

Abstract
The outer hair cell lateral membrane motor, prestin, drives the cell's mechanical response that underpins mammalian cochlear amplification. Little is known about the protein's structure-function relations. Here we provide evidence that prestin is a 10-transmembrane domain protein whose membrane topology differs from that of previous models. We also present evidence that both intracellular termini of prestin are required for normal voltage sensing, with short truncations of either terminal resulting in absent or modified activity despite quantitative findings of normal membrane targeting. Finally, we show with fluorescence resonance energy transfer that prestin-prestin interactions are dependent on an intact N-terminus, suggesting that this terminus is important for homo-oligomerization of prestin. These domains, which we have perturbed, likely contribute to allosteric modulation of prestin via interactions among prestin molecules or possibly between prestin and other proteins, as well.
INTRODUCTION
A key evolutionary step in the development of high-frequency acoustic sensitivity in mammals appears to have been the recruitment of an anion transporter family member, prestin (SLC26A5) (1), to enable amplification of passive basilar membrane motion by outer hair cells (OHCs) (2). When transfected into nonauditory cells, this voltage-dependent, integral membrane protein presents all of the known biophysical attributes expressed by the native OHC lateral membrane motor, including voltage and tension sensitivity (1,3–6). These preserved attributes confirm the identity of prestin as the prime component of the lateral membrane motor. Nevertheless, we and others have noted some differences in the electromechanical activity of transfected prestin compared to that of the native OHC motor, and suggested that subunit interactions might be required for amplification of prestin's activity (6,7).
The molecular conformation or state of prestin, which underpins OHC electromotility and drives cochlear amplification, can be deduced from “gating charge” measures, or measures of the cell's voltage-dependent, nonlinear capacitance (NLC). Using these measures, the control of prestin's voltage-dependent activity has recently been linked to prestin's affinity for anions, in particular chloride ions (9,10). It has been suggested that these ions function either as extrinsic voltage sensors (9), or as allosteric modulators that influence the protein's steady-state energy profile (10,11). In this series of experiments, we extend our concept of allosteric modulation of prestin to include modulation via protein-protein interactions.
METHODS
Preparing mutants and transient transfection
Truncations of the N-terminus were achieved by introducing start codons before the indicated amino acid positions by polymerase chain reaction (PCR) amplification. Similarly, C-terminal truncations were obtained by introducing stop codons after the indicated positions. Yellow (YFP) or cyan (CFP) fluorescent protein fusion proteins were made by introducing the fluorescent proteins at the C-terminus of the prestin. In the case of the C-terminal truncations the truncated protein was fused directly to the N-terminus of YFP without an intervening stop codon. Amplification by PCR was done using Hi-Fidelity DNA polymerase (Roche, Indianapolis, IN). The parameters were 10 cycles of 94°C for 40 s, 55°C for 2 min, and 68°C for 6 min; and 30 cycles of 94°C for 40 s, 55°C for 2 min, and 68°C for 6 min, with 20 s extension for every cycle. A clone of gerbil prestin served as the template. Amplified products were purified, ligated into PCDNA 3.1 topo TA (Invitrogen, Carlsbad, CA) or into eYFPN1 (Clontech, Palo Alto, CA). Each of the clones generated were sequenced to exclude the possibility of PCR-generated errors. Transient cotransfection into Chinese hamster ovary (CHO) cells was achieved with Lipofectamine (Invitrogen) in accordance with the manufacturer's recommendations.
Fluorescence resonance energy transfer
Fluorescence resonance energy transfer (FRET) was determined using a Zeiss 510 with a meta-analyzer. CHO cells transiently transfected with prestin-YFP and prestin-CFP constructs (at a 2:3 ratio, to counter a lower CFP quantum yield and CFP's more diffuse emission spectrum) were analyzed 24–48 h after transfection. Emission from 10 defined regions of interest of each cell, detected by the meta-analyzer (in 10-nm increments) while excited at 458 nm were recorded before and after photobleaching at 514 nm. Photobleaching at 514 nm was done with the 40W argon laser set at maximum for a period of 1 min. FRET efficiency was defined as (Ea − Eb) × 100/Ea, where Ea was the CFP emission (473–494 windows) after photobleaching and Eb the CFP emission before photobleaching, and calculated as previously described (12). In determining FRET efficiency in unbleached controls, CFP emission values obtained 1 min after determining the initial CFP emission were defined as Ea (a period of 1 min was the interval of photobleaching). To minimize CFP photobleaching while determining its emission, the laser intensity was set at 5% while measuring CFP emission. Although there was some minimal CFP bleaching during photobleaching of YFP, the effect was the same across all the sample combinations tested where YFP photobleaching was performed. Thus, the increase in FRET efficiency observed in these bleaching experiments was found despite the slight decrease in CFP fluorescence after YFP bleaching. Thus, we cannot rule out that a relatively slight increase in FRET efficiency was masked in the observed decrease in efficiency found with start 21 prestin-YFP and prestin-CFP (see Fig. 5). Nevertheless, each group would be affected similarly.
FIGURE 5.

FRET demonstrated by acceptor (YFP) photobleaching confirms prestin-prestin interactions and suggests that the N-terminus is important in these interactions. The figure shows a CHO cell transfected with prestin-CFP (A and D) and prestin-YFP (B and E) before (A–C) and after (D–F) photobleaching YFP (514 line of the Argon laser) in the right half of the cell. (C and F) Merged images of A and B, and D and E, respectively. As is evident, there is an increase in CFP emission (D and F) concomitant with a decrease in YFP emission (E and F) in the right half of the cell. (G) The graph shows a quantitative estimate of FRET efficiencies after photobleaching (7). The efficiency of FRET is compared between CHO cells transfected with prestin-CFP together with prestin-YFP without (first bar) and with (second bar) acceptor photobleaching, prestin-CFP together with start 21 prestin-YFP (third bar), and prestin-CFP together with stop 709 prestin-YFP (fourth bar). A normal decrease in FRET efficiency in the absence of YFP photobleaching (−7.39 ± 2.189 (mean ± SE), n = 3 cells) is converted to an increase in FRET efficiency with photobleaching in prestin-CFP and prestin-YFP (4.64 ± 0.63, n = 5), and prestin-CFP and stop 709 prestin-YFP (4.67 ± 1.54, n = 4), confirming interactions between normal prestin molecules, and C-terminally truncated prestin molecules and normal prestin. The absence of an increase in FRET efficiency with start 21 prestin-YFP and prestin-CFP (−1.94 ± 1.57, n = 4 cells) confirms an absence of interaction between the N-terminally truncated prestin molecules and normal prestin. A one-way ANOVA between these groups demonstrated a significant difference between unbleached normal prestin and bleached normal prestin (p < 0.002) or stop 709 prestin (p < 0.002), but no difference between unbleached normal prestin and start 21 prestin. Similarly, there was a significant difference between FRET efficiencies in normal prestin and start 21 prestin (p < 0.05) transfected cells. Also noteworthy was that FRET efficiency at the membrane (6.5 ± 1.5, n = 3) was not significantly different from elsewhere in the cell.
Immunofluorescence staining
Affinity-purified rabbit polyclonal antibodies against two peptides corresponding to extracellular domains of prestin, GGKEFNERFKEKLPAPI (274–290) and KELANKHGYQVDGNQEL (359–375), respectively, were purchased from Genemed Synthesis (San Francisco, CA). Live OHCs or transfected CHO cells were incubated separately with these primary antibodies and then subsequently fixed. Primary antibody was detected using a fluorescent-labeled secondary antibody. Specificity of antibody labeling was established by preadsorbtion of the primary antibody with the corresponding peptide. Cells that were transfected with empty vector and stained in an identical fashion served as negative controls. Specific staining was not observed with either of these procedures.
Fluorescence-activated cell sorting analysis
CHO cells transfected with the different prestin-YFP constructs were labeled with affinity-purified anti-prestin antibodies. Live cells were disassociated and incubated with antibody in phosphate-buffered saline, 1% bovine serum albumin, and 0.5% fetal calf serum at 4°C. The primary antibody was detected using a biotinylated anti-rabbit antibody (Molecular Probes, Eugene, OR) followed by streptavidin conjugated to Alexa 647. The cells were fixed briefly in 1% paraformaldehyde before analysis. Fluorescence-activated cell sorting (FACS) analysis was carried out using FlowJo software. Stained cells were analyzed on a FACSCalibur TM (Becton Dickson Immunocytometry Systems, Franklin Lakes, NJ). Forward and side scatter profiles were used to exclude dead cells. From 30,000 to 50,000 events were collected for analysis. Cells transfected with empty vector stained with the prestin antibodies and prestin-YFP stained with rabbit IgG served as negative controls for the primary and secondary antibodies, respectively.
Nonlinear capacitance
Cells were recorded by whole-cell patch-clamp configuration at room temperature using an Axon 200A amplifier (Axon Instruments, Foster City, CA), as described previously (13,14). The bath solution contained (in mM) TEA 20, CsCl 20, CoCl2 2, MgCl2 1.47, Hepes 10, NaCl 99.2, and CaCl2.2H2O 2, pH 7.2, and the pipette solution contained (in mM) CsCl 140, EGTA 10, MgCl2 2, and Hepes 10, pH 7.2. These conditions ensured that prestin-chloride interactions were maximal (9,10). Osmolarity was adjusted to 300 ± 2 mOsm with dextrose. Command delivery and data collections were carried out with a Windows-based whole-cell voltage-clamp program, jClamp (Scisoft, Ridgefield, CT), using an NI PCI 6052E interface (National Instruments, Austin, TX).
Capacitance was evaluated with a continuous high-resolution 2-sine wave technique fully described elsewhere (14,15). Capacitance data were fitted to the first derivative of a two-state Boltzmann function (13):
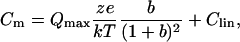 |
(1) |
where
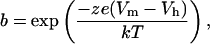 |
(2) |
Qmax is the maximum nonlinear charge transfer, Vh is the voltage at peak capacitance or half-maximal nonlinear charge transfer, Vm is membrane potential, Clin is linear capacitance, z is valence, e is electron charge, k is Boltzmann's constant, and T is absolute temperature. Gating charge currents were obtained with a standard P/−5 procedure (13).
RESULTS
CHO cells transfected with the gene for normal prestin show large gating currents and NLC, similar to that in OHCs (see Fig. 2 B). This signature of prestin's electromechanical activity was quantified through estimates of Vh (the voltage at half-maximal motor charge transfer), z (the charge valance), and Qmax (total charge moved) that were derived from fits with two-state Boltzmann functions (13). With our intracellular and extracellular solutions, average normal values for Vh, z, and Qmax (mean ± SE) were −102.4 ± 1.8 mV, 0.67 ± 0.02, and 130 ± 20 fC, respectively (n = 30). Nonlinear charge density (coulombs per pF of linear capacitance) averaged 4.8 ± 0.6 fC/pF. Additionally, we were able to measure NLC in prestin that had been fused with YFP at its C-terminus (n = 20). This fusion construct of prestin provides electrical properties statistically indistinguishable from normal prestin; fusion constructs of mutant prestin that we made (see below) similarly remained unaffected by the tag. The assessment of this YFP fusion construct permitted not only quantitative electrical evaluations, but both qualitative and quantitative evaluations of membrane targeting via confocal microscopy and FACS, respectively.
FIGURE 2.

Confocal images show CHO cells transfected with prestin-YFP constructs into which the HA epitope was inserted into position 168 (A–F) and position 371 (G–I). The HA tags were detected with a mouse anti-HA epitope and Alexa 647-conjugated antimouse antibody (A, D, and G). (B, E, and H). YFP images. (C, F, and I) Merged images. Cells in A–C and G–I were stained live, and cells in D-F were fixed and permeabilized with detergent. As is evident, the detection of the HA epitope at position 168 required that the cells be permeabilized, whereas that at position 371 was detectable in live cells without permeabilization. These results suggest that position 168 of prestin in these constructs lies on the intracellular surface, whereas position 371 lies on the extracellular surface. (J) Substitution of the two potential N-glycosylation asparagine residues with glutamine residues does not result in a change in molecular weight, indicating that prestin is not glycosylated at these two residues. CHO cells were transfected with constructs of prestin fused to poly-His V5 epitope tag at its C-terminus (lane A) and prestin N163Q + N166Q double mutant fused to a poly-His V5 epitope tag at its C-terminus (lane B). Normal and mutated prestin were purified from lysed CHO cells on a Ni column and separated by PAGE (8%). The gel was blotted on to polyvinylidene difluoride and probed with an anti-V5 antibody/horseradish peroxidase-conjugated secondary antibody followed by enhanced chemiluminescence detection. As is evident, prestin migrates at its predicted molecular weight of 80 kD, as does the N163Q + N166Q double mutant.
To better understand structure-function relationships of prestin, we sought to refine our working knowledge of prestin topology (Fig. 1). The current membrane topology model of prestin posits 12 transmembrane (TM) domains (9,16). This model is, in part, based on placing two potential N glycosylation sites on the extracellular surface of the protein (17). In our modeling of prestin's TM regions, we abandoned this constraint based on several pieces of information. First, potential N glycosylation sites are common and often nonspecific, being present even in frank intracellular proteins, including two potential N glycosylation sites that are within prestin's intracellular C-terminus (603 and 736). Second, in contradistinction to the results of Matsuda et al. (17), we find no significant effects of glycosylation site mutations (N163Q + N166Q) or glycosidase F treatment on the voltage dependence of NLC in transfected CHO cells (control (n = 28)/mutant (n = 10)/enzyme (n = 13): Vh = −100.2 ± 1.76/−95.9 ± 3.92/−100.1 ± 3.61; t-tests, p = 0.26/0.98). Furthermore, the molecular mass of both prestin and doubly mutated prestin (N163Q and N166Q) were indistinguishable at 80 kD (Fig. 2 J), this also being in contradistinction to the results of Matsuda et al. (17). In the absence of that constraint, we are left with the observation, supported by epitope tagging and/or antibody staining of live and permeabilized cells, that the N- and C-termini are likely intracellular (9,16), results that we too have since replicated. Consequently, the molecule has an even number of TM domains, and based on an analysis of prestin's structure with a number of topology prediction programs (TMPred, DAS, TMAP, TMHMM, and Predict Protein), we modeled prestin to contain 10 TM domains (Fig. 1 A). Our 10-TM model has a membrane topology the reverse of that previously proposed by Deak et al. (18); that is, segments on the intracellular surface in our model would lie on the extracellular surface of the cell in their model. We tested the model by immunostaining live outer hair cells and prestin-transfected CHO cells using antibodies raised to two peptides that are predicted, based on this model, to lie on the extracellular surface of the cell. The two peptides GGKEFNERFKEKLPAPI (274–290) and KELANKHGYQVDGNQEL (359–375) lie between TM domains 5 and 6, and 7 and 8 in our model (they would lie on the intracellular surface of the protein in the existing topology models (9,16,18)). We found live staining and fixed, nonpermeabilized staining of these cells with these two antibodies, confirming that the two peptides lie on the extracellular surface (Fig. 1 B). We also hemagglutinin (HA)-tagged prestin at residue 168, which proved functional, and were not able to find live staining by immunocytochemistry or FACS, but were able to immunocytochemically observe the tag after permeabilization (Fig. 2, A–F). HA tagging of residue 371 did not produce functional prestin (no NLC) and rarely showed membrane targeting; however, in those few rare cells live staining was observed (Fig. 2, G–I). We were unable to produce HA tagging of residue 281.
FIGURE 1.

(A) Model of prestin's transmembrane topology that posits 10 transmembrane domains with intracellular amino and carboxy termini of the protein. Shown are the positions (red circles) of individual truncations of the protein that we tested. A cartoon of the crystal structure of the C-terminus of prestin modeled on the bacterial protein SpoIIAA, to which it shows strong homology, is shown. The positions of two peptides that would lie on the extracellular surface of the protein between transmembrane domains 4 and 5 (peptide A) and 6 and 7 (peptide B) are indicated. (B) Affinity-purified antibodies against these peptides (ApA and ApB) were used to stain both live guinea pig OHC (upper panel of confocal images) and live CHO cells transfected with prestin (lower panel). These results, together with other data (see text), confirm that these antigens lie on the extracellular surface, and support the 10-transmembrane model.
Our 10-TM model of prestin indicates that the amino and carboxy termini before residue 94 and after residue 498 reside intracellularly, that is, outside the lipid bilayer and beyond where a functional voltage sensor should reside. We used site-directed C-terminal and N-terminal truncations to evaluate the contribution of these intracellular moieties to prestin 's voltage-dependent activity, as we did above with normal prestin. Successive truncations of the C- and N-terminus resulted in a graded decrease and eventual elimination of nonlinear charge movement (Fig. 2). In truncations that remained functional, voltage sensitivity (z) did not vary far from control values, indicating that the truncated protein's voltage sensor remained relatively unaltered. On the other hand, Vh varied among truncations, especially that of the N-terminal, and likely reflects structural influences on the steady-state energy profile that controls the voltage range over which the motors operate. Additionally, since the total charge movement Qmax decreased, with z remaining normal, it appears that the number of functional proteins decreased. Stops N-terminal to residue 712 and starts C-terminal to residue 20 were functionally lethal, indicating catastrophic alterations in protein structure and/or absent or abnormal interactions with other potential intracellular subunits.
To rule out absent surface expression of these lethal truncations as a cause of a loss in NLC we confirmed proper targeting to the plasma membrane by live antibody staining of mutated prestin molecules in transfected cells. Similar patterns of antibody staining appear in both control and nonfunctional mutations. Additionally, several of the truncated mutations were evaluated with a YFP fusion construct. In each case, the results were the same as the ones we obtained with green fluorescent protein coexpression experiments, and each successfully targeted the plasma membrane (Fig. 3, E–G). The only exception was the full C-terminus truncation (stop 498), which was nonfunctional and confined intracellularly (Fig. 3 H). Full truncations of the C-terminus are not expected to properly target the membrane (D. Oliver, University of Freiburg, and J. Zheng, Northwestern University, personal communications, AOR meeting, 2004). Finally, to allay our concerns over the poor statistical power of confocal microscopy, we used FACS in large populations of cells to confirm that membrane targeting was normal (Fig. 4). In these studies, the correspondence of fluorescence intensities in controls and mutants indicates that equal numbers of prestin molecules reach the plasma membrane in normal and mutant transfections. Thus, the proper targeting of functional and nonfunctional truncated mutations indicates that there is a decrease in the number of functional motors, not a decrease in the number of prestin molecules residing within the plasma membrane.
FIGURE 3.

Serial truncations of the amino and carboxy termini of prestin result in a loss of NLC. (A) Nonlinear capacitance as a function of C-terminal truncations. A stop codon at 712 abolishes NLC. (B) Examples of gating currents induced by a voltage step showing a decrease in magnitude corresponding to reductions in NLC. (C) Nonlinear capacitance as a function of N-terminal truncations. A start codon at 21 abolishes NLC. (E–G) Confocal images of CHO cells transfected with prestin-YFP, start 21 prestin-YFP, and stop 709 prestin-YFP, respectively, showing plasma membrane targeting of these constructs. (H) A confocal image of a CHO cell transfected with stop 498 prestin-YFP that does not target the membrane (and corroborates work by other workers in the field; Jing Zheng, Northwestern University, and Dominik Oliver, University of Freiburg, personal communication, AOR meeting, 2004). These results confirm that the loss in NLC with these truncations is not due to absent targeting to the surface membrane.
FIGURE 4.

Quantification of prestin surface expression in transfected CHO cells by flow cytometry. CHO cells transfected with prestin-YFP fusion constructs (normal prestin, start 21, and stop 709) were disassociated and stained live with two affinity-purified rabbit polyclonal antibodies to two peptides expressed on the extracellular surface of the protein (upper panels: antibody to peptide A = GGKEFNERFKEKLPAPI (aa 274–290); middle panels: antibody to peptide B = KELANKHGYQVDGNQEL (aa 359–375)). The primary antibody was detected using a biotinylated anti-rabbit antibody (Molecular Probes) and streptavidin conjugated to Alexa 647. All antibody staining was done in phosphate-buffered saline and 0.5% bovine serum albumin at 4°C. The cells were then fixed in 4% paraformaldehyde before flow cytometric analysis. Shown are contour plots with outliers of YFP intensity (Phycoerythrin window) on the x axis and prestin intensity (Alexa 647) on the y axis of live stained cells (identified by forward and side scatter). As is evident, a subpopulation of YFP positive cells, recognized by both antibodies, is present in those cells transfected with the three prestin-YFP fusion constructs (normal prestin, start 21, and stop 709) and not in those cells transfected with empty vector alone stained with anti-prestin antibodies to peptides A and B, unstained prestin-YFP transfected cells, or prestin-YFP transfected cells stained with rabbit IgG (lower panels). Both the intensity of staining and the percentage of cells within the boxed area recognized by the antibodies are similar in cells transfected with full-length prestin (4.4% and 4.3% for anti-peptides A and B, respectively), start 21 (2.6% and 3.9%), and stop 709 (3.2% and 3.3%). The three control groups contained an insignificant number of cells labeled with the antibodies. Differences in number of cells indicate variability in expression efficiency among experiments hovering around 3–6%. These efficiencies are obtained by other workers in the field (Dominik Oliver, personal communication). These quantitative analyses are evidence that the absence in NLC in these two truncations is not due to aberrant surface expression.
One possible mechanism for the reduction of functional motors is an interference with obligatory homomeric interactions among prestin molecules. To evaluate the possibility that prestin may function as multimers, we investigated whether individual prestin molecules could closely interact with each other using FRET. Fig. 5 shows that indeed close interactions (<100 Å) occur between fluorescently tagged neighboring C-terminals of prestin; additionally, controls with the fluorescently tagged Slo K channel do not show interactions with prestin. Restricting the area of interest to the plasma membrane produced the same FRET response, as larger cellular areas. Consistent with this data, chemical cross-linking of purified poly-his tagged prestin yielded a minimal molecular mass of 160 kD, suggesting that prestin formed dimers. To determine whether the intracellular termini were required for these interactions, we used FRET to identify homomultimerization of two prestin truncations, start 21 prestin-YFP and stop 709 prestin-YFP, each of which was physiologically nonfunctional, but targeted the membrane successfully. In contrast to stop 709 prestin-YFP, which showed FRET when cotransfected with normal prestin-CFP, start 21 prestin-YFP did not show FRET when cotransfected with normal prestin-CFP. The differences in FRET efficiencies obtained before and after acceptor photobleaching were statistically significant (see Fig. 5), indicating that the N-terminus of prestin is important for the formation of multimers, and the loss in NLC is possibly a result of an inability to form multimers. In contrast, since the C-terminal truncation that resulted in a loss of NLC was still able to form homomultimers, we speculate that the C-terminus interacts with other proteins that are important for generating NLC.
DISCUSSION
The discovery of prestin (19) identified a molecule that likely is the foundation of the mammalian auditory system's nonlinear amplificatory mechanism (20). How this protein works is key to understanding the remarkable ability of OHCs to respond mechanically to electrical stimuli, discovered some 20 years ago (21). Our observations on the functional consequences of short terminal truncations indicate that this protein does not work alone, but requires interacting partners to drive OHC motor activity. Though some molecular manipulations can interfere with proper membrane targeting (for example, full C-terminal truncations of prestin were not delivered to the plasma membrane), we combined techniques with low and high statistical power, namely confocal microscopy and FACS, respectively, to unambiguously confirm uncompromised membrane residence.
In previous work we demonstrated that intracellular chloride altered the operating voltage range of prestin, shifting Vh to more negative voltages (10). This observation was incongruent with a simple extrinsic voltage sensor scheme for anions previously proposed (9). Additionally, we showed that the valence of substituted anion did not, as expected, alter the valence of charge movement (11), further supporting our conclusion that anions serve as allosteric modulators rather than voltage sensors. However, although allosteric effects can be mediated by ion interactions with proteins, e.g., chloride with Cl channels (22) or Ca with K channels (23), conformational alterations can additionally arise from protein-protein/peptide/domain interactions (24). Thus, our present data, which show a requirement for homomeric interactions among prestin's N-termini for normal charge movement, indicate that motor behavior is also allosterically controlled by these interactions. Indeed, we have previously interpreted the motors' sensitivity to prior voltage conditions as resulting from prestin-prestin interactions within the bilayer (6,14). Thus, we argued that prestin interactions may underlie the shift in the steady-state energy profile that results from prepulse perturbations, possibly through viscoelastic relaxations of prestin-induced membrane tension. Clearly, our new data open the possibility that these voltage-dependent viscoelastic effects could arise from interactions between homomeric components of prestin motor complexes, as well as among the complexes.
The topology of SLC26 transporter family members is not well delineated, with divergent models predicted possessing 10–14 TM domains (25). For prestin, a 12-TM domain model had been proposed, but our data do not support this model. Instead, we propose a 10-TM model, with the antibody-identified loops 5 (aa274–290) and 7 (aa359–375) residing extracellularly, as opposed to intracellularly as in previous models. Interestingly, Adler et al. (26), using antibody labeling, also placed region 274–290 on an extracellular loop. Additionally, the original 12-TM model of Zheng et al. (16,17) has recently been modified by that group with a reentrant loop to move a putative cGMP phosphorylation site at residue 238 onto the intracellular aspect of the protein (18)—a location already predicted by our model. Taken together, evidence for our 10-TM model is strong and dramatically changes our view of the landscape over which intracellular constituents can interact with prestin.
Acknowledgments
We thank Peter Dallos and colleagues for the gerbil prestin clone, and Margaret Mazzucco for technical help.
Financial support was provided by the National Institutes of Health's National Institute on Deafness and Other Communication Disorders grants DC 000273 (J.S.S.) and K08 DC05352 (D.N.).
References
- 1.Zheng, J., W. Shen, D. Z. He, K. B. Long, L. D. Madison, and P. Dallos. 2000. Prestin is the motor protein of cochlear outer hair cells. Nature. 405:149–155. [DOI] [PubMed] [Google Scholar]
- 2.Liberman, M. C., J. Gao, D. Z. He, X. Wu, S. Jia, and J. Zuo. 2002. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature. 419:300–304. [DOI] [PubMed] [Google Scholar]
- 3.Ludwig, J., D. Oliver, G. Frank, N. Klocker, A. W. Gummer, and B. Fakler. 2001. Reciprocal electromechanical properties of rat prestin: the motor molecule from rat outer hair cells. Proc. Natl. Acad. Sci. USA. 98:4178–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meltzer, J., and J. Santos-Sacchi. 2001. Temperature dependence of non-linear capacitance in human embryonic kidney cells transfected with prestin, the outer hair cell motor protein. Neurosci. Lett. 313:141–144. [DOI] [PubMed] [Google Scholar]
- 5.Santos-Sacchi, J., and E. Navarrete. 2002. Voltage-dependent changes in specific membrane capacitance caused by prestin, the outer hair cell lateral membrane motor. Pflugers Arch. 444:99–106. [DOI] [PubMed] [Google Scholar]
- 6.Santos-Sacchi, J., W. Shen, J. Zheng, and P. Dallos. 2001. Effects of membrane potential and tension on prestin, the outer hair cell lateral membrane motor protein. J. Physiol. 531:661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong, X. X., and K. H. Iwasa. 2004. Tension sensitivity of prestin: comparison with the membrane motor in outer hair cells. Biophys. J. 86:1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reference deleted in proof.
- 9.Oliver, D., D. Z. He, N. Klocker, J. Ludwig, U. Schulte, S. Waldegger, J. P. Ruppersberg, P. Dallos, and B. Fakler. 2001. Intracellular anions as the voltage sensor of prestin, the outer hair cell motor protein. Science. 292:2340–2343. [DOI] [PubMed] [Google Scholar]
- 10.Rybalchenko, V., and J. Santos-Sacchi. 2003. Cl− flux through a non-selective, stretch-sensitive conductance influences the outer hair cell motor of the guinea-pig. J. Physiol. 547:873–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rybalchenko, V., and J. Santos-Sacchi. (2003). Allosteric modulation of the outer hair cell motor protein prestin by chloride. In Biophysics of the Cochlea: From Molecules to Models. A. Gummer, editor. World Scientific Publishing, Singapore. 116–126.
- 12.Karpova, T. S., C. T. Baumann, L. He, X. Wu, A. Grammer, P. Lipsky, G. L. Hager, and J. G. McNally. 2003. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. J. Microsc. 209:56–70. [DOI] [PubMed] [Google Scholar]
- 13.Santos-Sacchi, J. 1991. Reversible inhibition of voltage-dependent outer hair cell motility and capacitance. J. Neurosci. 11:3096–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santos-Sacchi, J., S. Kakehata, and S. Takahashi. 1998. Effects of membrane potential on the voltage dependence of motility-related charge in outer hair cells of the guinea-pig. J. Physiol. (Lond.). 510:225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos-Sacchi, J. 2004. Determination of cell capacitance using the exact empirical solution of partial differential Y/partial differential Cm and its phase angle. Biophys. J. 87:714–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng, J., K. B. Long, W. Shen, L. D. Madison, and P. Dallos. 2001. Prestin topology: localization of protein epitopes in relation to the plasma membrane. Neuroreport. 12:1929–1935. [DOI] [PubMed] [Google Scholar]
- 17.Matsuda, K., J. Zheng, G. G. Du, N. Klocker, L. D. Madison, and P. Dallos. 2004. N-linked glycosylation sites of the motor protein prestin: effects on membrane targeting and electrophysiological function. J. Neurochem. 89:928–938. [DOI] [PubMed] [Google Scholar]
- 18.Deak, L., J. Zheng, A. Orem, G. G. Du, S. Aguinaga, K. Matsuda, and P. Dallos. 2005. Effects of cyclic nucleotides on the function of prestin. J. Physiol. 563:483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng, J., L. D. Madison, D. Oliver, B. Fakler, and P. Dallos. 2002. Prestin, the motor protein of outer hair cells. Audiol. Neurootol. 7:9–12. [DOI] [PubMed] [Google Scholar]
- 20.Santos-Sacchi, J. 2003. New tunes from Corti's organ: the outer hair cell boogie rules. Curr. Opin. Neurobiol. 13:459–468. [DOI] [PubMed] [Google Scholar]
- 21.Brownell, W. E., C. R. Bader, D. Bertrand, and Y. de Ribaupierre. 1985. Evoked mechanical responses of isolated cochlear outer hair cells. Science. 227:194–196. [DOI] [PubMed] [Google Scholar]
- 22.Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, J. P., Jr., J. R. Balser, and P. B. Bennett. 2001. A novel extracellular calcium sensing mechanism in voltage-gated potassium ion channels. J. Neurosci. 21:4143–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim, W. A. 2002. The modular logic of signaling proteins: building allosteric switches from simple binding domains. Curr. Opin. Struct. Biol. 12:61–68. [DOI] [PubMed] [Google Scholar]
- 25.Mount, D. B., and M. F. Romero. 2004. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 447:710–721. [DOI] [PubMed] [Google Scholar]
- 26.Adler, H. J., I. A. Belyantseva, R. C. Merritt, G. I. Frolenkov, G. W. Dougherty, and B. Kachar. 2003. Expression of prestin, a membrane motor protein, in the mammalian auditory and vestibular periphery. Hear. Res. 184:27–40. [DOI] [PubMed] [Google Scholar]