

Abstract
Kinetics of compaction on single DNA molecules are studied by fluorescence videomicroscopy in the presence of 1), Xenopus egg extracts and 2), purified nucleosome reconstitution systems using a combination of histones with either the histone chaperone Nucleosome Assembly Protein (NAP-1) or negatively charged macromolecules such as polyglutamic acid and RNA. The comparison shows that the compaction rates can differ by a factor of up to 1000 for the same amount of histones, depending on the system used and on the presence of histone tails, which can be subjected to post-translational modifications. Reactions with purified reconstitution systems follow a slow and sequential mechanism, compatible with the deposition of one (H3-H4)2 tetramer followed by two (H2A-H2B) dimers. Addition of the histone chaperone NAP-1 increases both the rate of the reaction and the packing ratio of the final product. These stimulatory effects cannot be obtained with polyglutamic acid or RNA, suggesting that yNAP-1 impact on the reaction cannot simply be explained in terms of charge screening. Faster compaction kinetics and higher packing ratios are reproducibly reached with extracts, indicating a role of additional components present in this system. Data are discussed and models proposed to account for the kinetics obtained in our single-molecule assay.
INTRODUCTION
In eukaryotes, nuclear DNA is organized in chromatin. Its basic unit, the nucleosome, is composed of 145–147 basepairs of nucleosomal DNA wrapped around an octamer of core histones (one tetramer (H3-H4)2 and two dimers (H2A-H2B)). The nucleosomes are regularly spaced along the DNA with a repeat length of ∼200 bp and form a nucleofilament that represents the first level of chromatin folding (1). Higher levels of chromatin compaction involve additional proteins, many of which are unknown, to achieve the highest degree of condensation found in mitotic chromosomes. It is now established that nucleosomal structure and dynamics play a key role in genome expression and maintenance. Posttranslational modifications of selected residues of the histone amino-terminal tails have been involved in transcriptional regulation (2,3), and in the folding of the 30-nm fiber in vitro (4,5).
In vivo, the majority of chromatin assembly events occur immediately after DNA replication, when nucleosomes are transiently disrupted ahead of the replication fork and redistributed behind it onto daughter duplex DNA (6–9). Newly synthesized histones are incorporated to obtain the full nucleosome complement on nascent DNA. This involves the initial deposition of histones H3 and H4, followed by the incorporation of two dimers (H2A-H2B) to form the nucleosome core particle (10). The transfer and the ordered deposition of histone subcomplexes in the nucleus are promoted by a variety of assembly factors (11–14).
In vitro, reconstitution of nucleosomes can be achieved by dialyzing mixes of purified histones and DNA from high to low ionic strength solutions (1) or, alternatively, by adding negatively charged macromolecules such as polyglutamic acid or RNA (15–17). Indeed, almost any molecule that could screen the cationic charge of histones was found to facilitate the loading of histones onto DNA in vitro and to prevent aggregation. However, these methods, which enable nucleosomal particle reconstitution, generally yielded close-packed, irregular nucleosome arrays that do not correspond to the physiological regularly spaced nucleosomal arrays found within the cell nucleus.
Major insights into our understanding of chromatin assembly processes were gained from in vitro studies using extracts derived from Xenopus eggs (18,19) or Drosophila embryos (20–22). These extracts are highly efficient to support chromatin assembly on DNA templates that can be replicated, repaired, and transcribed in the same system and thus closely mimic physiological cellular conditions. Given that they contain large amounts of histones, nucleosome assembly factors, and histone chaperones, they provide a convenient physiological system to assay nucleosome formation and identify regulatory components.
Regularly spaced nucleosomal arrays can be reconstituted using purified histones at ionic strength considered physiological provided that the histones are complexed with a histone chaperone protein (8). In this respect, NAP-1 (nucleosome assembly protein 1) was used extensively (23). NAP-1 has been isolated from Drosophila extracts (24) in complex with H2A and H2B, and from human cells in complex with H2A (25), and it has thus been considered as an (H2A-H2B) histone chaperone in vivo. In vitro, NAP-1 from Drosophila (dNAP-1), HeLa cells (AP-1), and yeast (yNAP-1) can stimulate nucleosome reconstitution, interacting not only with the (H2A-H2B) dimer but also with the (H3-H4)2 tetramer (26–29).
The emergence of single-molecule micromanipulation offers an attractive approach to investigate chromatin assembly on individual molecules in real time. This strategy makes it possible to analyze kinetics and forces involved in the assembly (30–32) as well as chromatin mechanical properties (33,34). A first estimation of the kinetic parameters of assembly with Xenopus egg extracts under physiological conditions was obtained, strikingly showing that this process is faster than estimated from conventional biochemical assays (30). Optical tweezers experiments confirmed that assembly with extracts was force-dependent, and that the process could be stalled at ∼10 pN (32). The influence of the tension on the rate of assembly and disassembly was also studied with purified compounds using magnetic tweezers (31).
We present here results obtained with a new videomicroscopy system, whose characteristics are significantly improved in comparison to that described in our previous studies (30). We provide a refined kinetic analysis of chromatin assembly with Xenopus egg extracts, and compare it directly to the reconstitution kinetics obtained with purified components, namely native histones (purified from Drosophila embryos) or recombinant Xenopus histones WT (wild-type) and tailless (lacking the N-terminal amino-acids), in the presence or absence of the histone chaperone yNAP-1. Finally, to address more specifically the mechanism of action of yNAP-1, we compare these results with the kinetics of reconstitution mediated by two anionic polymers, polyglutamic acid (PGA) and RNA.
MATERIALS AND METHODS
Extracts preparation
Xenopus egg extracts were prepared as previously described (35). Briefly, eggs were collected from Xenopus females in 0.1 M NaCl. Incubation in 2% cysteine solution in 0.1 M NaCl allowed removal of the jelly coat. After extensive washes in 0.1 NaCl, eggs were rinsed in extraction buffer (10 mM HEPES-KOH, pH 7.8, 70 mM KCl, 5% sucrose, 0.5 mM DTT, and protease inhibitors) and transferred in chilled centrifugation tubes. Upon removing excess buffer, the eggs were subjected to low-speed crushing at 10,000 × g for 30 min at 4°C. The middle phase was collected by inserting a glass Pasteur pipette through the upper yellow lipid layer, and clarified by ultracentrifugation at 150,000 × g for 1 h at 4°C in Ultra-Clear tubes (Beckman-Coulter, Fullerton, CA). A syringe with a needle was used to pierce the tube and to collect the clear ooplasmic fraction corresponding to the extract, which was immediately aliquoted, frozen in liquid nitrogen and stored at −80°C. For each new extract preparation, after conductimetry verification, the chromatin assembly activity was tested by supercoiling and micrococcal nuclease digestion assays (36).
Histones and yNAP-1 preparation
Native histones from Drosophila were purified to homogeneity from postblastoderm Drosophila embryos by hydroxylapatite fractionation of chromatin (37). Recombinant histones were expressed individually from BL21 pLysS bacteria transformed with wild-type Xenopus histone cDNA cloned in pET-3b vectors (Novagen, San Diego, CA), and purified from inclusion bodies under denaturing conditions as described in works by Luger and colleagues (38,39). Tailless histones (lacking the N-terminal tail) were obtained as above by using truncated histone cDNA generated by PCR (Δ1–19 for H4, Δ1–26 for H3, Δ1–12 for H2A, Δ1–26 for H2B (40)). Purified H3-H4-H2A-H2B histones (WT or tailless) were then mixed at a 1/1/1/1 stoichiometry in the denaturing buffer (7 M guanidin-HCl, 20 mM Tris-HCl, pH 7.5, 10 mM DTT freshly added) and dialyzed progressively against the refolding buffer (2M NaCl, 10 mM Tris-Hcl, pH 7.5, 1 mM EDTA, pH 8, 5 mM β-mercapto-ethanol) to reconstitute histone octamers. These octamers were then purified from aggregates and H3-H4 tetramers by gel filtration and stored at −20°C. They were used as the source of histones in our experiment to ensure the correct histone ratio in (H3-H4)2 and H2A-H2B dimers for forming nucleosomal core particles.
The yNAP-1 was expressed from BL21 pLysS bacteria transformed with the yNAP-1 cDNA cloned into the pET28a vector (Novagen) to add a His tag to the N-terminal. Protein were purified with Ni-NTA resin according to manufacturer's instruction (Qiagen, Valencia, CA), and stored at −80°C.
The protocols were optimized to avoid any contamination, either by nucleic acids or high molecular weight histone-associated proteins. Protein purity of preparations was carefully checked by spectrophotometry and SDS-PAGE, and no contamination was detected. The concentration in histones of the different solutions was then properly estimated. Therefore, the comparison of the kinetics obtained with the different assembly systems used in the same concentration appears to be relevant.
yNAP-1-, PGA-, and RNA-histone combination
Nucleosome assembly using yNAP-1, PGA (Sigma, Saint Quentin Fallavier, France), and RNA (Roche Diagnostics, Meylan, France) as histone chaperones was achieved by first making a chaperone-histone mix (41) that was then injected in the chamber using siliconized pipette tips and tubes. Purified yNAP-1 (freshly diluted after thawing in 10 mM HEPES-KOH, pH 7.8, 10 mM KCl, 1.5 mM MgCl2, 10% glycerol, 1 mM DTT) was mixed with 350 ng of histone octamer to yield a yNAP-1/octamer molar ratio of 0.6:1 or 6:1. After vigorous vortexing, this mix was diluted two times with buffer A (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 0.05% NP40 (Sigma) and 0.1 μg/μL chicken ovalbumin (Sigma)). For polyglutamic acid and RNA, we used a PGA/octamer mass ratio of 5:1 as described in Stein (42) and an RNA/octamer mass ratio of 2:1 as described in Nelson et al. (16). The above buffer was used to dilute PGA or RNA. We incubated these mixes for at least 30 min at room temperature before diluting them in buffer A (complemented with 0.25 mg/mL bovine serum albumin (Sigma) to avoid nonspecific interactions and 2% β-mercapto-ethanol (Sigma) as an oxygen scavenger) to the appropriate histone final concentration and injecting them into the chamber to follow nucleosome assembly.
Preparation of streptavidin end-modified λ-dimers
In this study, we used dimers of λ-phage DNA (48.5 kb; Roche) to be able to resolve the packing ratio achieved with the different assembly systems (see Results). These DNAs were constructed by annealing and ligating a 22-mer biotinylated oligonucleotide (Eurogentec, Seraing, Belgium) with T4 DNA ligase (Roche). The molecule was then ligated to another unmodified λ-DNA to produce a dimer (97 kb). The dimers were coupled to streptavidin (Roche), aliquoted, and stored at −20°C. Before each experiment, DNA (1 ng/μL) was incubated for at least 1 h with the bis-intercalating fluorescent dye YOYO-1 iodide (Molecular Probes; Eugene, OR) with a stoichiometry of 1 molecule to every 8 bp. To assess the effect of the fluorescent labeling on the extract assembly kinetics, we decreased the intercalation ratio of YOYO-1 from 1:8 down to 1:24 and did not observe any change in the experiment (Supplementary Material).
Preparation of biotin-coated coverslips
Coverslips (24 × 40 mm) were prepared using the method reported by Perret et al. (43) and Merkel et al. (44), with some modifications. In brief, the coverslips were soaked in 2% aminopropyltriethoxysilane (Sigma), 4% H20, 0.8% glacial acetic acid (Sigma), and 93.2% methanol (Sigma) for at least 3 h at room temperature. Next, a mixture of amine-reactive poly(ethylene glycol) (PEG), consisting of a molar ratio of 1:10 SPA-PEG5000 and SPA-PEG3400-biotin (Nektar, San Carlos, CA) diluted at a concentration of 2 mg/mL, was incubated with the coverslips in a carbonate buffer (pH 8.5) for at least 2 days. We chose to use a PEG coating because of its known resistance to nonspecific interactions (45).
Microfluidic device and videomicroscopy
We used the set-up described in Bancaud et al. (46), an improved version of the prototype developed by Ladoux et al. (30). Briefly, chips were prepared by “soft lithography” poly(dimethylsiloxane) (PDMS) technology (47). We used a master composed of a positive relief of SU-8 resin (MicroChem, Newton, MA) on a glass wafer made by photolithography. PDMS was made with Sylgard 184 silicone elastomer kit (Dow Corning, Midland, MI) used in standard conditions. This polymer was cured on the wafer to form a cell with the impression of the master. The cell was punched with holes and sealed with the coverslip functionalized with PEG-biotin as described in the previous paragraph. The channels were 500 μm wide and 165 μm high. They were designed following a new geometry, in the shape of a T with one inlet (I) and two outlets (O1 and O2), as depicted in Fig. 1. The two outlets were connected by silicone tubing to an aspiring syringe pump (KD Scientific, Holliston, MA). A dual-channel pinch electrovalve (NResearch, West Caldwell, NJ) switched the flow between channels.
FIGURE 1.

Schematic of the experimental apparatus. Syringe pump (1), electro-valve (2), inlet channel (I), outlet channel 1 (O1), and outlet channel 2 (O2). (A) Biological samples are aspirated in microchannel O1 until the concentration is homogeneous at the T-junction. (B) The electrovalve switches the aspiration to channel O2 where the objective is positioned. (C) Example of compaction of an individual DNA molecule in time in the presence of 2 ng/μL of native histones.
The chip was mounted on an inverted microscope (Zeiss, Oberkochen, Germany) with laser excitation at 488 nm (Coherent, Santa Clara, CA) equipped with an oil objective (×100, 1.4 NA (Olympus, Melville, NY)). Images were collected using an intensified CCD camera (Lhesa, Les Ulis, France). They were digitized and analyzed in real time. DNA length was measured from the digitized images by using an automated algorithm written in NIH IMAGE (National Institutes of Health, Bethesda, MD).
Nucleosomal formation on immobilized DNA
First, DNA was incubated in channel O2 for 10 min. During the incubation, we prepared Xenopus egg extracts or a purified histone/histone chaperone (yNAP-1, PGA, or RNA) combination. The protein source was then aspirated through channel O1 until a constant concentration was reached at the T-junction (Fig. 1 A). The flow was switched to channel O2 and acquisition simultaneously started (Fig. 1 B). To minimize photoinduced damages, the laser excitation was periodically shuttered at a frequency of 1 Hz throughout the experiment. The shear rate was set at 20 s−1, corresponding to a Weisenberg number (Wi) of 30 for λ-dimers (48). Wi represents the relative effect of hydrodynamic versus Brownian forces on the whole molecule. Based on simulations (48), one could evaluate the maximum tension exerted on the DNA (i.e., at its attachment point) to ∼1 pN. Since the tension decreases along the molecule to zero at the free end, these experiments were typically carried out in the sub-pN range. In this situation (Wi <80), it was demonstrated by Ladoux et al. (30) that the kinetics of Xenopus egg extract assembly are independent of the shear rate and the measurement of the end-to-end length of the molecule gives access to the kinetic parameters. All the kinetic curves are averages over two to six experiments, each including 1–3 individual DNA molecules.
An example of a single DNA behavior after immersion in 2 ng/μL of native histones is shown in Fig. 1 C. The length of the DNA molecule decreased and reached a plateau in ∼60 s. This DNA retraction proceeded to a final end-to-end distance of ∼4–5 μm.
Data treatment
In an ideal kinetic experiment, all the components involved in the reaction should instantaneously be mixed together, and most models for interpreting biochemical kinetics, such as the Michaelis-Menten one, are based on this assumption. In the single-molecule experiment described here, “instantaneous mixing” means that, in the vicinity of the DNA molecule, the concentration of all proteins involved in the assembly immediately rises from zero to its nominal value and remains constant, i.e., follows a step function. In reality, however, mixing always takes a finite time. In batch experiments, it takes typically from a few seconds to a few minutes, during which the concentrations are inhomogeneous through the sample. In our experiment, the protein-containing solution is brought to the DNA molecule by a hydrodynamic flow. The protein concentration front then follows the Taylor-Aris dispersion regime, which results from the combination of an inhomogeneous velocity profile (Poiseuille profile) and molecular diffusion. This can lead to a rather large effective “mixing time”, typically tens of seconds in our configuration. Nevertheless, as the flow is laminar, this dispersion is very reproducible. Taking advantage of this feature, we developed a new method, which allowed us to quantitatively access the real kinetics, corresponding to the arrival of a step function of protein concentration (46). Our approach is briefly discussed in the following paragraph.
Since concentration front spreading increases with the distance traveled, we first minimized the distance by which proteins were advected in the microchannel. To do so, we built up a three-channel set-up, as described in the Microfluidic device section. With this geometry, a typical experiment was carried out as follows: 1), we first flowed the histones in channel O1 until a steady-state concentration of proteins was established at the T-junction; 2), then we switched the flow to channel O2 in a time <1 s. We chose to observe molecules anchored close to the T-junction (typically between 2 and 4.5 mm). For a shear rate of 20 s−1, the proteins typically reach the DNA molecules in 5 s, which also provides the order of magnitude of the front spreading (46).
This improved resolution was not sufficient to completely eliminate distortion of the results due to front spreading on the timescale of the kinetics we wished to observe (a few seconds). Consequently, we developed a new approach to 1), directly measure the time- and space-dependent profile of protein concentration at the vicinity of the surface, and 2), compensate for it in the data analysis. Briefly, the protein profile was obtained by carrying out the same experiment as above with a fluorescent marker advected in the flow, in combination with a dye strongly absorbing at the excitation wavelength to probe specifically the front close to the surface (see Appendix A1).
By appropriate kinetic modeling and knowing the protein concentration profile (see Appendix A2), one can determine the real kinetics of virtually any DNA condensation reaction (corresponding to the response to an instantaneous step of concentration), with the condition that each step in the kinetics is a first-order process. To do so, the compactions were plotted as a function of a pertinent timescale expressed as:
 |
(1) |
where f is the time-dependent normalized concentration profile of the proteins, measured ab initio at the longitudinal position x where the studied DNA is tethered, and t is the real experimental time. The fitting of the compensated data gives direct access to the kinetic parameters (see Appendix A2).
Kinetic framework
In vivo (49) and in vitro (50,51) data support a sequential process for histone deposition including at least three steps: 1), (H3-H4)2 are first loaded on the DNA with a kinetic constant k1 (this reaction could involve either dimers of H3-H4 (52) to form a tetramer onto the DNA or a preformed tetramer if it is available, as observed in several reconstitution systems); 2), a first H2A-H2B dimer binds to one of the two available sites on the (H3-H4)2 tetramer/DNA complex with a constant k2; 3), a second H2A-H2B dimer binds to the remaining site (k2′ = k2/2, because only one site among the two initial ones remains available). All these reactions are supposed to be first-order processes.
As demonstrated in Ladoux et al. (30), this model predicts an evolution of DNA length as a sum of exponentials, with time constants depending on the parameters k1 and k2 (see demonstration in Appendix A2). These parameters will be accessible simultaneously only if the three processes depicted above occur on similar timescales. Otherwise, only the kinetically limiting step will be observed and the decay will appear as a single exponential.
RESULTS
Kinetics of assembly and packing ratio achieved in Xenopus egg extracts
We measured the kinetics of DNA compaction as an indication of chromatin assembly with several different dilutions of Xenopus egg extracts (initial histone concentration of ∼80–100 ng/μL), namely 1:50, 1:100, 1:150, and 1:300. The analysis of the real-time compaction on single molecules showed an irregular process with successions of randomly distributed fast and slow assembly sequences (Supplementary Material). The averaged kinetics over a large number of molecules was smoother. The most suitable fit for these averaged data was achieved with a single-step process, as shown in Fig. 2. From the fitting, we evaluated the kinetic constant of the overall assembly reaction to be k = (6.4 ± 2.0) × 107 mol−1 L s−1.
FIGURE 2.

Chromatin assembly kinetics with Xenopus egg extracts appears as a one-step process. Xenopus egg extracts were diluted to 1:50, 1:100, 1:150, and 1:300 (from left to right on the graph) in a buffer containing 1 mM MgCl2 and 0.5 mM ATP. Each curve is fitted with a single exponential, and only one fitting parameter is adjusted for all the curves.
We could also measure the packing ratio, which represents the initial length of the DNA divided by that of the final product at the end of the compaction. Initially, the DNA molecules were stretched to a length of ∼25 μm. After a few seconds, we reproducibly observed a final length of 1 μm, corresponding to the optical resolution of fluorescence microscopy. Thus, we concluded that the packing ratio was at least 25. Interestingly, an order of magnitude for this parameter in physiological chromatin can be obtained from electron microscopy (53): the linear nucleosome density in a 30-nm fiber was found to be six to seven nucleosomes per 11 nm. Assuming that nucleosomes are evenly repeated every 200 bp, 11 nm of a 30-nm fiber should contain ∼1300 bp (200 × 6.5). Since 11 nm of DNA corresponds to 32 bp, we deduce a packing ratio of ∼40 (1300/32), in keeping with our experimental measurements.
Compaction kinetics with a purified nucleosome reconstitution system “histones + yNAP-1”
We used recombinant Xenopus histones with yNAP-1 as a chaperone under conditions that allow full nucleosome reconstitution (0.6:1 yNAP-1/octamer molar ratio), as verified by supercoiling assays (41). We worked with three different concentrations of histones (3, 2, and 1.4 ng /μL). The kinetic curves obtained under these conditions significantly differed from those with the Xenopus egg extracts (Fig. 3 A). The DNA retraction observed in our reaction is thought to reflect nucleosome reconstitution rather than DNA aggregation with positively charged proteins for the following reasons:
FIGURE 3.

Nucleosome reconstitution with native, WT, and tailless recombinant histones with yNAP-1 follows a three-step kinetics. (A) Nucleosome reconstitution with WT recombinant histones for three different concentrations (3, 2, and 1.4 ng/μL) with a yNAP-1/octamer molar ratio of 0.6:1. The curves are fitted with a three-step kinetics model (solid lines). (B) Fitting of the reconstitution curve (3 ng/μL of WT recombinant histones) with a one-step kinetic model (dashed line) or a three-step kinetic model (solid line). (C) Comparison of the reconstitution kinetics with WT recombinant (1), native (2) and tailless recombinant (3) histones. The histone concentration is set at 2 ng/μL and the yNAP-1/octamer molar ratio is 0.6:1.
First, the data could not be fitted with a single exponential, and a three-step mechanism yielded a much better accuracy (Fig. 3 B). The resulting kinetic constants k1 and k2 (see Kinetic framework) were determined (Table 1). In contrast, several studies of DNA condensation by protamine, a model protein containing a large number of positive residues and known to condense DNA in a structureless manner, revealed single exponential kinetics (54). Importantly, we reinvestigated protamine-induced compaction in Bancaud et al. (46) with the same set-up and experimental conditions as those used in our study and obtained results similar to those of Brewer et al. (54).
TABLE 1.
Kinetics constants, assembly rates, and packing ratios measured for the different nucleosome reconstitution systems
| Combination used in the reconstitution assay | k1 106 (s mol/L)−1 | k2 106 (s mol/L)−1 | Assembly rate* (μm s−1) | Packing ratio |
|---|---|---|---|---|
| Recombinant histones + yNAP-1 (1:0.6) | 9 ± 3 | 22 ± 10 | 3.4 ± 0.5 | 7 ± 1 |
| Recombinant histones + yNAP-1 (1:6) | 27 ± 10 | 22 ± 10 | 6 ± 1 | 12 ± 2 |
| Recombinant histones − yNAP-1 | 10 ± 3 | 17 ± 5 | 2.8 ± 0.5 | 8 ± 1 |
| Native histones + yNAP-1 (1:0.6) | 4 ± 2 | 8 ± 2 | 1.5 ± 0.5 | 5.5 ± 0.6 |
| Native histones − yNAP-1 | 2.0 ± 0.8 | 5 ± 1 | 0.7 ± 0.2 | 5.0 ± 0.5 |
| Tailless histones + yNAP-1 (1:0.6) | 0.9 ± 0.4 | 2 ± 1 | 0.5 ± 0.1 | 4.8 ± 0.5 |
| Tailless histones − yNAP-1 | 1.0 ± 0.4 | 2 ± 1 | 0.6 ± 0.1 | 4.8 ± 0.5 |
| Native histones + PGA (1:5) | — | — | 0.010 ± 0.005 | Not accessible |
| Native histones + RNA (1:2) | — | — | 0.02 ± 0.01 | Not accessible |
The assembly rate is defined as the steepest slope of the assembly curves.
Second, we previously carried out experiments in which DNA was exposed to histones purified by acid precipitation and not isolated from reconstituted octamers (55). We observed there a nonregular compaction involving a chaotic sequence of brutal shortening events and pauses likely to correspond to DNA-histone aggregation. This is very different from the regular shortening observed with histones purified from octamers (see Histones and yNAP-1 preparation) and chaperones. The compaction ratio and kinetics obtained with bulk acid-purified histones were also much less reproducible from one molecule to the other. Consequently, the three-step, regular reaction observed in this experiment is unlikely to correspond to a nonspecific interaction or a formation of aggregates.
Third, in experiments with purified histones, the length of the molecules always decreased to a reproducible plateau that was well resolved optically (as seen in Fig. 6, curve 2, for wild-type recombinant histones). Therefore, our direct measure of the packing ratio, namely 7 for recombinant histones (Table 1), is in agreement with the formation of an array of nucleosomes (see Discussion). This result is strikingly different from the condensation induced by protamine, in which the packing ratio exceeds 25 (46).
FIGURE 6.

Packing ratio is >25 with extracts and ∼6–7 with purified proteins. The assembly with extracts (1) at 2 ng/μL yields a compact structure (folding ratio >25). The length of the chromatin fiber assembled with yNAP-1 + WT recombinant (2), yNAP-1 + native (3), and yNAP-1 + tailless recombinant (4) histones, at a concentration of 2 ng/μL, reaches a plateau (seen only for WT recombinant histones because of the time axis truncation) from which we deduce the folding ratio. For native histones, it takes 30 s to reach the final length and for tailless histones 50 s.
Fourth, our experiments were carried out in conditions similar to those used in Ruiz-Carillo et al. (56), in which nucleosome reconstitution was achieved directly at physiological ionic strength. Our characteristic histone dilution (1 ng/μL in octamer) was comparable to the one in the study by Ruiz-Carillo and co-workers (56): ∼0.6 ng/μL injected every minute.
Finally, the single molecule experiments presented here use DNA molecules which are isolated at separated positions on the surface (typically one molecule per 10–100 μm2) and cannot interact with each other. This prevents DNA-DNA attractions mediated by polycations that can lead to macroscopic aggregation in bulk experiments, even when the attractive energy at the level of one nucleosome is weak (our experiment is kinetically equivalent to an infinite dilution of DNA, and it is thus much less prone to aggregation than reactions in solution).
To evaluate whether the nature of the histones could influence kinetics, we also used native histones isolated from Drosophila embryos. The reaction was twice as slow as with recombinant Xenopus histones (Fig. 3 C, curves 1 and 2, and Table 1). Again, these data were rather well fitted with the three-step model, and the constants k1 and k2 could be determined (Table 1). In addition, we measured a packing ratio of 5.5 with Drosophila native histones, inferior to that obtained when using recombinant WT Xenopus histones (Table 1).
The effect of histone tails on kinetics was examined by using recombinant Xenopus histones (H3, H4, H2A, and H2B) lacking their N-terminal tails (tailless histones). We compared the assembly of WT and tailless histones associated with yNAP-1 at a histone concentration of 2 ng/μL. The rate of assembly was dramatically decreased with the tailless histones. It was reduced by a factor of ∼7 as compared to the WT proteins (Fig. 3 C, curves 1 and 3; Table1), and we measured a packing ratio that was 1.5 times smaller (Table 1).
How does yNAP-1 affect the reconstitution?
We then investigated the role of yNAP-1 in the reconstitution kinetics by performing the same experiments without the chaperone. First, when we used native Drosophila histones without yNAP-1, the kinetics was twice as slow as when the chaperone was added at a 0.6:1 yNAP-1/octamer molar ratio, highlighting a facilitated assembly mediated by yNAP-1 (Fig. 4 A and Table 1). Moreover, we observed a slight (∼10%), yet reproducible, increase of the packing ratio in the presence of the chaperone (Table 1). This is consistent with supercoiling assays in which the amount of supercoiling increases with the concentration of yNAP-1 (41). Surprisingly, when WT or tailless recombinant Xenopus histones were used, we found that yNAP-1 at the same concentration (0.6:1 yNAP-1/octamer) did not speed up the assembly nor increase the packing ratio (Fig. 4 B and Table 1). To examine whether yNAP-1 is involved in the nucleosome reconstitution with recombinant Xenopus histones, we added the chaperone in large excess (6:1 yNAP-1/octamer molar ratio). Under this condition, both reaction kinetic constants and packing ratio were increased (Fig. 4 B, dashed line, and Table 1).
FIGURE 4.

Role of yNAP-1 in the kinetics of nucleosome reconstitution. (A) Assembly kinetics with native histones (2 ng/μL) in the presence (diamonds) or absence (dotted line) of yNAP-1. (B) Assembly kinetics with WT recombinant histones (2 ng/μL) in the absence (orange) or presence of yNAP-1 at a molar ratio of 0.6:1 (blue) or 6:1 (dash-dotted line). Assembly kinetics with tailless recombinant histones with yNAP-1 at a molar ratio of 0.6:1 (green) or without yNAP-1 (red).
It is important to note that all the kinetic curves obtained without the chaperone were still properly fitted by a three-step mechanism. Therefore, the arguments that supported a successful reconstitution of nucleosomes in the previous paragraph are still valid when yNAP-1 is not added with histones: 1), we observed a multistep kinetics; 2), we measured a relatively low packing ratio; and 3), we performed the experiments in similar conditions of dilution for the DNA molecules and for the octamers as in Ruiz-Carillo et al. (56), where the authors succeeded in reconstituting nucleosomes without any chaperone. Moreover, supercoiling bulk experiments carried out with the same assembly systems and conditions as ours showed that assembly occurred even when yNAP-1 was not added, but to a lesser extent than with the chaperone (57).
Finally, since negatively charged polyelectrolytes have been reported to facilitate in vitro nucleosome reconstitution, we studied the role of PGA and RNA in the assembly with native histones (2 ng/μL). We found that both PGA and RNA dramatically slowed down the kinetics (Fig. 5, curves 4 and 5, and Table 1). Hence, contrary to yNAP-1, these negatively charged molecules do not assist the reconstitution from the kinetic point of view, notwithstanding the fact that they may indirectly “help” reconstitution in bulk experiments by preventing multimolecular DNA-histone aggregation. The reaction rate was so slow in this case that the decay during the experiment was not large enough to discriminate between a single exponential and multiexponential behavior.
FIGURE 5.

PGA and RNA do not facilitate nucleosome reconstitution. Comparison of the reconstitution with extracts (1) or native histones (2ng/μL) + [yNAP-1 – 0.6:1 molar ratio (2), no chaperone (3), RNA – 2:1 mass ratio (4), PGA – 5:1 mass ratio (5)].
Compaction rate
The compaction rate (defined as the steepest slope of the compaction curves) associated with each reconstitution system is reported in Table 1 (see also Fig. 6). We measured 26.5 μm/s with extracts (1:50) and 3.4, 1.5, and 0.5 μm/s, for WT recombinant, Drosophila native, and tailless recombinant histones, respectively, with yNAP-1 (histones at a concentration of 2 ng/μL). The compaction rate dramatically decreased to 0.01 and 0.02 μm/s in the reconstitution using native histones (2 ng/μL) with PGA or RNA.
DISCUSSION
Nucleosome reconstitution with purified proteins
The kinetics of nucleosome reconstitution with purified proteins can be fitted with a three-step process. Interestingly, the constant k1 (Table1), which is associated with the tetramer deposition step in a kinetic model assuming a first deposition of the (H3-H4)2 tetramer, followed by sequential deposition of two H2A-H2B dimers, seems to be the limiting step with all the purified proteins used. We also find that the assembly is roughly twice as fast with WT recombinant Xenopus histones as with histones purified from Drosophila embryos (native histones). Native histones undergo posttranslational modifications such as acetylation, which neutralizes positive charges carried by the lysine residues of the tail domain. Since recombinant Xenopus histones are expressed in bacteria (which is an organism that does not possess the posttranslational modification activities found in eukaryotic cells), charged residues cannot be posttranslationally modified. Given that histone/DNA interaction in the nucleosome is mainly driven by electrostatics, our result is consistent with the fact that native histones are potentially not as positively charged as WT recombinant Xenopus histones. In addition, this interpretation is strongly supported by our experiment with the tailless recombinant Xenopus histones. These proteins lack the positively charged residues located in the N-terminal tails and thus can be seen from an electrostatic aspect as highly acetylated histones. Indeed the kinetic constants obtained with the tailless recombinant Xenopus histones are much lower than those measured with WT recombinant Xenopus and native Drosophila histones (Table 1).
Recently, chromatin assembly with chicken erythrocyte core histones and yNAP-1 was probed with a magnetic tweezers set-up (31). The role of the tension was inspected and an initial assembly rate at zero force of ∼100 nm/s was deduced for a histone concentration of 10 ng/μL. By extrapolation of our data, we measure much higher assembly rates, namely 17 and 7.5 μm/s, respectively, with WT recombinant and native histones. Although they did not use the same histones (31), we would expect their measurement to be in the same order of magnitude as ours. This discrepancy may arise at least in part from the fact that their buffer solution contained 150 mM NaCl, whereas our experiment was carried out at 50 mM NaCl. Electrostatic interactions were more strongly screened in their system, consistent with a lower assembly rate. Unfortunately, we could not perform our experiment at the same ionic strength as used in the Leuba et al. experiment (31) (which is closer to physiological environment), because the quantum yield of YOYO-1 strongly decreases at high salt concentration. Still, the discrepancy seems too large to be solely due to this factor 3 in the salt concentration, and it may also be due to an experimental artifact: the experimental description in Leuba et al. (31) does not mention the problem of the finite size of the concentration front discussed in the Data treatment section of this article. We determined that neglecting this effect can lead to an underestimate of kinetic rates by a factor of 10 or more, especially for processes occurring in the range of a few seconds to a few tens of seconds. Consequently, this could also be a cause of the apparent discrepancy between their results and ours.
Packing ratio of the assembled structure
Our set-up and the use of dimers of λ-DNA allows us to quantitatively measure the packing ratio obtained with each assembly system. More exactly, we can provide a lower limit of this factor with extracts (25) and measure it precisely with purified systems (between 4.8 and 12). Note that the packing ratio is obtained at very low forces (typically 0.1 pN) because the flow rate remains constant throughout the experiment and the hydrodynamic drag on the assembled fiber is much smaller than on the initial DNA. Previous micromanipulation studies carried out at 1 pN revealed a packing ratio of 8 with Xenopus egg extracts (32). Yet, the sub-pN regime has not been addressed so far in single-molecule studies. The difference between their results and ours suggests that 1 pN is sufficient to significantly decondense native chromatin.
The packing ratio of chromatin strongly depends on the nucleosomal organization and density along the fiber. It is therefore important to give a relevant order of magnitude for this parameter in our experimental conditions (reconstitution with purified systems). Earlier investigations of chromatin architecture in low salt (10 mM NaCl) demonstrated that it formed a 10-nm nucleofilament without 3D organization (58). A comparable structure was found when chromatin assembled in 150 mM NaCl using purified histones with yNAP-1 was imaged by atomic force microscopy (AFM) on λ-DNA (31). Unfortunately, these authors did not evaluate per se the packing ratio of these structures. Nevertheless, using their AFM images obtained with chromatin reconstituted on 5S tandemly repeated positioning sequences (31), it is possible to deduce an order of magnitude for this parameter ranging from R ∼3 to R ∼5. These results must be taken with caution because AFM requires a fixation step, which can significantly alter chromatin folding. However, the point may be that these orders of magnitude seem to be consistent with our experiments. Some of our packing ratios are slightly higher, but this is not unreasonable since the surface fixation process is expected to extend the nucleofilament rather than compact it.
Moreover, we can compute the theoretical packing ratio of an extended nucleofilament by means of two simple geometrical models. First, if we assume that two full turns of DNA are wrapped around each nucleosome, leading to 177 bp of DNA around the histone octamer (59), it becomes clear that each linker DNA contains 23 bp (200 − 177). Considering an entry/exit angle between 120° and 160° (60), the packing ratio should be at least 8.8. It is also possible to imagine that the nucleosomes are “open” (61) and the entry/exit DNAs do not cross. In this case, we suppose that 147 bp are wrapped around the nucleosome and the entry/exit angle remains ∼120°, leading to a lower limit for the packing ratio of 3.8.
Both models and AFM experiments give orders of magnitude for the packing ratios that are consistent with our own measurements. Also, the packing ratios obtained at the end of the assembly in our experiments are very reproducible, providing a further argument in favor of the formation of nucleosome arrays.
Moreover, our experiments also enabled us to compare the packing ratios obtained with different reconstitution systems. The role of core histone N-termini in chromatin folding has already been elucidated in vitro in the case of regularly spaced arrays with long-range interactions: 1), tailless nucleosome fibers were unable to fold extensively (4); and 2), linear hyperacetylated oligonucleosomes appeared in an extended conformation (62). This seems to be in agreement with our results: the global compaction significantly decreases (∼20%) in the presence of posttranslational modifications or even moreso (∼30%) with deletion of the histone tails (Table 1). Furthermore, the addition of yNAP-1 tends to increase the packing ratio in the case of native histones (with a 0.6:1 ratio) and WT recombinant histones (with a 6:1 ratio). This is not surprising since this chaperone favors nucleosome formation, as inferred from supercoiling assays (41,63) in which DNA supercoiling was shown to increase with the amount of NAP-1 used in the assembly reaction. It should be noted, however, that a detailed interpretation of packing ratios is rendered difficult by the fact that it combines two effects: the nucleosome density (average repeat length) and 3D organization. Ideally, it would be desirable to access the two types of information independently, but unfortunately the optical resolution of our experiment is not sufficient to see individual nucleosomes and, conversely, AFM has the desired resolution but involves a surface-fixation step that may alter the 3D structure of the fiber. We thus conclude that our results seem to confirm the current view of the effect of histone modifications and NAP-1-mediated chaperoning on compaction, but should be substantiated with more refined experimental approaches.
Role of yNAP-1
Our set-up enables us to detect quantitative effects of yNAP-1 on the kinetics of nucleosome reconstitution at a yNAP-1/histone octamer ratio of 0.6:1. The relative amount of yNAP-1 is somewhat smaller than that used in McBryant et al. (28) and McQuibban et al. (29), but it is sufficient to obtain nucleosome assembly on plasmid DNA (41). In these conditions, a fraction of the histone octamers is presumably not chaperoned and we anticipate a strong influence on the kinetics of the ratio yNAP-1/octamer. Nevertheless, our study aims at comparing the relative effect of yNAP-1 in the different assembly systems using the same yNAP-1/octamer ratio with each system. Therefore, the uncertainty associated with incomplete chaperoning should be minimized, since we use the same chaperoning system for all histones.
yNAP-1, in a molar ratio of 0.6 per octamer, accelerates nucleosome reconstitution with native Drosophila histones by a factor of 2. When using WT recombinant Xenopus histones, a ratio of 6:1 is required to similarly accelerate the reconstitution reaction. This difference may originate from the nature and the posttranslational modifications of histones. Indeed, quantifications of yNAP-1 interaction with WT recombinant histones showed that two copies bind to one dimer and four to one tetramer (28), whereas complexes with a yNAP-1/octamer molar ratio of 4:1 were formed when using histones purified from chicken erythrocyte (29). These data suggest, then, that yNAP-1 interaction with histones may change with histone modifications and, thus, could partially account for a requirement of 10- fold more yNAP-1 for the same amount of histones to achieve similar acceleration of the reconstitution reaction with nonposttranslationally modified WT Xenopus histones. Both kinetic constants (k1 and k2) are equally affected by yNAP1 when the reconstitution experiment is performed with native Drosophila histones (Table 1). Assuming that these constants represent the tetramer and dimer deposition steps, respectively, this suggests that the chaperone interacts not only with the dimers (H2A-H2B) but also with the tetramer (H3-H4)2, in agreement with several biochemical in vitro assays (28,29,63). Furthermore, the fitting of the kinetics when WT recombinant Xenopus histones are used with a 10-fold excess of yNAP-1 shows that the first constant k1 is strongly increased, in contrast to k2 (Table 1). Therefore, yNAP-1 could act principally on the tetramer deposition step, possibly because it has more affinity for (H3-H4)2, as demonstrated by McBryant et al. (28).
Since yNAP-1 is a highly negatively charged protein (64), its affinity for histones should strongly depend on electrostatics. Nevertheless, competition assays showed that poly-L-lysine and poly-L-arginine do not bind yNAP-1 as tightly as core histones, despite a higher linear charge density (28). Furthermore, we demonstrate that nucleosome reconstitution with native histones and PGA or RNA occurs at a rate two orders of magnitude lower than with yNAP-1 (Table 1). This reveals that PGA and RNA act as “chaperones” in vitro because they strongly screen electrostatic interactions between histones and DNA and avoid massive aggregation. However, they do not seem to accelerate nucleosome reconstitution, in contrast to the chaperone yNAP-1. Thus, the chaperoning effect of yNAP-1 is likely to result from a balance between two opposite effects: 1), physical interaction with histones, and 2), release onto the DNA for nucleosome formation. Consequently, an overly strong interaction between a negatively charged molecule and the core histones could be insufficient to enhance nucleosome assembly, and might even be deleterious by preventing transfer of the histones onto the DNA. Importantly, the effect of PGA and RNA should strongly depend on the ionic strength, and we would expect the kinetic constants to change at higher salt concentration. As previously mentioned, however, experiments at higher ionic strength are not possible in our set-up, at least with the surface treatment, labeling, and grafting strategy we are presently using.
Taken together, these considerations, we would like to suggest the following hypothesis to explain the accelerating effect of yNAP-1. At low ionic strength and in our protein concentration range, >99% of the tetramers (H3-H4)2 are dissociated into dimers (H3-H4). Since the free energy of formation of (H3-H4)2 is measured to be ΔG° = −7.2 kcal/mol (1), the resultant association constant is ∼1.105 M−1, consistent with the value 5.104 M−1 in Sperling and Wachtel (65). As yNAP-1 is known to interact with (H3-H4)2, it may stabilize the tetrameric form. The formation of tetrasomes (DNA-(H3-H4)2 complex) should then be accelerated when the chaperone is present. Furthermore, the interaction of yNAP-1 with the (H2A-H2B) dimers tends to lower the nonspecific interactions of (H2A-H2B) with DNA, leading to a faster formation of the nucleosomal structure. These combined effects may increase the kinetic constants and the packing ratio as they both facilitate nucleosome reconstitution.
Chromatin assembly with Xenopus egg extracts
The optimization of the single-molecule assembly assay described here with Xenopus egg extracts reveals that the kinetics is 12 times faster than previously reported (30). Under the improved experimental set-up (observation of the very early steps of the compaction, low tension, and no torsional constraint on the molecule), the averaged compaction curve appears to follow a one-step kinetic scheme. This contrasts with the three-step process previously proposed, although we did also observe here a more irregular behavior on individual compaction events. Consequently, the observed compaction is probably not the consequence of a simple, one-step stochastic process. Rather, the kinetics may be dominated by a series of stochastically distributed rate-limiting steps, resulting in an apparent single-exponential decrease only when averaged.
Histones, in any case, are a key component of the observed retraction, since no compaction occurred when histones and assembly capacity were titrated out (30). Moreover, thanks to the use of λ-phage DNA dimers, we could measure more accurately the packing ratio, and find a value of at least 25 for chromatin assembled in extracts, which is compatible with the formation of a higher-order structure. Finally, the AFM experiments reported in Ladoux et al. (30) indicate that, in conditions similar to those used in this work, the first step of compaction is the assembly of a “beads on a string” array of nucleosome-like particles, and that this step is followed by the formation of a more compact array.
Extracts contain numerous assembly factors that facilitate chromatin formation (50). In the purified systems, we demonstrated that yNAP-1 accelerates the kinetics. Consequently, since the cell must assemble chromatin rapidly and evenly, we would expect that the presence in cellular extracts of several chaperones and of a much more complete subset of proteins could account for the 10 times faster compaction and the increased packing ratio compared to the purified systems. One may also expect that other chromatin-associated proteins such as high-mobility group proteins (66) are likely to contribute to this high packing ratio, since they are known to be present in large amounts in Xenopus egg extracts and to be involved in the formation of a higher-order organization.
More experimental work combining physical and biological approaches will be necessary to progress further in the understanding of the compacting mechanism, particularly in cellular extracts.
CONCLUSIONS AND PROSPECTIVE REMARKS
We have quantitatively characterized chromatin assembly kinetics with different systems at rates extending down to ∼1 s, a resolution not accessible using conventional biochemical assays. We highlight differences in assembly rates of up to three orders of magnitude with comparable histone concentrations, depending on the cofactor and on the nature of histones used. Our experiments confirm that yNAP-1 tends to facilitate nucleosome formation. Nonetheless, its function in our assay cannot be explained solely in terms of charge screening.
NAP-1 was found to interact with the dimers (H2A-H2B) in vivo and to participate in the assembly of nucleosomes. However, it has been demonstrated that this chaperone can also favor the release of (H2A-H2B) from the nucleosomal structure (67). Obviously, all the modes of action of this chaperone observed in our in vitro experiment may not be relevant to in vivo situations, where other assembly and regulation factors are at play. In any case, the exact mechanism of action of NAP-1 in mediating the interaction of core histones with DNA still deserves further investigation and clarification, in particular in the cellular context. Our set-up could help to gain further insight in this area. For instance, one can contemplate the study of specific mutations of yNAP-1 and of their impact on nucleosome reconstitution kinetics. Importantly, the function of other histone chaperones (11) could be tested and compared with this set-up. The precise role of each histone tail could also be evaluated within the kinetic approach described here with specific mutations of the amino acid residues, mimicking posttranscriptional modifications such as acetylation or phosphorylation.
Finally, this work also raises new challenging questions about assembly in cellular extracts. In our experiments, packing ratios compatible with higher levels of chromatin organization, such as the “30-nm fiber”, are reached within a few seconds. Assembly of DNA into such a complex and regular organization within that amount of time would be surprising. In fact, the rapid collapse could be interpreted as a condensation into a “soft” state, in which the further reorganization necessary to yield a highly organized chromatin could be performed efficiently with the help of chromatin remodeling factors, aided by the high chromatin density (in a process reminiscent of the old idea of the “molten globule” for protein folding). Testing this idea, however, will be an experimental challenge; it will require further improvement in molecular imaging to reach the necessary spatial and temporal resolutions. We hope that future developments will enable us to achieve this ultimate goal.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Prof. P. Becker for critical comments and Pascal Silberzan for the use of the clean room at the Institut Curie.
A.B. and G.W. thank the French Ministry of Research and Technology for the predoctoral fellowship Allocation Couplée. This work was partly supported by the Centre National de la Recherche Scientifique, Ministère de l'Éducation Nationale, de la Recherche et de la Technologie ACI Dynamique et Réactivité des Assemblages Biologiques and Institut Curie cooperative program Physics of the Cell. The laboratory of G.A. is supported by la Ligue Nationale contre le Cancer.
APPENDIX A1: PROBING THE SURFACE CONCENTRATION
Experimental strategy
We use as a fluorescent probe casein coupled to fluorescein (Sigma). This makes the protein concentration easy to measure in our experiment. In addition, this protein is oligomeric, and should have a diffusion constant comparable with that of chaperoned histones. Its excitation wavelength is 490 nm and emission is at 540 nm. To probe the local concentration close to the surface, we add a nonfluorescent dye, Orange G (Sigma), strongly absorbing at the excitation wavelength (490 nm) and weakly at the emission one (540 nm) (46). When a solution of fluorescent casein is in the field of the objective, the intensity collected by the detector, 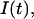 is the convolution of the concentration of fluorescent proteins with the Beer-Lambert absorption profile,
is the convolution of the concentration of fluorescent proteins with the Beer-Lambert absorption profile,
 |
(2) |
with R a numerical factor accounting for the quantum yield of the fluorophore, c(z,t) the local concentration of fluorescent casein, and ζ the penetration length of the excitation beam.
When ζ was <5 μm, we demonstrated that Eq. 2 could be simplified (46):
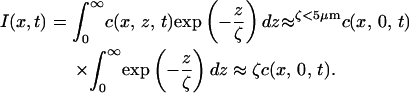 |
(3) |
Since the intensity is linearly related to the concentration at the walls, we have a fairly accurate measure of protein surface concentration.
From one experiment to another, the DNA molecules are located at different positions downstream from the T-junction. We thus recorded the concentration profiles at different positions along the channel (from 2 to 4.5 mm from the T-junction) to measure protein arrival for any experimental situation.
APPENDIX A2: KINETIC MODELING
The raw kinetics are strongly biased by the longitudinal position along the channel (Fig. 7 A). We expect the true biological processes to be independent of this position and we will now demonstrate that the surface concentration is responsible for this artifact.
FIGURE 7.

(A) Raw compaction curves obtained with extracts at two different positions along the channel (solid circles, x = 2 mm; shaded crosses, x = 3 mm). The two curves appear different because the extracts take more time to arrive in the vicinity of the molecule located further in the channel. The plots of the corresponding casein concentration profiles are given as dotted lines (x = 2 mm, solid; x = 3 mm, shaded). (B) The same compaction curves as in A after data treatment: the normalized length is plotted versus the pertinent time τ (for the solid curve, τ(2 mm,t) ; for the shaded curve, τ(3 mm,t)).
Incorporating the concentration front into the kinetics
In the following, we assume that the assembly is a single-step reaction where A and B react to produce C (Eq. 4) (this result can be generalized to more complex kinetic framework, see below).
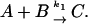 |
(4) |
In our experiment, A is the fraction of naked DNA, immobilized on the surface at the longitudinal position x, and B are the flowing proteins that compact DNA and whose concentration is time-dependent. We define very generally f(x,t) as the normalized concentration of B, i.e., 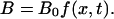 Finally,
Finally, 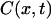 is the fraction of the molecule that has been condensed (ratio of DNA bp involved in chromatin versus total number of bp).
is the fraction of the molecule that has been condensed (ratio of DNA bp involved in chromatin versus total number of bp).
The evolution of the concentration of A is given by the kinetic equation
 |
(5) |
Since the DNA molecule is not compacted when the experiment begins, the initial conditions read 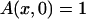 and
and  It becomes
It becomes 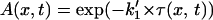 with
with  and
and 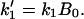 Moreover, the fraction of the molecule compacted at time t is given by
Moreover, the fraction of the molecule compacted at time t is given by
 |
(6) |
Let us now examine the relationship between the observed retraction of the molecule and 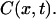 During the assembly, the length of a DNA located at the longitudinal position x can be written as
During the assembly, the length of a DNA located at the longitudinal position x can be written as
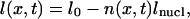 |
(7) |
where l0 is the initial length, n(x,t) the number of assembled nucleosomes, and lnucl the length compacted in one nucleosome. Since the compaction is associated with nucleosome formation, we have n(x,t) = C(x,t) × nf, with nf the number of nucleosomes assembled at the end of the reaction.
Finally, we should introduce the inverse of the packing ratio of the assembled structure, defined as 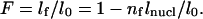 Therefore, combining Eqs. 6 and 7, it becomes
Therefore, combining Eqs. 6 and 7, it becomes
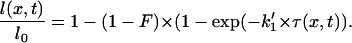 |
(8) |
Hence, the decrease of DNA length is a function of the “pertinent” timescale τ(x,t) multiplied by the kinetic parameter k1′.
Finally, to directly take the concentration front into account in the fit, we define a new function g (46), representing the normalized length of the molecule as a function of τ(x,t), which depends only on the kinetic parameters:
 |
(9) |
As shown in Fig. 7 A, the raw kinetics appear very different at 2 and 3 mm from the T-junction. The two curves collapse (Fig. 7 B) when we plot g as a function of the pertinent timescale, and the fit of the function g gives access to the kinetic parameter  (46).
(46).
Assembly kinetics with purified proteins
We consider a three-step model, in which a tetramer is first deposited, with kinetic constant k1, and two dimers are then deposited sequentially on two equivalent sites, with kinetic constant k2 (see Kinetic framework section; 10,30). Applying Eq. 6 to this model, one can demonstrate that
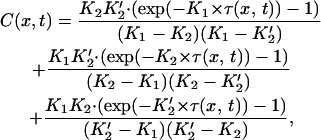 |
(10) |
with K1 = k1 × [(H3-H4)2]max, K2 = k2 × [(H2A-H2B)]max, and K2′ = K2/2.
Eqs. 8 and 10 hold that the packing ratio, as well as the kinetic parameters, can be deduced from fitting the data.
References
- 1.Van Holde, K. E. 1988. Chromatin. Springer, New York.
- 2.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science. 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 3.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Nature. 403:41–45. [DOI] [PubMed] [Google Scholar]
- 4.Carruthers, L. M., and J. C. Hansen. 2000. The core histone N termini function independently of linker histones during chromatin condensation. J. Biol. Chem. 275:37285–37290. [DOI] [PubMed] [Google Scholar]
- 5.Dorigo, B., T. Schalch, K. Bystricky, and T. J. Richmond. 2003. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 327:85–96. [DOI] [PubMed] [Google Scholar]
- 6.Randall, S. K., and T. J. Kelly. 1992. The fate of parental nucleosomes during SV40 DNA replication. J. Biol. Chem. 267:14259–14265. [PubMed] [Google Scholar]
- 7.Gruss, C., J. Wu, T. Koller, and J. M. Sogo. 1993. Disruption of the nucleosomes at the replication fork. EMBO J. 12:4533–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolffe, A. P. 1998. Chromatin Structure and Function. Academic Press, San Diego.
- 9.Krude, T. 1999. Chromatin replication: finding the right connection. Curr. Biol. 9:394–396. [DOI] [PubMed] [Google Scholar]
- 10.Kaufman, P. D., and G. Almouzni. 2000. DNA replication, nucleotide excision repair, and nucleosome assembly. In Chromatin Structure and Gene Expression. Frontiers in Molecular Biology, 2nd ed. J. Workman and S. Elgin, editors. Oxford University Press, Oxford. UK, 24–48
- 11.Loyola, A., and G. Almouzni. 2004. Histone chaperones, a supporting role in the limelight. Biochim. Biophys. Acta. 1677:3–11. [DOI] [PubMed] [Google Scholar]
- 12.Ito, T. 2003. Nucleosome assembly and remodelling. Curr. Top. Microbiol. Immunol. 274:1–22. [DOI] [PubMed] [Google Scholar]
- 13.Verreault, A. 2003. Histone deposition at the replication fork: a matter of urgency. Mol. Cell. 11:283–284. [DOI] [PubMed] [Google Scholar]
- 14.Tyler, J. K. 2002. Chromatin assembly. Cooperation between histone chaperones and ATP-dependent nucleosome remodeling machines. Eur. J. Biochem. 269:2268–2274. [DOI] [PubMed] [Google Scholar]
- 15.Stein, A., J. P. Whitlock, and M. Bina. 1979. Acidic polypeptides can assemble both histones and chromatin in vitro at physiological ionic strength. Proc. Natl. Acad. Sci. USA. 76:5000–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson, T., R. Wiegand, and D. Brutlag. 1981. Ribonucleic acid and other polyanions facilitate chromatin assembly in vitro. Biochemistry. 20:2594–2601. [DOI] [PubMed] [Google Scholar]
- 17.Sobolewski, C. H., H. H. Klump, and G. G. Lindsey. 1993. A novel nucleosome assembly procedure (with a little help from pectin). FEBS Lett. 318:27–29. [DOI] [PubMed] [Google Scholar]
- 18.Laskey, R. A., A. D. Millis, and N. R. Morris. 1977. Assembly of SV40 chromatin in a cell-free system from Xenopus eggs. Cell. 10:237–243. [DOI] [PubMed] [Google Scholar]
- 19.Almouzni, G., and M. Méchali. 1988. Assembly of spaced chromatin promoted by DNA synthesis in extracts from Xenopus eggs.. EMBO J. 7:665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson, T., T. S. Hsieh, and D. Brutlag. 1979. Extracts of Drosophila embryos mediate chromatin assembly in vitro. Proc. Natl. Acad. Sci. USA. 11:5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soeller, W. C., S. J. Poole, and T. Kornberg. 1988. In vitro transcription of the Drosophila engrailed gene. Genes Dev. 2:68–81. [DOI] [PubMed] [Google Scholar]
- 22.Becker, P. B., and C. Wu. 1992. Cell-free system for assembly of transcriptionally repressed chromatin from Drosophila embryos. Mol. Cell. Biol. 12:2241–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fyodorov, D. V., and J. T. Kadonaga. 2003. Chromatin assembly in vitro with purified recombinant ACF and NAP-1. Methods Enzymol. 371:499–515. [DOI] [PubMed] [Google Scholar]
- 24.Ito, T., M. Bulger, R. Kobayashi, and J. T. Kadonaga. 1996. Drosophila NAP-1 is a core histone chaperone that functions in ATP-facilitated assembly of regularly spaced nucleosomal arrays. Mol. Cell. Biol. 16:3112–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang, L., S. S. Loranger, C. Mizzen, S. G. Ernst, C. D. Allis, and A. T. Annunziato. 1997. Histones in transit: cytosolic histone complexes and diacetylation of H4 during nucleosome assembly in human cells. Biochemistry. 36:469–480. [DOI] [PubMed] [Google Scholar]
- 26.Ishimi, Y., M. Kojima, M. Yamada, and F. Hanaoka. 1987. Binding mode of nucleosome-assembly protein (AP-I) and histones. Eur. J. Biochem. 162:19–24. [DOI] [PubMed] [Google Scholar]
- 27.Ishimi, Y., and A. Kikuchi. 1991. Identification and molecular cloning of yeast homolog of nucleosome assembly protein I which facilitates nucleosome assembly in vitro. J. Biol. Chem. 266:7025–7029. [PubMed] [Google Scholar]
- 28.McBryant, S. J., Y. J. Park, S. M. Abernathy, P. J. Laybourn, J. K. Nyborg, and K. Luger. 2003. Preferential binding of the histone (H3–H4)2 tetramer by NAP1 is mediated by the amino-terminal histone tails. J. Biol. Chem. 278:44574–44583. [DOI] [PubMed] [Google Scholar]
- 29.McQuibban, G. A., C. N. Commisso-Cappelli, and P. N. Lewis. 1998. Assembly, remodeling, and histone binding capabilities of yeast nucleosome assembly protein 1. J. Biol. Chem. 273:6582–6590. [DOI] [PubMed] [Google Scholar]
- 30.Ladoux, B., J.-P. Quivy, P. Doyle, O. du Roure, G. Almouzni, and J.-L. Viovy. 2000. Fast kinetics of chromatin assembly revealed by single-molecule videomicroscopy and scanning force microscopy. Proc. Natl. Acad. Sci. USA. 97:14251–14256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leuba, S. H., M. A. Karymov, M. Tomschik, R. Ramjit, P. Smith, and J. Zlatanova. 2003. Assembly of single chromatin fibers depends on the tension in the DNA molecule: magnetic tweezers study. Proc. Natl. Acad. Sci. USA. 100:495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennink, M. L., L. H. Pope, S. H. Leuba, B. G. de Grooth, and J. Greve. 2001. Single chromatin assembly using optical tweezers. Single Mol. 2:91–97. [Google Scholar]
- 33.Cui, Y., and C. Bustamante. 2000. Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc. Natl. Acad. Sci. USA. 97:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brower-Toland, B. D., C. L. Smith, R. C. Yeh, J. T. Lis, C. L. Peterson, and M. D. Wang. 2002. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl. Acad. Sci. USA. 99:1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Almouzni, G. 1998. Assembly of chromatin and nuclear structures in Xenopus egg extracts. In Chromatin. A Practical Approach. H. Gould, editor. Oxford University Press, New-York. 195–218.
- 36.Gaillard, P. H., D. Roche, and G. Almouzni. 1999. Nucleotide excision repair coupled to chromatin assembly. Methods Mol. Biol. 119:231–243. [DOI] [PubMed] [Google Scholar]
- 37.Simon, R. H., and G. Felsenfeld. 1979. A new procedure for purifying histone pairs H2A + H2B and H3 + H4 from chromatin using hydroxylapatite. Nucleic Acids Res. 6:689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luger, K., T. J. Rechsteiner, A. J. Flaus, M. M. Waye, and T. J. Richmond. 1997. Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 272:301–311. [DOI] [PubMed] [Google Scholar]
- 39.Luger, K., T. J. Rechsteiner, and T. J. Richmond. 1999. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304:3–19. [DOI] [PubMed] [Google Scholar]
- 40.Luger, K., and T. J. Richmond. 1998. DNA binding within the nucleosome core. Curr. Opin. Struct. Biol. 8:33–40. [DOI] [PubMed] [Google Scholar]
- 41.Clapier, C. R., G. Langst, D. F. Corona, P. B. Becker, and K. P. Nightingale. 2001. Critical role for the histone H4 N terminus in nucleosome remodeling by ISWI. Mol. Cell. Biol. 21:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stein, A. 1989. Reconstitution of chromatin from purified components. Methods Enzymol. 170:585–603. [DOI] [PubMed] [Google Scholar]
- 43.Perret, E., A. Leung, A. Morel, H. Feracci, and P. Nassoy. 2002. Versatile decoration of glass surfaces to probe individual protein protein interaction and cellular adhesion. Langmuir. 18:846–854. [Google Scholar]
- 44.Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 397:50–53. [DOI] [PubMed] [Google Scholar]
- 45.Sofia, S. J., V. Premnath, and E. W. Merril. 1998. Poly(ethyleneoxide) grafted to silicon surfaces: grafting density and protein adsorption. Macromolecules. 31:5059–5070. [DOI] [PubMed] [Google Scholar]
- 46.Bancaud, A., G. Wagner, K. D. Dorfman, and J.-L. Viovy. 2005. Measurement of the surface concentration for bioassay kinetics in microchannels. Anal. Chem. 77:833–839. [DOI] [PubMed] [Google Scholar]
- 47.Whitesides, G., E. Ostuni, T. Shuichi, J. Xingyu, and D. Ingber. 2001. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 3:335–373. [DOI] [PubMed] [Google Scholar]
- 48.Doyle, P. S., B. Ladoux, and J.-L. Viovy. 2000. Dynamics of a tethered polymer in shear flow. Phys. Rev. Lett. 84:4769–4772. [DOI] [PubMed] [Google Scholar]
- 49.Worcel, A., S. Han, and M. L. Wong. 1978. Assembly of newly replicated chromatin. Cell. 15:969–977. [DOI] [PubMed] [Google Scholar]
- 50.Dilworth, S. M., S. J. Black, and R. A. Laskey. 1987. Two complexes that contain histones are required for nucleosome assembly in vitro: role of nucleoplasmin and N1 in Xenopus egg extracts. Cell. 51:1009–1018. [DOI] [PubMed] [Google Scholar]
- 51.Ruiz-Carrillo, A., and J. L. Jorcano. 1978. Nucleohistone assembly: sequential binding of histone H3–H4 tetramer and histone H2A–H2B dimer to DNA. Cold Spring Harb. Symp. Quant. Biol. 42:165–170. [DOI] [PubMed] [Google Scholar]
- 52.Tagami, H., D. Ray-Gallet, G. Almouzni, and Y. Nakatani. 2004. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 116:51–61. [DOI] [PubMed] [Google Scholar]
- 53.Gerchman, S. E., and V. Ramakrishnan. 1987. Chromatin higher-order structure studied by neutron scattering and scanning transmission electron microscopy. Proc. Natl. Acad. Sci. USA. 84:7802–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brewer, L. R., M. Corzett, and R. Balhorn. 1999. Protamine-induced condensation and decondensation of the same DNA molecule. Science. 286:120–123. [DOI] [PubMed] [Google Scholar]
- 55.Ladoux, B. 2000. Etude par vidéomicroscopie de fluorescence de molécules individuelles d'intérêt biologique en interaction. PhD thesis, Universite Paris VII, Paris, France.
- 56.Ruiz-Carrillo, A., J. L. Jorcano, G. Eder, and R. Lurz. 1979. In vitro core particule and nucleosome assembly at physiological ionic strength. Proc. Natl. Acad. Sci. USA. 76:3284–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clapier, C. 2001. Déterminants critiques pour la reconnaissance du substrat par l'ATPase de remodelage de la chromatine ISWI. PhD thesis, Université Joseph Fourier, Grenoble, France.
- 58.Leuba, S. H., G. Yang, C. Robert, B. Samori, K. van Holde, J. Zlatanova, and C. Bustamante. 1994. Three-dimensional structure of extended chromatin fibers as revealed by tapping-mode scanning force microscopy. Proc. Natl. Acad. Sci. USA. 91:11621–11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zlatanova, J., S. H. Leuba, and K. van Holde. 1998. Chromatin fiber structure: morphology, molecular determinants, structural transitions. Biophys. J. 74:2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leuba, S. H., C. Bustamante, K. van Holde, and J. Zlatanova. 1998. Linker histone tails and N-tails of histone H3 are redundant: scanning force microscopy studies of reconstituted fibers. Biophys. J. 74:2830–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Lucia, F., M. Alilat, A. Sivolob, and A. Prunell. 1999. Nucleosome dynamics. III. Histone tail-dependent fluctuation of nucleosomes between open and closed DNA conformations. Implications for chromatin dynamics and the linking number paradox. A relaxation study of mononucleosomes on DNA minicircles. J. Mol. Biol. 285:1101–1119. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Ramirez, M., C. Rocchini, and J. Ausio. 1995. Modulation of chromatin folding by histone acetylation. J. Biol. Chem. 30:17923–17928. [DOI] [PubMed] [Google Scholar]
- 63.Nakagawa, T., M. Bulger, M. Muramatsu, and T. Ito. 2001. Multistep chromatin assembly on supercoiled plasmid DNA by nucleosome assembly protein-1 and ATP-utilizing chromatin assembly and remodeling factor. J. Biol. Chem. 276:27384–27391. [DOI] [PubMed] [Google Scholar]
- 64.Fujii-Nakata, T., Y. Ishimi, A. Okuda, and A. Kikuchi. 1992. Functional analysis of nucleosome assembly protein, NAP-1. The negatively charged COOH-terminal region is not necessary for the intrinsic assembly activity. J. Biol. Chem. 267:20980–20986. [PubMed] [Google Scholar]
- 65.Sperling, R., and E. J. Wachtel. 1981. The histones. Adv. Protein Chem. 34:1–60. [DOI] [PubMed] [Google Scholar]
- 66.Nightingale, K., S. Dimitrov, R. Reeves, and A. P. Wolffe. 1996. Evidence for a shared structural role for HMG1 and linker histones B4 and H1 in organizing chromatin. EMBO J. 15:548–561. [PMC free article] [PubMed] [Google Scholar]
- 67.Ito, T., T. Ikehara, T. Nakagawa, W. L. Kraus, and M. Muramatsu. 2000. p300-mediated acetylation facilitates the transfer of histone H2A–H2B dimers from nucleosomes to a histone chaperone. Genes Dev. 14:1899–1907. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.