- 14.Joti, Y., M. Nakasako, A. Kidera, and N. Go. 2002. Nonlinear temperature dependence of the crystal structure of lysozyme: correlation between coordinate shifts and thermal factors. Acta Crystallogr. D Biol. Crystallogr. 58:1421–1432. [DOI] [PubMed] [Google Scholar]
- 15.Idiyatullin, D., I. Nesmelova, V. A. Daragan, and K. H. Mayo. 2003. Heat capacities and a snapshot of the energy landscape in protein GB1 from the pre-denaturation temperature dependence of backbone NH nanosecond fluctuations. J. Mol. Biol. 325:149–162. [DOI] [PubMed] [Google Scholar]
- 16.Brooks III, C. L. 1998. Simulations of protein folding and unfolding. Curr. Opin. Struct. Biol. 8:222–226. [DOI] [PubMed] [Google Scholar]
- 17.Bu, Z., J. Cook, and D. J. Callaway. 2001. Dynamic regimes and correlated structural dynamics in native and denatured α-lactalbumin. J. Mol. Biol. 28:865–873. [DOI] [PubMed] [Google Scholar]
- 18.Masson, P., C. Cléry, P. Guerra, A. Redslob, C. Albaret, and P. L. Fortier. 1999. Hydration change during the aging of phosphorylated human butyrylcholinesterase: importance of residues aspartate-70 and glutamate-197 in the water network as probed by hydrostatic and osmotic pressures. Biochem. J. 343:361–369. [PMC free article] [PubMed] [Google Scholar]
- 19.Ollis, D. L., E. Cheah, M. Cygler, B. Dijkstra, F. Frolow, S. M. Franken, M. Harel, S. J. Remington, I. Silman, and J. Schrag. 1992. The α/β hydrolase fold. Protein Eng. 5:197–211. [DOI] [PubMed] [Google Scholar]
- 20.Silman, I., and J. Sussman. 2005. Acetylcholinesterase: “classical” and “non-classical” functions and pharmacology. Curr. Opin. Pharm. 5:293–302. [DOI] [PubMed] [Google Scholar]
- 21.Nicolet, Y., O. Lockridge, P. Masson, J. C. Fontecilla-Camps, and F. Nachon. 2003. Crystal structure of human butyryl cholinesterase and of its complexes with substrate and products. J. Biol. Chem. 278:41141–41147. [DOI] [PubMed] [Google Scholar]
- 22.Darvesh, S., D. A. Hopkins, and C. Geula. 2003. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 4:131–138. [DOI] [PubMed] [Google Scholar]
- 23.Grunwald, J., D. Marcus, Y. Papier, L. Raveh, Z. Pittel, and Y. Ashani. 1997. Large-scale purification and long-term stability of human butyrylcholinesterase: a potential bioscavenger drug. J. Biochem. Biophys. Meth. 34:123–135. [DOI] [PubMed] [Google Scholar]
- 24.Ashani, Y., and S. Pistinner. 2004. Estimation of the upper limit of human butyrylcholinesterase dose required for protection against organophosphates toxicity: a mathematically based toxicokinetic model. Toxicol. Sci. 77:358–367. [DOI] [PubMed] [Google Scholar]
- 25.Quinn, D. M. 1987. Acetylcholinesterase: enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 87:955–979. [Google Scholar]
- 26.Sussman, J., M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker, and I. Silman. 1991. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 253:872–879. [DOI] [PubMed] [Google Scholar]
- 27.Shen, T., K. Tai, R. H. Henchman, and J. A. McCammon. 2002. Molecular dynamics of acetylcholinesterase. Acc. Chem. Res. 35:332–340. [DOI] [PubMed] [Google Scholar]
- 28.Tai, K., T. Shen, U. Borjesson, M. Philippopoulos, and J. A. McCammon. 2001. Analysis of a 10-ns molecular dynamics simulation of mouse acetylcholinesterase. Biophys. J. 81:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilson, M. K., T. P. Straatsma, J. A. McCammon, D. R. Ripoll, C. H. Faerman, P. H. Axelsen, I. Silman, and J. L. Sussman. 1994. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science. 263:1276–1278. [DOI] [PubMed] [Google Scholar]
- 30.Wlodek, S. T., T. W. Clark, L. R. Scott, and J. A. McCammon. 1997. Molecular dynamics of acetylcholinesterase dimer complexed with tacrine. J. Am. Chem. Soc. 119:9513–9522. [Google Scholar]
- 31.Gnagey, A. L., M. Forte, and T. L. Rosenberry. 1987. Isolation and characterization of acetylcholinesterase from Drosophila. J. Biol. Chem. 262:13290–13298. [PubMed] [Google Scholar]
- 32.Masson, P., S. Adkins, P. Gouet, and O. Lockridge. 1993. Recombinant human butyrylcholinesterase G390, the fluoride-2 variant, expressed in Chinese hamster ovary cells, is a low affinity variant. J. Biol. Chem. 268:14329–14341. [PubMed] [Google Scholar]
- 33.Marcel, V., L. Gagnoux-Palacios, C. Pertuy, P. Masson, and D. Fournier. 1998. Two invertebrate acetylcholinesterases show activation followed by inhibition with substrate concentration. Biochem. J. 329:329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Millard, C. B., G. Kryger, A. Ordentlich, H. M. Greenblatt, M. Harel, M. L. Raves, Y. Segall, D. Barak, A. Shafferman, I. Silman, and J. L. Sussman. 1999. Crystal structures of aged phosphorylated acetylcholinesterase: nerve agent reaction products at the atomic level. Biochemistry. 38:7032–7039. [DOI] [PubMed] [Google Scholar]
- 35.Masson, P., and J. Goasdoue. 1986. Evidence that the conformational stability of “aged” organophosphate-inhibited cholinesterase is altered. Biochim. Biophys. Acta. 14:304–313. [DOI] [PubMed] [Google Scholar]
- 36.Ellman, G. L., K. D. Courtney, V. Andres, and R. M. Featherstone. 1961. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7:88–95. [DOI] [PubMed] [Google Scholar]
- 37.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- 38.Chaabihi, H., D. Fournier, Y. Fedon, J. P. Bossy, M. Ravallec, G. Devauchelle, and M. Cerutti. 1994. Biochemical characterization of Drosophila melanogaster acetylcholinesterase expressed by recombinant baculoviruses. Biochem. Biophys. Res. Commun. 203:734–742. [DOI] [PubMed] [Google Scholar]
- 39.Estrada-Mondaca, S., and D. Fournier. 1998. Stabilization of recombinant Drosophila acetylcholinesterase. Protein Exp. Purific. 12:166–172. [DOI] [PubMed] [Google Scholar]
- 40.Gabel, F. 2005. Protein dynamics in solution and powder measured by incoherent elastic neutron scattering: the influence of Q-range and energy resolution. Eur. Biophys. J. 34:1–12. [DOI] [PubMed] [Google Scholar]
- 41.Guinier, A., and G. Fournet. 1955. Small Angle Scattering of X-Rays. John Wiley & Sons, New York, London.
- 42.Bée, M. 1988. Quasielastic Neutron Scattering. Adam Hilger, Bristol and Philadelphia.
- 43.Combet, J., B. Frick, O. Losserand, M. Gamon, and B. Guerard. 2000. Simultaneous diffraction and inelastic scattering on the backscattering instrument IN16. Phys. B Cond. Matt. 283:380–385. [Google Scholar]
- 44.Gabel, F., M. Weik, B. P. Doctor, A. Saxena, D. Fournier, L. Brochier, F. Renault, P. Masson, I. Silman, and G. Zaccai. 2004. The influence of solvent composition on global dynamics of human butyrylcholinesterase powders: a neutron scattering study. Biophys. J. 86:3152–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masson, P., P. Gouet, and C. Cléry. 1994. Pressure and propylene carbonate denaturation of native and “aged” phosphorylated cholinesterase. J. Mol. Biol. 238:466–478. [DOI] [PubMed] [Google Scholar]
- 46.Tai, K., T. Shen, R. H. Henchman, Y. Bourne, P. Marchot, and J. A. McCammon. 2002. Mechanism of acetylcholinesterase inhibition by fasciculin: a 5-ns molecular dynamics simulation. J. Am. Chem. Soc. 124:6153–6161. [DOI] [PubMed] [Google Scholar]
- 47.Frauenfelder, H., F. Parak, and R. D. Young. 1988. Conformational substates in proteins. Annu. Rev. Biophys. Biophys. Chem. 17:451–479. [DOI] [PubMed] [Google Scholar]
- 48.Zidek, L., M. V. Novotny, and M. J. Stone. 1999. Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nat. Struct. Biol. 6:1118–1121. [DOI] [PubMed] [Google Scholar]
- 49.Tidor, B., and M. Karplus. 1994. The contribution of vibrational entropy to molecular association. The dimerization of insulin. J. Mol. Biol. 238:405–414. [DOI] [PubMed] [Google Scholar]
- 50.Fischer, S., J. C. Smith, and C. S. Verma. 2001. Dissecting the vibrational entropy change on protein/ligand binding: burial of a water molecule in bovine pancreatic trypsin inhibitor. J. Phys. Chem. B. 105:8050–8055. [Google Scholar]
- 51.Fitter, J. 2003. A measure of conformational entropy change during thermal protein unfolding using neutron spectroscopy. Biophys. J. 84:3924–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tarek, M., D. A. Neumann, and D. J. Tobias. 2003. Characterisation of sub-nanosecond dynamics of the molten globule state of lactalbumin using neutron scattering and molecular dynamics simulations. Chem. Phys. 292:435–443. [Google Scholar]
- 53.Cléry, C., F. Renault, and P. Masson. 1995. Pressure-induced molten globule state of cholinesterase. FEBS Lett. 370:212–214. [DOI] [PubMed] [Google Scholar]
- 54.Weingard-Ziade, A., F. Ribes, F. Renault, and P. Masson. 2001. Pressure- and heat-induced inactivation of butyrylcholinesterase: evidence for multiple intermediates and the remnant inactivation process. Biochem. J. 356:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaccai, G. 2000. How soft is a protein? A protein dynamics force constant measured by neutron scattering. Science. 288:1604–1607. [DOI] [PubMed] [Google Scholar]
- 56.Cordone, L., M. Ferrand, E. Vitrano, and G. Zaccai. 1999. Harmonic behavior of trehalose-coated carbon-monoxy-myoglobin at high temperature. Biophys. J. 76:1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsai, A. M., D. A. Neumann, and L. N. Bell. 2000. Molecular dynamics of solid-state lysozyme as affected by glycerol and water: a neutron scattering study. Biophys. J. 79:2728–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai, A. M., T. J. Udovic, and D. A. Neumann. 2001. The inverse relationship between protein dynamics and stability. Biophys. J. 81:2339–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Privalov, P. L. 1982. Stability of proteins. Proteins which do not present a single cooperative system. Adv. Protein Chem. 35:1–104. [PubMed] [Google Scholar]
- 60.Saxena, A., L. Raveh, Y. Ashani, and B. P. Doctor. 1997. Structure of glycan moieties responsible for the extended circulatory life time of fetal bovine serum acetylcholinesterase and equine serum butyrylcholinesterase. Biochemistry. 36:7481–7489. [DOI] [PubMed] [Google Scholar]
- 61.Kronman, C., B. Velan, D. Marcus, A. Ordentlich, S. Reuveny, and A. Shafferman. 1995. Involvement of oligomerization, N-glycosylation and sialylation in the clearance of cholinesterases from the circulation. Biochem. J. 311:959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nachon, F., Y. Nicolet, N. Viguié, P. Masson, J. C. Fontecilla-Camps, and O. Lockridge. 2002. Engineering of a monomeric and low-glycosylated form of human butyrylcholinesterase. Eur. J. Biochem. 269:630–637. [DOI] [PubMed] [Google Scholar]
- 63.Mutero, A., and D. Fournier. 1992. Post-translational modifications of Drosophila acetylcholinesterase. In vitro mutagenesis and expression in Xenopus oocytes. J. Biol. Chem. 267:1695–1700. [PubMed] [Google Scholar]
- 64.Velan, B., C. Kronman, A. Ordentlich, Y. Flashner, M. Leitner, S. Cohen, and A. Shafferman. 1993. N-glycosylation of human acetylcholinesterase: effects on activity, stability and biosynthesis. Biochem. J. 296:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudd, P. M., H. C. Joao, E. Coghill, P. Fiten, M. R. Saunders, G. Opdenakker, and R. A. Dwek. 1994. Glycoforms modify the dynamic stability and functional activity of an enzyme. Biochemistry. 33:17–22. [DOI] [PubMed] [Google Scholar]
- 66.Mer, G., H. Hietter, and J. F. Lefevre. 1996. Stabilization of proteins by glycosylation examined by NMR analysis of a fucosylated proteinase inhibitor. Nat. Struct. Biol. 3:45–53. [DOI] [PubMed] [Google Scholar]