

Abstract
We introduce time-resolved infrared spectroscopy as a powerful method to study the kinetics of RNA folding and unfolding transitions. A laser-induced temperature jump is used to initiate a perturbation in the RNA structure. A probe laser, tuned to a specific infrared absorption of the RNA, is then used to monitor the subsequent relaxation kinetics. A 10-ns pump pulse permits the investigation of fast, nanosecond events. In this work we probe two vibrational transitions, one at 1620 cm−1 and one at 1661 cm−1. The former transition reports mainly on the dynamics of A and U interactions, the latter is attributed to mainly G and C interactions. Our results reveal three distinct kinetic phases for each vibrational transition probed. We propose two models to describe the data. In one mechanism, the unfolded state partitions into two separate populations; each is conformationally biased to proceed via one of two distinct pathways. In an alternative model, folding proceeds through a series of sequentially populated intermediates. In both cases, the first step in the proposed folding mechanism is rate limiting (hundreds of microseconds) and involves a collapse into incorrectly folded intermediate populations. Two faster kinetic phases (tens of microseconds and hundreds of nanoseconds) follow in which the intermediate populations undergo localized reorganizational motions in the search for native contacts.
INTRODUCTION
RNA is a vital biological macromolecule that plays an essential role in many biological functions (e.g., transcription and translation). It is commonly known that a biopolymer like RNA must fold into a precise three-dimensional conformation for it to properly function. The manifold of conformational space that even a modestly sized RNA molecule can sample is astronomically large—so large that a random search for the precise biologically active structure would be impossible, taking billions upon billions of years to sample all possible configurations (1). Yet most RNAs fold completely in seconds or less. Clearly, a very specific mechanism and/or pathway is followed. For example, the search could be biased to a small subset of possible conformations. It is the goal of any RNA folding research to uncover the phenomena that are at the heart of these mechanisms or pathways and to explore the energy landscape that determines the RNA structure and folding dynamics.
There is a limit to the amount of information that can be obtained by studying folding under equilibrium conditions. Time-resolved experiments on RNAs approaching equilibrium provide the next level of detail. In keeping with this line of thought, experiments that access faster timescales will provide even more information. Generally speaking, RNA secondary structure forms on a timescale of hundreds of microseconds, whereas the formation of tertiary structure occurs on a millisecond timescale (2,3). What about the motions that precede these core folding events? These, too, must be studied to obtain a more thorough understanding of RNA folding.
RNA folding differs fundamentally from protein folding. The intramolecular interactions within a protein are relatively weak and folding is largely driven by hydrophobic collapse. Any intermediates that form along the folding pathway are unstable and short-lived. In contrast, the intramolecular interactions in RNA (basepairing and basestacking) are very strong and are ultimately responsible for driving folding. Quasistable intermediates form readily and early in the folding pathway (4–6). To more fully characterize these intermediates and gain a more thorough understanding of early events in RNA folding, experiments that combine faster time resolution with improved structural sensitivity must be employed.
Perhaps the most significant impediment to the study of fast RNA dynamics is the problem of initiating folding/unfolding on sufficiently short timescales. A particularly powerful approach that has been used previously to study folding kinetics in various nucleic acids is to use a laser-induced temperature jump (7–11). A temperature jump (T-jump) thermally perturbs the equilibrium of a sample on a timescale that is faster than the molecular dynamics of interest, allowing the ensuing relaxation dynamics to be observed. A pulsed laser is tuned to a wavelength that corresponds to a vibrational mode of the solvent. The pulse deposits enough energy into the laser interaction volume to raise the temperature of the irradiated region by as much as 25°C in a time that corresponds to the pulse width of the laser (in water, thermalization of the absorbed energy occurs within 50 ps).
The use of infrared (IR) absorption spectroscopy to monitor the relaxation kinetics can be especially informative. In addition to fast time resolution, the folding events can be monitored with a high degree of structural specificity. By using IR, the intrinsic properties of the molecule can be exploited and specific regions or groups within the sample can be probed. This eliminates the need for nonnative probes to be introduced. For example, in proteins, folding is followed by monitoring the transient absorption changes between 1610 and 1680 cm−1, the frequency range corresponding to the amide-I bond (12–14). Similarly, the IR modes of RNA such as the C=N, C=C, and C=O stretches of the nucleotide bases have been shown to be sensitive to specific conformational changes (e.g., basepairing) that occur upon RNA folding at equilibrium (15). Although widely used to study protein folding, laser induced T-jump combined with IR detection has never been used to study RNA folding. The primary objective of the work presented here is to explore structurally specific fast folding dynamics in RNA as well as to demonstrate the utility of T-jump IR to the study of RNA folding.
We have chosen yeast tRNAphe as our focus. It is extremely well characterized and it possesses elements of the most common structural motifs such as turns and hairpins (16). These attractive features provide the opportunity to observe the dynamics of elements that are common to all RNAs and make yeast tRNAphe well suited for the work presented here.
MATERIALS AND METHODS
Sample preparation
Yeast phenylalanine tRNA (tRNAphe) was obtained from Sigma (St. Louis, MO) and used as received. The experiments were carried out multiple times, each from a different sample lot of RNA. Consistent results were obtained for all of the replicate trials. Contributions from impurities (if present) are therefore considered insignificant. All buffer reagents and the tRNAphe were lyophilized three times against D2O to remove any labile protons before sample preparation. All experiments reported here were done at tRNAphe concentrations of ∼10 mg/mL in deuterated 100 mM phosphate buffer, pD 7.2. The buffered samples were heated and allowed to slowly return to room temperature to ensure that the tRNA was fully equilibrated before any experimentation.
The same sample cells were used for both the static Fourier transform infrared (FTIR) spectroscopy and the time-resolved work. The cells are custom-made and are comprised of two CaF2 windows separated by a 63-μm-thick Teflon spacer that defines the optical pathlength of the cell. The Teflon spacers were cut so that each cell would have two compartments, one for the sample (buffer and tRNAphe) and one for the reference (buffer alone). Using this design, reference data could be taken under nearly identical conditions as the sample data. The CaF2 cell is held together in a two-piece, custom-made copper holder. The copper holder is directly mounted to a thermally controlled block to control the temperature of the sample. The cell temperature is monitored via an externally mounted thermocouple and maintained to within ±0.1°C for all data presented here. The samples were allowed to equilibrate for ∼20 min at each temperature before spectra were recorded or time-resolved data was taken.
Temperature jump setup
The T-jump pulse is generated from a Q-switched Nd:YAG operating at 10 Hz and 1.064 μm. The nominal pulse width is 10 ns. The pulses are Raman shifted in H2 at 175 psi to generate 1.9-μm pulses at ∼20 mJ each. D2O absorbs ∼20% of the 1.9-μm radiation and deposits enough energy to the medium to induce T-jumps of up to 20°C in <20 ns (essentially instantaneous given the time resolution of our detection setup). The tRNAphe does not absorb any of the 1.9-μm radiation; thus, photo damage and other complicating photophysical processes are avoided.
The T-jump experiment is essentially a pump-probe technique; the 1.9-μm pulse pumps the sample that is subsequently probed by a continuous-wave diode laser operating at the desired frequency. The transient absorption is detected with a mercury cadmium telluride detector that has a rise time of ∼20 ns. The analog signal is processed with a 200-MHz digitizer and recorded on a computer. The cell is mounted on a linear translation stage so that sample and reference transients can be recorded without removing and replacing the cell. All transients reported here are averaged over 2000 laser shots. The relaxation kinetics of the tRNA are obtained by subtracting the reference signal from the sample signal. The beam diameter of the pump pulse is adjusted so that it is ∼5× the diameter of the probe beam. This is done to minimize fluctuations due to drift and to ensure a uniform heating profile for the probed volume. The magnitude of the T-jump is controlled by rotating a polarizer to attenuate the 1.9-μm beam. The response time of the system is on the order of 20 ns. As such, data were fit from 100 ns without deconvolution. Cooling of the heated volume begins ∼1 ms after the heating pulse, thereby limiting the long-time resolution of the experiment.
RESULTS
Equilibrium spectroscopy
The spectra of folded and melted tRNAphe are shown in Fig. 1 a along with thermal difference spectra, Fig. 1 b. The vibrational transitions in the 1600–1750 cm−1 region are due to C=C, C=O, and C=N stretching vibrations of the nucleic acid bases (17,18). These are the groups directly involved in basepairing interactions. Changes in the local interactions between hydrogen-bonded groups that occur as the RNA folds or unfolds, result in a redistribution of electron density, manifesting in changes in the IR spectra. Consequently, these transitions are highly sensitive to the configuration and basepairing interactions of the tRNA molecule. Although the spectra do indeed show the expected high degree of sensitivity, the broad, overlapping features make interpretation difficult. However, inspection of the thermal difference spectra reveal the most significant structurally induced spectral changes, including two broad, hyperchromic features at 1620 and 1661 cm−1 and a broad, hypochromic feature at 1680 cm−1.
FIGURE 1.

FTIR (a) and thermal difference spectra (b) of tRNAphe. (a) Folded (dashed line) and unfolded (solid line); the symbols mark the positions of the main vibrational modes and their assignments. (b) The low-temperature spectrum is subtracted from all other spectra. The arrows indicate the direction of spectral changes with increasing temperature. Temperatures ranged from 5.5 to 86.6°C in ∼5°C increments. Absorption changes at 1620 cm−1 correspond to A and U interactions and the changes at 1661 cm−1 correspond to G and C interactions. All of the spectral features in this wavelength range are due to C=N, C=C, and C=O stretching vibrations of the nucleic acid bases.
In addition to the broad, overlapping transitions, the presence of posttranscriptionally modified bases that are native to tRNAs could further complicate spectral interpretation (16). There are 11 different nonnative bases in tRNAphe that comprise 14 of the 76 total bases. With the exception of wybutosine, the modifications do not have spectral signatures in the 1600–1750 cm−1 region and account for only four of the 20 basepairs. Wybutosine accounts for only 1.3% of the total number of bases. Therefore, the affects (if any) of the modified bases are expected to be negligible and will not complicate the results.
Spectral interpretation was aided by taking the second derivative of the FTIR spectra (data not shown) to uncover otherwise indistinguishable features. Quantitative analysis was made possible by the early work of Thomas (19) and mode assignments are from the previously published work of others (17,18). The most prominent feature in the spectra of both folded and unfolded tRNA is a sharp band near 1620 cm−1. This mode is attributed to a strong C=N stretching vibration of adenine with weak contributions from a cytosine in-plane ring vibration. Upon formation of a basepair with uracil, the C=N stretch of adenine is shifted to higher wavenumbers (1632 cm−1) and reduces in intensity, thereby giving rise to the positive difference feature at 1620 cm−1. The formation of a cytosine-guanine basepair has little impact on the position and intensity of the cytosine ring vibration and does not contribute significantly to the 1620-cm−1 difference feature. At higher frequencies, the broad peak centered around 1660 cm−1 is mostly due to the combined contributions of a guanine C6=O stretch at 1669 cm−1 and a cytosine C2=O stretch at 1652 cm−1. The guanine C6=O stretch shifts to 1688 cm−1 when basepaired with cytosine whereas the cytosine C2=O transition is shifted to 1650 cm−1, thus giving rise to the positive difference feature at 1660 cm−1. There is also a minor contribution of a uracil C4=O stretch at 1657 cm−1 that shifts to 1673 cm−1 when basepaired with adenine. In summary, the 1620-cm−1 difference feature is mostly sensitive to interactions involving the bases A and U whereas the 1660-cm−1 feature is predominantly due to interactions involving the bases G and C. The shift in the guanine C6=O stretch gives rise to the negative difference feature at 1680 cm−1. Henceforth, the transition at 1620 cm−1 will be referred to as νAU and the transition at 1661 cm−1 will be νGC.
Equilibrium melting
At the equilibrium limit, the data can be treated as a two-state system. Intermediates are short-lived and are not significantly populated at equilibrium. The absorbance, A, is proportional to the fraction unfolded, f, which is related to the equilibrium constant, Keq, according to,
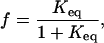 |
(1) |
where Keq is equal to exp(−ΔGU/RT). Because ΔG(T) = ΔH(T) − TΔS(T), ∂ΔH/∂T = ΔCp, and T(∂S/∂T) = Cp, the temperature dependence of the entropy and enthalpy can be calculated and used to express the Gibbs free energy according to the Gibbs-Helmholtz relationship as follows,
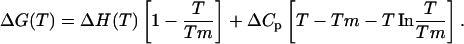 |
(2) |
Therefore, the absorption as a function of temperature can be expressed accordingly,
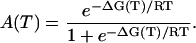 |
(3) |
The melt curves in Fig. 2 were simultaneously fit directly to Eq. 3 using a global fitting algorithm to obtain the relevant thermodynamic parameters. Fitting the curves simultaneously facilitates a more reliable, and statistically meaningful, comparison.
FIGURE 2.

Thermal melting curves for data collected at 1620 cm−1 (○) and 1661 cm−1 (□). Solid lines are a direct fit to the Gibbs-Helmholtz equation (Eq. 3).
The thermodynamic parameters for unfolding, ΔHU, ΔSU, and ΔCp, for the data collected at νAU are 21.6 ± 1.1 kcal/mol, 65.2 ± 3.4 cal/mol/K, and 0.39 ± 0.08 kcal/mol/K. The same parameters for the data collected at νGC are 31.7 ± 1.9, 95.5 ± 5.8, and 0.66 ± 0.15. The melting temperatures (in °C) are 58.1 ± 0.6 and 58.8 ± 0.4 for the data at νAU and νGC, respectively. The positive enthalpy and entropy of unfolding for both vibrational transitions probed are consistent with the disruption of secondary structure. Additionally, a heat capacity change is also observed for both transitions, signaling a redistribution of solvent molecules as the exposed bases are solvated. To well within experimental error, the melting temperatures for each transition are the same.
The initial temperature used for the kinetic experiments was chosen so that the majority of the tRNA molecules are folded before the T-jump. In our experiments, the initial temperature was held constant at 47°C, where >98% of the total population is folded. After a 20°C T-jump, the equilibrium is shifted in favor of the unfolded structure, resulting in a folded population of <1%. Under these conditions, the rate of unfolding, ku, dominates the relaxation time, 1/τ = kf + ku.
Singular value decomposition analysis
Factor analysis of the equilibrium melting data by singular value decomposition (SVD) was used to more fully characterize the equilibrium data. If the data contain contributions from more than one unique species, SVD can provide a means to identify this and inferences can be made regarding the nature of the components. The array of temperature-dependent spectra constitute an m × n matrix (m > n), A, where the m rows correspond to melting profiles at a specific wavelength and the n columns are complete spectra at a single temperature. The spectrum at any given temperature is a linear combination of the spectral contributions of each unique chemical species. SVD factorization provides a method to extract from the original data matrix the individual component spectra as well as to determine the relative contribution from each. The process produces three matrices, U, S, and VT, the product of which gives the original data matrix, A. S is an n × n diagonal matrix containing the singular values (i.e., the relative abundance of each component), U is an m × n matrix that contains the basis spectrum of each component, and VT is an n × n matrix containing the temperature dependence of each component (i.e., melting behavior).
The first three singular values account for 90% of the trace of the S matrix. This implies that there are three significant species that contribute to the overall data. A more rigorous and objective method to determine the number of significant components is to compare the autocorrelation values of the individual U and VT vectors. The autocorrelation values are calculated as follows,
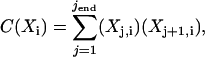 |
(4) |
where the indices j and i denote row and column assignments, respectively, for either the U or VT matrix. It has been previously shown that an autocorrelation value of 0.8 corresponds to a vector having a signal/noise ratio of 1 (20). By calculating the autocorrelation values for the U and VT matrices, a completely objective criterion can be used to determine the significant species. Using C(Xi) ≥ 0.8 as a cutoff, only the first two components can be considered significant (data not shown). Although nine components of the U matrix meet the required criterion, only two components of the VT matrix are significant. This difference is due to the greater dimensionality of the U matrix (one row for each wavelength, m = 416) compared to the VT matrix (one row for each temperature, n = 16). Because the three matrices are not independent entities, the more conservative conclusion of two components is the most accurate.
Time-resolved spectroscopy
The initial temperature is held constant whereas the magnitude of the T-jump is varied. Transients are recorded for each incremental T-jump and the final temperature achieved in each experiment is used for all temperature-dependent kinetic analyses. Selected transients and fit functions for both νAU and νGC are shown in Fig. 3. The kinetics monitored at νAU fit well to a triexponential model. The phases are well separated in time spanning hundreds of nanoseconds to hundreds of microseconds and the amplitudes contribute more or less equally to the total decay, the slowest phase contributing the least. Similar fits were obtained using a triexponential model for the data collected at νGC. Using the three-exponential model, the two fast phases had roughly the same amplitudes and time constants as the νAU parameters, whereas the slow phase was ∼5% slower than the corresponding νAU phase. However, recall that the difference feature at νGC has a small contribution from A and U interactions. To separate the G and C kinetics from the residual affects of the A and U kinetics, a fourth exponential was added to the model. At each final temperature, the fourth time constant was set equal to the corresponding relaxation rate monitored at νAU. Effectively, this “subtracts” the A and U contribution from νGC leaving only the G and C component.
FIGURE 3.

Selected data sets with exponential fits; 1620 cm−1 (○) and 1661 cm−1 (□). The data have been normalized for comparison. For clarity, every 20th data point is shown out of a total of 2048 collected data points. The data at 1620 cm−1 is fit to a three-exponential model and the data at 1661 cm−1 is fit to a four-exponential model. See text for further details.
The rationale for this warrants additional explanation. First, because the two fast kinetic phases are very similar for both frequencies, it is assumed that the major differences are present only in the slow phase; hence, this was the parameter to fix. The resultant fits yielded the same parameters for the two fast exponentials. The slow kinetic phase was substantially slower than the rate of the fixed parameter and the amplitude was split equally between the two, arguing for a physically meaningful solution. As added proof, the regression analysis was performed while letting all parameters vary. The amplitudes were nearly the same as they were using the previous method and the time constants were always well within 10% of the previous values. If the data at νAU were fit to a four-exponential model, invariably, one of the exponentials would be physically unreasonable; hence, the three-exponential model is justified. A stretched exponential model was also considered, but the results showed little correlation.
An understanding of the nature of the three kinetic phases is necessary to further describe the kinetic mechanism in terms of physically meaningful events. A more detailed analysis of the transition state thermodynamics will facilitate the interpretation. From transition state theory we know that
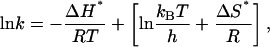 |
(5) |
where kB is Boltzmann's constant and h is Planck's constant. Provided there is no change in heat capacity (there is no indication of curvature in the plots), a plot of lnk vs. 1/T is and has a slope equal to ΔH*/R. The y intercept is equal to the quantity in brackets from which ΔS* can be calculated. The term, kBT/h is the Arrhenius frequency factor. At 330 K, this term is on the order of 1012 s−1, a rate that is only valid for gas phase reactions. A reasonable value for solution phase reactions is the more conservative estimate of 108 s−1 (21). Assuming a linear relationship, it is easy to show that y intercepts greater than ln108 give a positive ΔS*, whereas intercepts less than ln108 result in a negative ΔS*.
Fig. 4 shows plots of the kinetic data in Arrhenius coordinates. For the slow phase, the data at νAU is slightly faster than the νGC data. Additionally, the temperature dependence shows that this phase corresponds to an activated process for both probe wavelengths. It is also clear that ΔS* > 0 for this phase, because the y intercepts clearly exceed ln108 (most likely, the intercepts are also in excess of ln1012, the upper most limit). The magnitude of the activation barriers and the favorable increase in entropy is consistent with the melting of secondary structure (2).
FIGURE 4.

Arrhenius plots of observed rates. A and U interactions (1620 cm−1, ○); G and C interactions (1661 cm−1, □). (a) k1 (upper part) and k2 (lower part) data correspond to nonactivated processes; (b) the slow kinetic phase, k3, for AU and GC basepairs. Overall the A and U interactions are faster than the G and C interactions. Both correspond to activated processes. The solid lines in all of the plots are fits to a linear Arrhenius model.
The activation barriers for k1 and k2 are quite weak (ΔH* ≈ 0); based on their lack of temperature dependence. Linearly extrapolating to the y intercept suggests a negative entropic term, although this is difficult to quantify. The lack of enthalpic barrier argues that there is no net formation or net disruption of structure on these timescales, although an entropic barrier would indicate that there is a reduction in configurational or translational degrees of freedom of solvent molecules and counterions as they adjust to accommodate the melting tRNA. If this is the case, we would expect the plots would deviate from linearity and show a curvature. The curvature would be the result of a nonzero change in heat capacity. Our data do not span a wide enough temperature range to confirm this notion. As in the case of the slow phase, the data at νAU and νGC can also be separated kinetically for both of the fast phases. Although this distinction is subtle, the best-fit lines show that it is statistically valid. The T-jump experiment is subject to greater noise at faster timescales and may conceal more prominent differences.
DISCUSSION
The primary objective of this work is twofold. First, we wanted to demonstrate the utility of time-resolved IR to study RNA folding. Secondly, we wanted to attain a more thorough understanding of RNA folding dynamics by accessing faster timescales and by probing with structural specificity. We anticipated that this combination would uncover new phenomena. Our results suggest two possible folding mechanisms for tRNA. One possible interpretation is that folding proceeds via multiple pathways, whereas another feasibility is that the tRNA folds through a series of sequentially populated intermediates. Each will be discussed in more detail below.
Before discussing the folding mechanisms a brief digression is in order. The early work of Crothers showed that secondary structure forms on a timescale of hundreds of microseconds and that tRNA does not possess a significant degree of tertiary structure under low ionic strength conditions ([Na+] < 0.017 M) (2). Based on this early work, experimental conditions (low ionic strength), and our data (microsecond, activated kinetic phase), we surmise that the slow phase of our data corresponds to the formation of secondary structure and that native tertiary contacts are negligible.
The multiple pathway model is best envisioned from an energy landscape perspective. At temperatures above the melting temperature, the landscape partitions into two metastable minima separated by a barrier that is large enough to prohibit interconversions between them. Each minimum is further composed of many related unfolded configurations. Each of these configurations is separated by a relatively small barrier so that a flux of related structures is continuously evolving. Each pathway is confined to a “channel” in the energy landscape (22). Three kinetic phases imply two intermediates. In the data presented here, each phase can further be separated into two distinct events (data at νAU and at νGC), defining two pathways governed by six rate constants, 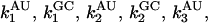 and
and 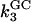 (see Scheme 1) where UAU and UGC are the different populations of unfolded ensembles, F is the folded ensemble, and
(see Scheme 1) where UAU and UGC are the different populations of unfolded ensembles, F is the folded ensemble, and 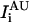 and
and 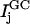 are the intermediates. One pathway is observed when probing at a wavelength that reports predominantly G and C interactions (kinetic data recorded at νGC) whereas the other is observed when probing at a wavelength that typifies predominantly A and U interactions (kinetic data recorded at νAU). Alternatively, the unfolded state comprises a single ensemble of extended structures and folding proceeds through a series of sequentially populated intermediates (see Scheme 2). In this scheme, GC and AU basepairs form consecutively and the intermediates that result have different amounts of each.
are the intermediates. One pathway is observed when probing at a wavelength that reports predominantly G and C interactions (kinetic data recorded at νGC) whereas the other is observed when probing at a wavelength that typifies predominantly A and U interactions (kinetic data recorded at νAU). Alternatively, the unfolded state comprises a single ensemble of extended structures and folding proceeds through a series of sequentially populated intermediates (see Scheme 2). In this scheme, GC and AU basepairs form consecutively and the intermediates that result have different amounts of each.
SCHEME1.

SCHEME2.

Perhaps the strongest argument for both models comes from the equilibrium data and not the kinetic data. The thermodynamic parameters that are derived from the equilibrium experiments (ΔH, ΔS, ΔCp) are state functions. Hence, their values depend only on the initial and final states of the system and are independent of the pathway between them. The differences between the thermodynamic parameters we calculated, for the melt data at νAU versus νGC, are statistically significant. These differences imply that the initial and final states are not the same and support the multiple pathway model. The model outlined in Scheme 1 indicates that the initial states are different and that both pathways converge upon a common final structure.
An alternate interpretation of the equilibrium data supports the sequential folding mechanism. The argument in the preceding paragraph assumes that both melt curves reach a plateau at high temperatures and that unfolding is complete. However, a closer inspection of the melt curves in Fig. 2 could argue that this is not the case. The melt curve corresponding to AU basepairs (circles, Fig. 2) has a fairly sharp transition and has a well-defined plateau at high temperatures indicating that melting is complete. However, the melt curve corresponding to GC basepairs (squares, Fig. 2) does not reach a clearly defined limit as in the AU data making the case that unfolding is not yet complete. At the high-temperature limit, enough energy has been supplied to disrupt all of the AU basepairs (two hydrogen bonds each) but not enough to disrupt all of the GC basepairs (three hydrogen bonds each). Hence, the thermodynamic differences could be due to a small amount of GC basepairs that persist at this upper temperature limit. Accordingly, folding proceeds through a series of sequentially populated intermediates where AU and GC basepairs form at slightly different rates.
The vectors from the VT matrix constitute melting profiles of individual components; the two most significant are shown in Fig. 5. Using a global regression analysis the melting profiles were simultaneously fit to Eq. 3 in the same manner as the raw melt curves. The results of the fitting are comparable to the fit parameters from the raw data melt curves. The analysis of the raw data shows the melting behavior at a single specific wavelength, whereas the analysis using the factored data is distinctly different. The melting profiles from the SVD are derived from all wavelengths considered equally and are far less vulnerable to errors. This would argue that the thermodynamic parameters are a global feature of the tRNA melting and are not unique to a particular basepairing interaction. As such, these findings would be in greater support of the multiple pathway model than the alternative sequential scheme.
FIGURE 5.

First two row vectors of the VT matrix representing melting profiles for each component. The circles denote the most significant component and the squares denote the next most significant component. The solid lines are fits to Eq. 3.
Despite the fact that a kinetic mechanism cannot be confirmed, the nature of the individual kinetic phases can still be discussed. Regardless of the mechanism, each set of rate constants (i.e., k1 for GC and AU, k2 for GC and AU, etc.) would most likely arise from analogous motions. Large-scale motions must be preceded by smaller scale, local fluctuations. The local fluctuations represent molecular “attempts” at finding the appropriate conditions that permit the larger-scale motion. Consider a basepairing interaction. Before the basepair can break, the local environment must be poised to form new hydrogen bonds and ionic interactions that stabilize the unpaired base. If a hydrogen bond is supplied with enough energy to break, but the surrounding conditions do not present a favorable alternative, the hydrogen bond will reform without a permanent break. In this case, the energy that was used to break the bond is nearly exactly cancelled by the energy liberated when the bond reforms, hence the process is incomplete and the net activation enthalpy is equal to zero. Similarly, if a basepair breaks and both form basepairs with other bases, the energies also cancel out (at least to within the resolution of our data). Thus, the fast kinetic phases, k1 and k2, are most likely due to local basepairing reorganizations or unsuccessful attempts at breaking secondary structural contacts as the collapsed tRNA “samples” its configurational space. Most likely, these events occur in concert with a redistribution of solvent molecules or counterions. As the temperature is raised, counterions as well as solvent molecules begin to position themselves to solvate the soon-to-be-exposed hydrophobic bases. Likewise, there is an ordering of water molecules and counterions surrounding the negatively charged backbone. The barrier for k3 is enthalpic because energy must be supplied to disrupt secondary structural contacts. The equilibrium thermodynamics are consistent with this model indicating the disruption of structure and an increase in disorder. The nonzero ΔCp at equilibrium further supports the assertion that solvent molecules and counterions are involved in the unfolding of the tRNA.
The conclusions we have drawn up to this point provide two consistent, albeit conflicting, descriptions of the data. Different thermodynamics are observed depending upon the probe frequency; these results have been confirmed through SVD factor analysis. The thermodynamic differences could result from incomplete melting or unique unfolded ensembles. From the kinetic data we see faster kinetics for one over the other and, more importantly, both sets of kinetic data are mutually exclusive. In other words, the kinetics observed at one frequency do not significantly affect the processes observed at the other frequency. This could have one of two interpretations. If the mechanism involves multiple pathways then the implication is that the interactions via one pathway are not present in the other. Of course it is quite reasonable to postulate that both types of interactions are present in both pathways, but that one dominates to the extent that the other can't be resolved kinetically. Because the IR absorption changes are proportional to the number of basepairs, this interpretation would indicate that one pathway involves mostly A and U interactions (with little contribution from G and C interactions) whereas the other pathway involves mostly G and C interactions with little contribution from A and U interactions. The implication is that the “AU” pathway must already have a significant proportion of native GC contacts and that the “GC” pathway already has a high proportion of native AU contacts. An alternative interpretation is that the formation of AU basepairs and GC basepairs occur consecutively at slightly different rates.
CONCLUSIONS
We have put forth two plausible models that describe the formation of tRNAphe secondary structure. In one mechanism, folding proceeds through multiple pathways. Each pathway is dominated by either A and U interactions or G and C interactions; both are spectroscopically distinguishable. In this model, the unfolded state is partitioned into two ensembles where each is conformationally biased to proceed via one of two pathways. An alternative interpretation of the data argues that GC and AU basepairs form at slightly different rates and that folding proceeds through a series of sequentially populated intermediates. In either case, the first step involves an initial collapse of the unfolded states (or state) into separate ensembles (or a single ensemble) composed of misfolded (or partially folded) intermediates on a timescale of hundreds of microseconds. The initial collapse is followed by two fast (tens of microseconds and hundreds of nanoseconds) reorganizational phases in which the intermediates sample their environment searching for appropriate native contacts. At this time, the data are insufficient to distinguish the two models.
Furthermore, we have shown that the infrared spectra of tRNAphe between 1600 and 1750 cm−1 is extremely rich with information and is exquisitely sensitive to conformational changes. The work presented here demonstrates the power of time-resolved IR spectroscopy to study RNA folding and establishes a framework upon which numerous experiments can be constructed. Its structural specificity and excellent time resolution will complement existing methods and open new doors to previously unexplored territory. Now that the technique of time-resolved IR has been established to study RNA folding, additional experiments, designed to distinguish the mechanisms postulated here, can be developed.
Acknowledgments
This work was made possible through funding from a Los Alamos National Laboratory Director's Funded Postdoctoral Fellowship (E.B.B.).
References
- 1.Levinthal, C. 1968. Are there pathways for protein folding? Journal de Chimie Physique et de Physico Chimie Biologique. 65:44–49. [Google Scholar]
- 2.Cole, P. E., and D. M. Crothers. 1972. Conformational changes of transfer ribonucleic acid. Relaxation kinetics of the early melting transition of methionine tRNA. Biochemistry. 11:4368–4374. [DOI] [PubMed] [Google Scholar]
- 3.Crothers, D. M., P. E. Cole, C. W. Hilbers, and R. G. Shulman. 1974. The molecular mechanism of thermal unfolding of Escherichia coli formylmethionine transfer RNA. J. Mol. Biol. 87:63–88. [DOI] [PubMed] [Google Scholar]
- 4.Russell, R., and D. Herschlag. 2001. Probing the folding landscape of the Tetrahymena ribozyme: commitment to form the native conformation is late in the folding pathway. J. Mol. Biol. 308:839–851. [DOI] [PubMed] [Google Scholar]
- 5.Treiber, D. K., M. S. Rook, P. P. Zarrinkar, and J. R. Williamson. 1998. Kinetic intermediates trapped by native interactions in RNA folding. Science. 279:1943–1946. [DOI] [PubMed] [Google Scholar]
- 6.Zarrinkar, P. P., and J. R. Williamson. 1994. Kinetic intermediates in RNA folding. Science. 265:918–924. [DOI] [PubMed] [Google Scholar]
- 7.Ansari, A., S. V. Kuznetsov, and Y. Q. Shen. 2001. Configurational diffusion down a folding funnel describes the dynamics of DNA hairpins. Proc. Natl. Acad. Sci. USA. 98:7771–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dewey, T. G., and D. H. Turner. 1979. Laser temperature jump study of stacking in adenylic-acid polymers. Biochemistry. 18:5757–5762. [DOI] [PubMed] [Google Scholar]
- 9.Dewey, T. G., and D. H. Turner. 1980. Laser temperature jump study of solvent effects on poly adenylic-acid stacking. Biochemistry. 19:1681–1685. [DOI] [PubMed] [Google Scholar]
- 10.Proctor, D. J., H. R. Ma, E. Kierzek, R. Kierzek, M. Gruebele, and P. C. Bevilacqua. 2004. Folding thermodynamics and kinetics of YNMG RNA hairpins: specific incorporation of 8-bromoguanosine leads to stabilization by enhancement of the folding rate. Biochemistry. 9:14004–14014. [DOI] [PubMed] [Google Scholar]
- 11.Williams, A. P., C. E. Longfellow, S. M. Freier, R. Kierzek, and D. H. Turner. 1989. Laser temperature-jump spectroscopic and thermodynamic study of salt effects on duplex formation by dGCATGC. Biochemistry. 28:4283–4291. [DOI] [PubMed] [Google Scholar]
- 12.Callender, R., and R. B. Dyer. 2002. Probing protein dynamics using temperature jump relaxation spectroscopy. Curr. Opin. Struct. Biol. 12:628–633. [DOI] [PubMed] [Google Scholar]
- 13.Dyer, R. B., F. Gai, and W. H. Woodruff. 1998. Infrared studies of fast events in protein folding. Acc. Chem. Res. 31:709–716. [Google Scholar]
- 14.Werner, J. H., R. B. Dyer, R. M. Fesinmeyer, and N. H. Andersen. 2002. Dynamics of the primary processes of protein folding: helix nucleation. J. Phys. Chem. B. 17:487–494. [Google Scholar]
- 15.Parker, F. S. 1971. Applications of Infrared Spectroscopy in Biochemistry, Biology, and Medicine. Plenum Press, New York, NY.
- 16.Jovine, L., S. Djordjevic, and D. Rhodes. 2000. The crystal structure of yeast phenylalanine tRNA at 2.0 angstrom resolution: cleavage by Mg2+ in 15-year old crystals. J. Mol. Biol. 301:401–414. [DOI] [PubMed] [Google Scholar]
- 17.Banyay, M., M. Sarkar, and A. Graslund. 2003. A library of IR bands of nucleic acids in solution. Biophys. Chem. 1:477–488. [DOI] [PubMed] [Google Scholar]
- 18.Tsuboi, M. 1969. Application of infrared spectroscopy to structure studies of nucleic acids. App. Spec. Rev. 3:45–90. [Google Scholar]
- 19.Thomas, G. J. 1969. Determination of base pairing content of ribonucleic acids by infrared spectroscopy. Biopolymers. 7:325–334. [DOI] [PubMed] [Google Scholar]
- 20.Henry, E. R., and J. Hofrichter. 1992. Singular value decomposition: application to analysis of experimental data. Methods Enzymol. 210:129–192. [Google Scholar]
- 21.Krieger, F., B. Fierz, O. Bieri, M. Drewello, and T. Kiefhaber. 2003. Dynamics of unfolded polypeptide chains as model for the earliest steps in protein folding. J. Mol. Biol. 5:265–274. [DOI] [PubMed] [Google Scholar]
- 22.Russell, R., X. W. Zhuang, H. P. Babcock, I. S. Millett, S. Doniach, S. Chu, and D. Herschlag. 2002. Exploring the folding landscape of a structured RNA. Proc. Natl. Acad. Sci. USA. 99:155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]