

Abstract
As an aggressive pathogen, Staphylococcus aureus poses a significant public health threat and is becoming increasingly resistant to currently available antibiotics, including vancomycin, the drug of last resort for gram-positive bacterial infections. S. aureus with intermediate levels of resistance to vancomycin (vancomycin-intermediate S. aureus [VISA]) was first identified in 1996. The resistance mechanism of VISA, however, has not yet been clarified. We have previously shown that cell wall thickening is a common feature of VISA, and we have proposed that a thickened cell wall is a phenotypic determinant for vancomycin resistance in VISA (L. Cui, X. Ma, K. Sato, et al., J. Clin. Microbiol. 41:5-14, 2003). Here we show the occurrence of an anomalous diffusion of vancomycin through the VISA cell wall, which is caused by clogging of the cell wall with vancomycin itself. A series of experiments demonstrates that the thickened cell wall of VISA could protect ongoing peptidoglycan biosynthesis in the cytoplasmic membrane from vancomycin inhibition, allowing the cells to continue producing nascent cell wall peptidoglycan and thus making the cells resistant to vancomycin. We conclude that the cooperative effect of the clogging and cell wall thickening enables VISA to prevent vancomycin from reaching its true target in the cytoplasmic membrane, exhibiting a new class of antibiotic resistance in gram-positive pathogens.
Since the emergence of methicillin-resistant Staphylococcus aureus (MRSA) in 1961, options for treatment of S. aureus infection have been significantly limited. Vancomycin remained the last resort for MRSA treatment until recent years. Therefore, the emergence of vancomycin-intermediate S. aureus (VISA) with a vancomycin MIC of 8 mg/liter in 1996 (23) evoked great concern among health care workers around the world. VISA is becoming prevalent worldwide: in Japan (23), France (8, 38), the United States (4, 43, 45), South Korea (26), the United Kingdom (25, 37), South Africa (13), Brazil (36), Greece (47), Germany (39), and China (31). The mechanism of resistance of VISA was not associated with any extant resistance genes and has been the subject of intensive research.
Vancomycin is a relatively large glycopeptide antibiotic (molecular weight, 1,485.7) derived from Nocardia orientalis (formerly known as Streptomyces orientalis) (1). It is active against most gram-positive bacteria, including streptococci, staphylococci, corynebacteria, clostridia, listeriae, and Bacillus species. Vancomycin does not interact with or block any enzyme involved in cell wall synthesis as do beta-lactam antibiotics; it physically blocks the important substrates for cell wall-synthesizing machinery, i.e., the d-alanyl-d-alanine residue (DDR) of lipid II precursor. Thereby, it inhibits utilization of the substrates by glycosyltransferase (a cell wall synthesis enzyme) to produce the nascent peptidoglycan chain (20). However, besides the lipid II murein monomer precursors, which are the real targets of vancomycin, the cell wall peptidoglycan of a single S. aureus cell is known to possess about 6.0 × 106 DDRs (19) to which vancomycin molecules could bind while penetrating the peptidoglycan layers. Thus, the residues in the cell wall serve as false targets of vancomycin, making vancomycin a somewhat inefficient drug in terms of maintaining an efficacious concentration around its real targets.
A transposon carrying a unique set of genes for vancomycin resistance has been identified in vancomycin-resistant enterococci (VRE) (50). In the presence of the vanA gene transposon Tn1546, enterococci can replace the DDR of peptidoglycan with d-Ala-d-Lac to prevent their cell wall components from being bound to vancomycin (7). In 2002, two highly vancomycin resistant S. aureus strains were isolated from U.S. patients. These strains had vancomycin MICs of 1,024 and 32 mg/liter and were found to have acquired the vanA gene transposon from a coexisting VRE strain (5, 6). However, in the case of VISA strains, neither the vanA gene nor its homologues have been identified (17, 22). Instead, a change in cell physiology due to accumulation of mutations has been implicated in their resistance (15, 21, 24). Many researchers have studied different VISA strains and reported altered expression of genes such as pbp2 (40), pbpD (14, 42), sigB (2, 17), ddh (3, 33), tcaA (32), and vraSR (28). Several sets of up- and down-regulated genes associated with vancomycin resistance, including regulators, have also been reported (10, 28, 34, 48). However, none of the genes could be attributed to VISA strains specifically. Despite this diversity in transcription profiling and uncertainties about the genes’ role in raising vancomycin resistance, our previous studies using electron microscopy revealed that all the VISA clinical strains have thickened cell walls as a common feature (9, 11, 17, 26, 36). Therefore, our current concern is whether this phenotypic feature, a thickened cell wall, is responsible for vancomycin resistance. We demonstrate here that it does contribute to resistance through a novel mechanism, i.e., by reducing the diffusion coefficient of vancomycin penetration through the cell wall peptidoglycan layers. The present study also proves the existence of cell wall clogging with cell wall-bound vancomycin in peptidoglycan layers, which we suggested soon after our characterization of a significant correlation between cell wall thickness and the level of vancomycin resistance in 2000 (12).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Strain Mu50 is a VISA strain isolated from a patient who responded poorly to vancomycin therapy (23). Strain MI is the second clinical VISA reported after Mu50 from the United States (45). All cultures were grown in brain heart infusion (BHI) broth (Difco, Detroit, MI) at 37°C with aeration, if not otherwise indicated. For each experiment, an overnight culture was diluted 100-fold in prewarmed fresh BHI broth and further incubated with aeration to ensure exponential-growth conditions before sampling. Cell growth was monitored by measuring the optical density of the culture at 600 nm (OD600) with a spectrophotometer (Phamacia LKB Biotechnology, Inc., Uppsala, Sweden).
Preparation of cells with different cell wall thicknesses and determination of vancomycin susceptibility.
Cells with different cell wall thicknesses were prepared as described previously (12). Briefly, Mu50 and MI cells were cultivated in BHI broth to an OD600 of 0.7 to 1.0 and washed twice with RMg- medium (a modified resting medium in which glucose was omitted from the reported ingredients) at 4,000 × g for 10 min to remove the BHI components. Resting medium (RM) (1 mM glycine, 1 mM d-glutamic acid, 0.5 mM dl-alanyl-dl-alanine, 0.3 mM dl-lysine, 1 mM MgCl2, 0.1 mM MnCl2, 0.17 mM uracil, 0.0082 mM nicotinamide, 80 mM K2HPO4, 0.003 mM thiamine, 28.8 mM glucose [pH 7.4]), initially reported by G. D. Shockman et al., was designed as a cell wall-synthetic medium that would not allow cell multiplication (41). To generate cells with different cell wall thicknesses, equal portions of the washed cells were further incubated separately in the following media: RM, RMg-, RMg- containing 30 mM l-glutamine or GlcNAc, and RM containing 30 mM l-glutamine. Incubation was performed at 37°C with gentle shaking (40 rpm) for 2 h. RM contains all the cell wall component amino acids and glucose as a carbon source, allowing cells to synthesize nascent peptidoglycan chains but preventing cell multiplication, while RMg- allows neither de novo cell wall biosynthesis nor cell multiplication (12). GlcNAc was used instead of glucose as a carbon source in some cases. l-Glutamine is one of the factors influencing cell wall thickness (12). The cell preparations resulting from this procedure were used to check cell wall thickness and vancomycin susceptibility. Cell wall thickness was examined by transmission electron microscopy (see Fig. 1A). To evaluate vancomycin susceptibility, the cell preparations were spun down and pellets were resuspended in prewarmed BHI medium in the presence or absence of 30 mg/liter vancomycin. Susceptibility was then judged by measuring the time required for regrowth (TRG) in the presence of vancomycin from growth curves recorded by a photorecording incubator (TN-261; ADVANTEC, Tokyo, Japan) at 37°C with 20-rpm shaking (12).
FIG. 1.

Thickened cell wall and level of vancomycin resistance. (A) Transmission electron microscopy images of representative cells with thin and thick cell walls prepared as described previously (12). The cell wall thickness, in nanometers (mean ± SD), is given under each image. (B and C) Growth curves in BHI medium without vancomycin (B) and with vancomycin at 30 mg/liter (C). Growth curves were recorded by measuring the OD600 with a photorecording incubator (TN-261; ADVANTEC, Tokyo, Japan) at 37°C. Vancomycin susceptibility was determined by measuring the time required for regrowth in BHI medium (12). No difference was seen between growth rates of cells with thin versus thick cell walls in the absence of vancomycin (B). In the presence of vancomycin, cells with thin cell walls showed delayed growth compared to cells with thick cell walls (C).
Transmission electron microscopy.
Preparation and examination of S. aureus cells by transmission electron microscopy were performed as described previously (12). Cells were morphometrically evaluated for cell wall thickness by using photographic images at a final magnification of ×30,000. Thirty cells of each strain with nearly equatorial cut surfaces were measured for cell wall thickness examination. The cell wall thickness was expressed as a mean ± standard deviation (SD).
Vancomycin bioassay and HPLC assay.
The microbiological assay (bioassay) for determination of the vancomycin concentration in the culture medium is carried out by a paper disk diffusion method with Bacillus subtilis strain ATCC 6633 as the indicator organism as described previously (12). The bioassay is calibrated by using reversed-phase high-performance liquid chromatography (HPLC) when necessary. In the HPLC assay, the column (CLC-NH2; particle diameter, 5 μm; pore diameter, 10 nm; inner diameter, 6 mm; length, 15 cm) is eluted with a linear gradient of 50% to 70% acetonitrile in 50 mM sodium phosphate (pH 2.8) containing 0.0005% sodium azide, with a 2-ml/min flow rate, within 20 min. The vancomycin peak is detected by absorption at 240 nm.
Determination of membrane-bound vancomycin levels.
Mu50 cell preparations with thin (thickness, 31.03 ± 3.27 nm; prepared with RMg-) and thick (thickness, 53.29 ± 5.21 nm; prepared with RM) cell walls (Fig. 1A) were used to determine membrane-bound vancomycin levels. Cells that were sampled at 0, 2.5, 5, 15, 30, and 60 min after exposure to vancomycin (at 30 mg/liter) in RMg- medium were washed twice with fresh RMg- medium on 0.45-μm filter membranes and used for protoplast preparation to extract the pure cytoplasmic membrane. Filter membranes with harvested cells were moved to 6 ml of digestion buffer (30% raffinose in 0.05 M Tris [pH 7.5] with 0.145 M NaCl) containing 1 mg of lysostaphin (Sigma Chemical Co., St. Louis, MO), 100 mg of DNase (Worthington Diagnostics, Freehold, NJ), and 1 mM phenylmethylsulfonyl fluoride. Cell mixtures were incubated for 30 min at 37°C with gentle shaking. Protoplasts were harvested by centrifugation at 1,000 × g for 30 min. The pellet was then washed gently with digestion buffer twice to remove cell wall debris by centrifugation as above. The harvested protoplasts were transferred to 5 ml of water, and the cells were further disrupted by three freeze-thaw repetitions, followed by gentle sonication with a discontinuous pulse (1 min of sonication and 2 min for cooling down of samples) for 10 min. After that, the cytoplasmic membrane was extracted by ultracentrifugation using a method described elsewhere (29). To verify the purity of the membrane, samples were taken during purification for morphology examination under electron microscopy (Fig. S1, available at http://www.staphylococcus.org/en/cui/Fig-S1.jpg). To release vancomycin from the membrane, the membrane pellet suspended in 50 μl of water was treated with an equal volume of 20% trichloroacetic acid for 30 min at 37°C. Following that, the suspension was neutralized by addition of 80 μl of saturated sodium bicarbonate and was filtered before the vancomycin concentration was determined. The membrane-bound vancomycin concentration was determined in triplicate on three independent occasions by both HPLC and bioassay.
Incorporation of d-[14C]glucose into the cell wall.
Mu50 cell preparations with thin and thick cell walls were exposed to vancomycin for different lengths of time (0, 2, 4, 15, and 30 min) and then washed twice with RMg- on a filter membrane (HAWP02500; Millipore Co., MA). Cells on the filter membrane were harvested, and the rate of incorporation of d-[14C]glucose into cell wall peptidoglycan was determined after purification of peptidoglycan as described previously (12). The experiment was performed in duplicate on three independent occasions, and results are expressed as means ± SDs.
Statistical analysis.
The statistical significance of the data was evaluated by the Student t test. Linear regression analysis was performed by using StatView J4.5 (Abacus Concepts, Inc., Berkeley, CA).
RESULTS AND DISCUSSION
Thickened cell wall and prolongation of vancomycin consumption.
Cells with different cell wall thicknesses were prepared using a single colony of strain Mu50 or MI (12). In order to observe the effect of cell wall thickness on vancomycin susceptibility, the TRG of each cell was measured in growth-supportive BHI medium containing 30 mg/liter of vancomycin. Results (Fig. 1; Table 1) show that the degree of cell wall thickness correlated well with the vancomycin resistance level as judged by the TRG: the thicker the cell wall, the faster the recovery from the growth-suppressive effect of vancomycin (correlation coefficients, 0.976 [P < 0.01] and 0.993 [P < 0.01] for Mu50 and MI, respectively). The vancomycin concentration in the BHI medium dropped to 5 mg/liter before cells started to regrow. This drop is caused by binding of vancomycin to the cell wall peptidoglycan (12).
TABLE 1.
Cell wall thickness and TRG in the presence of vancomycin
| Mediuma | Mu50
|
MI
|
||
|---|---|---|---|---|
| Cell wall thickness (nm)b | TRG (h)c | Cell wall thickness (nm) | TRG (h) | |
| RMg- | 31.03 ± 3.27 | 16.0 | 31.86 ± 2.13 | 15.0 |
| RM (RMg- plus 30 mM d-glucose) | 53.29 ± 3.01 | 3.5 | 61.47 ± 7.33 | 2.0 |
| RMg- plus 30 mM l-glutamine | 31.07 ± 1.75 | 17.0 | 31.86 ± 3.17 | 12.0 |
| RMg- plus 30 mM N-acetylglucosamine | 38.13 ± 3.49 | 11.0 | 50.80 ± 9.64 | 8.0 |
| RM plus 30 mM l-glutamine | 57.18 ± 3.18 | 4.5 | 65.36 ± 7.86 | 1.5 |
See Materials and methods.
Thirty cells of each cell culture with nearly equatorial cut surfaces (photo taken by transmission electron microscopy) were measured for the evaluation of cell wall thickness. Results are expressed as means ± standard deviations.
In the presence of 30 mg/liter vancomycin in BHI medium.
We then performed experiments to compare the time course of vancomycin consumption by Mu50 cells with thin and thick cell walls (see Fig. 1A). These cells were used in all subsequent experiments unless indicated otherwise. The cell preparations were incubated in glucose-free resting medium (RMg-) containing 30 mg/liter vancomycin, and then vancomycin consumption by these cells was determined by measuring the remaining vancomycin concentration in the medium. RMg- lacks essential amino acids and a carbon source, allowing neither de novo cell wall biosynthesis nor cell multiplication (see Materials and Methods). Thus, the amount of vancomycin consumed at each time point during the incubation is equal to the cumulative amount of vancomycin consumed by the initial number of cells with the initial cell wall thickness. The result given in Fig. 2A shows that the maximum vancomycin consumption and the time needed to reach that level (defined as the time required for consuming the maximum amount of vancomycin [TCMV]) were different for the two cell preparations. Cells with thin cell walls consumed vancomycin up to a maximum amount of 15 mg/liter in 4 min, while cells with thick cell walls consumed vancomycin up to 25 mg/liter in 60 min. The results were reproducible in five independent experiments, two with strain MI preparations and three with Mu50 preparations. A correlation of TCMV prolongation with the degree of cell wall thickness was also observed in a parallel experiment (data not shown). The amount of vancomycin consumption was not affected at all by altering the initial vancomycin concentration in the medium. The same total amounts of consumption were obtained for both cells with thin cell walls and cells with thick cell walls when initial vancomycin concentrations of 40, 50, and 60 mg/liter instead of 30 mg/liter were used for the experiment. In contrast, the TCMV became shorter according to the increase in the initial vancomycin concentration (data not shown). The reaction-diffusion equation, which will be discussed later, reproduced these findings well.
FIG. 2.

Comparison of vancomycin consumption and growth capability between cells with thin and thick cell walls. (A) Vancomycin consumption was compared by plotting vancomycin concentrations remaining in the medium as functions of incubation time. During the time the cells were incubated in RMg- containing 30 mg/liter vancomycin, 0.5 ml of cell preparations with thin and thick cell walls was taken out at selected time intervals to measure vancomycin consumption. Concentrations of vancomycin remaining in the medium and numbers of viable cells were determined up to 120 min as described in Materials and Methods. Arrows indicate the TCMV. The y axis indicates the vancomycin (VCM) concentration remaining in the medium. Symbols: circles, vancomycin concentrations; squares, cell numbers. Open symbols, cells with thin cell walls; solid symbols, cells with thick cell walls. Error bars fall within the symbol size and are therefore not shown except for open circles. (B and C) Growth capabilities of cells with thin (B) and thick (C) cell walls after exposure to vancomycin for various times. Portions (0.1 ml) of the cell preparations taken out for the experiment for which results are shown in panel A were inoculated in 10 ml prewarmed growth-supportive BHI medium, and growth curves were recorded as described for Fig. 1. Data in all panels are results of three independent experiments.
The prolonged consumption of vancomycin observed in cells with thick cell walls signifies the delay in complete saturation with vancomycin at DDRs present in both cell wall peptidoglycan layers and lipid II in the cytoplasmic membrane. If DDRs in lipid II were not completely saturated, peptidoglycan synthesis would not be suppressed completely, since DDRs of lipid II are the real targets of vancomycin. In agreement with this view, cells with thick cell walls retained a much greater capability for cell wall synthesis and cell multiplication than cells with thin cell walls after vancomycin exposure for various lengths of time. In an experiment parallel to that for determination of vancomycin consumption (Fig. 2A), we harvested cell preparations after they had been exposed to vancomycin for various lengths of time (0, 2, 4, 6, 8, 10, 15, 20, 30, 60, 90, and 120 min) in RMg- medium and measured their growth capabilities. Nearly equal numbers of the harvested cells were transferred to a drug-free BHI medium (which supports cell growth), and cell growth capabilities were determined by measuring the TRG from growth curves recorded by optical densitometer for cell preparations with thin (Fig. 2B) and thick (Fig. 2C) cell walls. For cell preparations with thin cell walls, vancomycin-mediated growth suppression was instantaneous, as reflected by a TCMV of 4 min (Fig. 2A), and cell growth did not resume until 5 h later (Fig. 2B). On the other hand, the greater growth capability of samples with thick cell walls was retained, as shown in Fig. 2C; all cells started to regrow by 5 h of incubation (faster regrowth). The experiment also showed that for both cells with thin cell walls and cells with thick cell walls, the TRG was correlated with the length of exposure to vancomycin: the longer the exposure, the longer it took for regrowth. This time dependence of vancomycin-mediated growth suppression is thought to reflect a gradual access of vancomycin to its real targets.
Since vancomycin is relatively large compared to the molecular mesh structure of peptidoglycan and has a strong binding affinity to DDRs of peptidoglycan, we suspected that the cell wall might be clogged by vancomycin molecules densely bound to DDRs within the peptidoglycan, thus building physical barriers to vancomycin penetration. If this is the case, disruption of the 3-dimensional structure of the cell wall peptidoglycan would nullify the effect of the barrier without causing any decrease in the amount of vancomycin binding. In the next experiment (Fig. 3), we treated the cells with lysostaphin prior to vancomycin exposure and then determined vancomycin consumption. Lysostaphin is a glycylglycine endopeptidase that specifically cleaves the pentaglycine cross-bridges of peptidoglycan without cutting off the vancomycin-binding targets (DDRs) from peptidoglycan. As expected, treated cells consumed amounts of vancomycin comparable to those consumed by untreated cells with intact peptidoglycan. However, the TCMV of the treated cells was shortened significantly, and the degree of TCMV shortening was dependent on the amount of lysostaphin used (Fig. 3A to E). Similar results were obtained by disruption of the cell wall by sonication (Fig. 3F). However, in this case, a concomitant decrease in vancomycin consumption was observed, probably due to the loss of some vancomycin-binding targets by physical destruction (49). This experiment suggests that the intact structure of the cell wall served as a physical barrier to vancomycin penetration.
FIG.3.

Intactness of 3-dimensional structure of cell wall and vancomycin consumption. Consumption of vancomycin (VCM) by cells with thin (circles) and thick (triangles) cell walls was compared after the cell walls were digested by various concentrations of lysostaphin (A to E) or mechanically disrupted by sonication (F). Vancomycin concentrations remaining in the medium were determined at selected time intervals during the incubation. Arrows indicate the TCMV.
Alterations in cell wall components, such as an increased nonamidated murein monomer in the cell wall peptidoglycan (4, 18), decreased PBP4 activity (44), and increased glycan chain length (27), have also been reported to correlate with vancomycin resistance. The nonamidated muropeptide is considered to contribute directly to the increased consumption of vancomycin, due to the fact that its binding affinity to vancomycin is greater than that of its amidated counterpart, by decreasing peptidoglycan cross-linkage (nonamidated murein monomer is an inefficient substrate for cross-bridge formation by penicillin-binding proteins) (12). It is noteworthy in this regard that a significant decrease in cell wall cross-linkage is also observed in a vancomycin-resistant mutant strain in vitro (44). However, the decrease in cell wall peptidoglycan cross-linkage does not make the cell resistant to vancomycin if it is not accompanied by a thickening of the cell wall, as demonstrated with the femC mutant strain BB589 (production of nonamidated muropeptide is a characteristic of the femC mutant BB589, in which Tn551 is inserted in the vicinity of the glutamine synthetase repressor gene [glnR], exerting a polar effect on the transcription of the glutamine synthetase gene [glnA] [16]). The contribution of a decrease in cell wall peptidoglycan cross-linkage becomes effective only when the strain has a thickened cell wall (12). Therefore, cell wall thickening is considered to be the prerequisite for vancomycin resistance.
Mathematical model of vancomycin consumption by S. aureus.
To explain the observed vancomycin consumption profiles of cells with thin and thick cell walls by the vancomycin clogging hypothesis, we formulated a mathematical model. Our basic idea is to calculate the competition between the diffusion (penetration) of vancomycin through the peptidoglycan layers and the binding reactions with their DDRs. We solved the following 1-dimensional coupled equations:
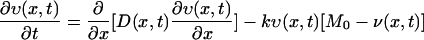 |
(1) |
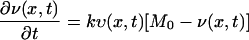 |
(2) |
where υ(x,t) and v(x,t) stand for the concentrations of free and bound vancomycin, respectively, M0 stands for the concentration of DDRs at the initial time in the cell wall, and k is the reaction rate for the binding of vancomycin and DDRs. The diffusion coefficient D(x,t) is generally a function of position (x) and time (t). The choice of k value and other details of the numerical calculation are given in the Appendix.
First, the diffusion coefficient is assumed to be a constant D like the reaction rate k. In Fig. 4A, we show the theoretical consumption curves of vancomycin for both cells with thin cell walls and cells with thick cell walls based on a normal reaction-diffusion model with a constant diffusion coefficient, together with the corresponding experimental data. The common diffusion constant D employed for both cells is 2.70 × 10−10 cm2/s. In this figure, we see that the curves fail to fit the short-time data for both cells with thin cell walls and cells with thick cell walls and that they also fail to fit the data around 15 min for cells with thick cell walls. Calculations showed that it is impossible to improve the fit by changing the diffusion constant D. When we increased the diffusion constant to improve the fit of the short-time behavior for both cell types, the deviation around 15 min for cells with thick cell wall cells became larger. When we decreased the diffusion constant to fit the data in this region, worse reproduction for short-time behavior for both cell types was found. It is therefore impossible to choose a value for diffusion constant D that yields better fit of calculated to experimental data for both cells with thin cell walls and cells with thick cell walls simultaneously.
FIG. 4.

Models of vancomycin consumption by cells with thin and thick cell walls. Model calculations (Calc) and comparisons with experimental data (Exp) are given. Two models were examined. (A) Vancomycin consumption curves for cells with thin and thick cell walls calculated using a standard reaction-diffusion equation with diffusion constant D and compared with experimental data. (B) Vancomycin consumption curves for cells with thin and thick cell walls calculated using a clogging model according to equation 3 and compared with experimental data. The diffusion constant D0 and clogging parameter β were determined as the best-fit values for these data. Error bars for experimental data points fall within the symbol size.
In order to improve the fit, especially for cells with thick cell walls, we studied the possibility of a cell wall-clogging effect. Such a possibility is naturally understandable, because the formation of a vancomycin-DDR bond in the cell wall will surely change the cell wall properties. Vancomycin is supposed to be 1.8 × 1.5 × 1.0 nm3 in size (30) and has a high dimerization constant, 1.0 × 104 M−1, for the vancomycin-ligand complex (51). In a calculation based on our experimental data, the vancomycin concentration in the cell wall was found to be about 5,600 times higher than that in the 30-mg/liter medium when all DDRs are bound to vancomycin. The total volume of saturated vancomycin in the cell wall amounts to about 21% of the entire volume of the Mu50 cell wall. The volume of 21% corresponds to 59% on the length scale (since 0.593 = 0.21). When 0.59 × 0.59 of one area unit of peptidoglycan mesh is occupied by vancomycin, a physical image of nearly complete clogging when all DDRs are saturated with vancomycin becomes realistic, since we have to take into account the other mesh materials that also work as obstacles to the transmission of incoming vancomycin. We therefore assume that the vancomycin diffusion coefficient decreases as a function of the density of the bond. In order to formulate this clogging effect, we treat the diffusion coefficient as a function of space and time in the following equation:
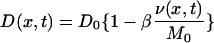 |
(3) |
where the diffusion constant D0 is the diffusion coefficient at time zero (i.e., the diffusion coefficient before the clogging process starts) and β stands for a new parameter that represents the strength of the clogging caused by the formation of the bond between vancomycin and DDRs. Since M0 is the original density of DDRs in the cell wall, a β value of 1 means that the diffusion is completely stopped when all DDRs are bound to vancomycin. On the other hand, a β value of zero means that there is no clogging effect. By introducing the new parameter β into the analysis, the value of the diffusion constant D0 can be chosen differently from the diffusion constant D used in the normal reaction-diffusion equation without clogging. Thus, we searched for a single set of parameters (D0 and β) to reproduce simultaneously the data for cells with thin and thick cell walls.
In Fig. 4B, we show the calculated results for cells with thin and thick cell walls where we chose the best parameter set, D0 = 2.60 × 10−9 cm2/s and β = 0.97, together with the corresponding experimental data. Comparing this figure with Fig. 4A, we see a significant improvement of the fit. Now the short-time behaviors for cells with thin and thick cell walls are improved, in addition to great improvement near 15 min for cells with thick cell walls. Thus, we could overcome the discrepancies observed in Fig. 4A, and we proved that the introduction of a clogging effect into the diffusion coefficient is necessary. The clogging reduces the value of the original diffusion coefficient to 1/33 of its starting value (D0) when all DDRs are bound to vancomycin. When we set β at zero, the TCMV value is reduced to almost 1/10 of the present value. A remarkable fact is that although the clogging effect is huge, it is only after a detailed analysis of the vancomycin consumption curves that its existence could be proven.
Clogging and its role in failure of treatment of VISA infection.
By using the formula given above for vancomycin diffusion, we now calculate the amount of bound vancomycin at the innermost segment (cytoplasmic membrane) with the same initial vancomycin concentration of 30 mg/liter that was used in Fig. 4, to evaluate vancomycin-mediated inhibition of peptidoglycan synthesis. The existence of the clogging effect can be seen more clearly in the time dependence of vancomycin-cytoplasmic membrane binding, as will be shown in the following. The broken line in Fig. 5A represents results calculated by the normal reaction-diffusion model (corresponding to Fig. 4A) for cells with thick cell walls. The theoretical curve shows a downward-convex shape, and the time constant for saturation is less than 17 min. The solid line in Fig. 5A represents results of calculation with the clogging model (corresponding to Fig. 4B) for cells with thick cell walls. In this case, the theoretical curve shows an upward-convex shape, and the time constant for saturation is about 27 min. To test which of the two models is more appropriate in terms of the time dependence of the binding of vancomycin to its targets in the cytoplasmic membrane, we proceeded to measure the membrane-bound vancomycin directly at various times after exposure of cells to vancomycin. Cells with thin and thick cell walls were prepared, and a portion of each cell preparation was harvested after exposure to vancomycin for 0, 2.5, 5, 15, 30, and 60 min to determine the amount of membrane-bound vancomycin. As shown in Fig. 5B, the amount of membrane-bound vancomycin molecules increased gradually with vancomycin exposure time in cells with thick cell walls, whereas it increased quickly in cells with thin cell walls. Figure 5B also shows that the amount of membrane-bound vancomycin in cells with thick cell walls is smaller than that in cells with thin cell walls during the time of observation. The upward-convex shape of the curve for cells with thick cell walls coincided with the theoretical curve with a clogging factor, and the time required for saturation was about 30 to 60 min, again consistent with the curve shown as a solid line in Fig. 5A. The observed delay of vancomycin saturation in the cytoplasmic membrane of the cells with thick cell walls strongly supports our hypothesis that a thickened cell wall acts as a barrier against vancomycin penetration, resulting in increased vancomycin resistance.
FIG. 5.

Comparison of mathematical calculations and experimental data for membrane-bound vancomycin. (A) Amount of bound vancomycin located at the innermost segment (cytoplasmic membrane) of cells with thick cell walls, calculated with both a standard reaction-diffusion equation (broken line) and a clogging model (solid line). (B) The amount of membrane-bound vancomycin was determined for both cells with thin cell walls and cells with thick cell walls. Cell preparations with thin (open circles) and thick (closed circles) cell walls were exposed to vancomycin for 2.5, 5, 15, 30, and 60 min and were used to determine membrane-bound vancomycin concentrations by microbiological assay and HPLC assay. Membrane-bound vancomycin levels increased in accordance with vancomycin exposure time, and the curve of the increment shows an upward-convex shape, which coincides with theoretical curves developed by introducing the clogging factor (solid line in panel A).
To evaluate the cell wall-producing capabilities of the cells with thin and thick cell walls used in the experiments discussed above in the presence of vancomycin, we further compared the degree of vancomycin-mediated inhibition of peptidoglycan synthesis at various times after cells were exposed to vancomycin. Cells with thin and thick cell walls were exposed to 30 mg/liter of vancomycin for various lengths of time in RMg- before they were transferred to RM. The latter medium, containing all the cell wall component amino acids and glucose as a carbon source, allows cells to synthesize nascent peptidoglycan chains but prevents cell multiplication. Cell wall-biosynthetic activity was determined by measuring the rate of d-[14C]glucose incorporation into the cell wall peptidoglycan fraction. Figure 6A and B illustrate the rate of d-[14C]glucose incorporation into cell wall peptidoglycan of cells with thin and thick cell walls, respectively. The time dependence of vancomycin-mediated inhibition of cell wall synthesis and the difference in this inhibition between cells with thin (Fig. 6A) and thick (Fig. 6B) cell walls were consistent with the time dependence of vancomycin-cytoplasmic membrane binding (Fig. 5B) and with the prolongation of the TRG (Fig. 1C and 2B and C) and TCMV (Fig. 2A).
FIG. 6.

Rate of incorporation of d-[14C]glucose into cell wall peptidoglycan. Incorporation rates were compared for cells with thin and thick cell walls after they were exposed to vancomycin. Cultures of cells with thin (A) and thick (B) cell walls, prepared similarly to those used for the experiments for which results are shown in Fig. 1, were exposed to 30 mg/liter vancomycin for 0, 2, 4, 15, and 30 min in RMg- medium. Determination of d-[14C]glucose incorporation into cell wall peptidoglycan was carried out after purification of cell wall peptidoglycan. Counts per minute were measured at the time points indicated. Results represent three independent experiments.
Finally, we evaluated the clogging effect under clinical circumstances. VISA strain Mu50 has a cell wall thickness of 35.02 nm, which is 1.64 times thicker than the cell wall of vancomycin-susceptible S. aureus (VSSA) and hence has about 1.64 times more peptidoglycan (11). Vancomycin is known to penetrate certain human tissues poorly. It is reported that a vancomycin concentration of 2.46 to 2.49 mg/liter is achieved in the sputa of patients and 5 mg/liter or less in an abscess following 2.0 g of multiple intravenous injections per day (35, 46). To simulate such clinical situations, we assumed the same cell wall structure for VISA and VSSA, and we performed the clogging model calculations with the same parameter values given in Fig. 4B. In this case, however, the vancomycin concentration in the medium was kept constant throughout the time course of vancomycin consumption. It was shown that when the concentration of vancomycin is fixed at 1 to 5 mg/liter, the time calculated by the present clogging model for vancomycin to saturate the cell membrane ranges from 16 to more than 60 min in VISA strain Mu50, about 3 times longer than in VSSA strain N315 (Fig. 7). We regard this delay caused by the thickened cell wall in Mu50 as the reason for poor response to vancomycin treatment, since it is obvious that the delay in the binding of vancomycin to the cytoplasmic membrane will result in continuation of bacterial growth. The results of the present study could explain the failure of treatment of VISA infection, and it could be justifiable to conclude that a cooperative effect of the clogging and cell wall thickening enables VISA to resist vancomycin. It should also be noted again that the clogging mechanism is related to the comparable sizes of a vancomycin molecule and a peptidoglycan mesh. This suggests a strategy for developing a new antibiotic that has a molecular size smaller than that of vancomycin but retains the function of DDR binding and cell wall biosynthesis inhibition.
FIG. 7.

Simulation for the saturation curve of bound vancomycin (VCM) in the innermost cell wall segments (cell membranes) of VISA strain Mu50 and vancomycin-susceptible strain N315. The time dependence of vancomycin saturation in the innermost cell wall layer was predicted with clogging model parameters (D0 = 2.60 × 10−9 cm2/s; β = 0.97 [see Fig. 4B]) for vancomycin-susceptible strain N315 (A) and VISA strain Mu50 (B). Cell wall thicknesses used for this calculations were 21.46 ± 2.25 nm for N315 and 35.02 ± 4.01 nm for Mu50 (11), and the vancomycin concentration in the medium was set as a constant.
In conclusion, we showed in this study that VISA resists vancomycin by means of a novel strategy that is different from any previously established drug resistance mechanism. Due to the clogging effect, a moderate thickening of the cell wall seen in clinical VISA strains causes a sufficient delay of vancomycin saturation in the cytoplasmic membrane. During the delay, VISA may have completed a new round of cell division and multiplication, nullifying the bactericidal function of vancomycin.
Acknowledgments
We thank Mitsuaki Yanagida, Patrick J. Loll, and Harald Labischinski for scientific discussions on the dimerization constant of the vancomycin-ligand complex, the 3-dimensional size of vancomycin, and the peptidoglycan structure, respectively.
This work was supported by a Grant-in-Aid for 21st Century COE Research and a Grant-in-Aid for Scientific Research on Priority Areas (13226114) from The Ministry of Education, Science, Sports, Culture and Technology of Japan.
APPENDIX
The formulation of the mathematical model for vancomycin consumption by S. aureus is as follows. The coupled differential equations 1 and 2 (see Results and Discussion) for υ(x,t) and v(x,t), the concentrations of free and bound vancomycin, respectively, inside the cell wall, are numerically solved by the finite difference method in the following way. As initial conditions, we use
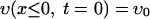 |
(A1) |
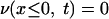 |
(A2) |
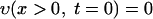 |
(A3) |
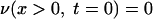 |
(A4) |
where x ≤ 0 means a location outside the cell wall, x > 0 means a location inside the cell wall, and υ0 is the initial concentration of vancomycin. We divide the cell wall into N equal-width segments where the x-direction is perpendicular to the surface. The boundary conditions at both sides of the wall are given by
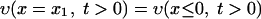 |
(A5) |
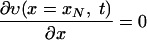 |
(A6) |
where x1 stands for the outermost segment of the cell wall and xN stands for the innermost segment when equations 1 and 2 are solved by the finite difference method. The first (Dirichlet-type) condition (equation A5) is the condition of fast equilibration of the active vancomycin concentration between the medium and the surface layer of the cell (x1). The second (Neumann-type) condition (equation A6) is the reflection condition at the innermost side of the cell wall (xN). Through the boundary condition (equation A5), the vancomycin provided to the surface layer diffuses into the cell wall, where the bonding with DDR occurs. The bonding with DDRs is represented by the loss term of vancomycin in equation 1, which is equal to the gain term in equation 2.
In order to solve the equations, it is necessary to know the parameters occurring in equations 1 and 2: the original concentration of DDRs in the cell wall (M0), the reaction rate k for the binding of vancomycin and DDRs, and the diffusion coefficient D(x,t). The following quantities specifying the experimental conditions for cells with thin (I) and thick (C) cell walls are used to calculate these parameters: number of cells per cubic centimeter, 7.96 × 108 (I) and 7.83 × 108 (C); average surface area (in square centimeters) per cell, 3.28 × 10−8 (I) and 3.72 × 10−8 (C); total surface area (in square centimeters) per cubic centimeter, 26.1 (I) and 29.1 (C); cell wall thickness (in centimeters), 3.10 × 10−6 (I) and 5.33 × 10−6 (C); total cell wall volume (in cubic centimeters) per cubic centimeter, 8.09 × 10−5 (I) and 15.5 × 10−5 (C). We evaluate M0 as follows. From the observed vancomycin consumption data, 15.59 (I) and 25.99 (C) mg/liter, after cell wall disruption with 20 mg of lysostaphin/liter (Fig. 3E), we obtain the DDR concentrations in solvent: 1.05 × 10−8 (I) and 1.75 × 10−8 (C) mol/cm3. Using the total cell wall volume given above, 8.09 × 10−5 (I) and 15.5 × 10−5 (C) cm3/cm3, we obtain M0 values of 1.30 × 10−4 (I) and 1.13 × 10−4 (C) mol/cm3. These values are used for M0 in equations 1 and 2.
The data of Fig. 3E are used in order to estimate the value of the reaction rate k. In the analysis of vancomycin consumption in disrupted cell walls, we can neglect the effect of diffusion in equations 1 and 2, and the concentrations of vancomycin υ and v are independent of x and functions only of time t. This amounts to simply solving the well-known equations of the second-order reaction rate. The solutions of these equations for υ and v can be analytically integrated as
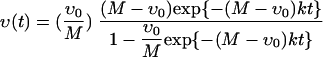 |
(A7) |
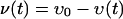 |
(A8) |
where υ0 is the original concentration of vancomycin in the medium (υ0 = 2.02 × 10−8 mol cm−3) and M is the concentration of DDRs in a solvent calculated from the vancomycin consumption: 1.409 × 10−8 mol cm−3 (I) and 1.749 × 10−8 mol cm−3 (C). With a reaction rate k of 1.0 × 107 cm3 mol−1 s−1, equation A7 gives vancomycin consumption data for disrupted cell walls consistently. The small change in this value causes no large effect in the following calculations. Therefore, we fix this value in the present calculation.
Finally, as for the diffusion coefficient D(x,t), the clogging effect is examined by using equation 3 (see Results and Discussion) where D0 is the constant diffusion coefficient of the unclogged cell wall and β is a new parameter that represents the strength of the clogging caused by the formation of the bond between vancomycin and DDRs. Since M0 is the original density of DDRs in the cell wall, a β value of 1 means that the diffusion is completely stopped when all DDRs are bound to vancomycin. On the other hand, a β value of zero means no clogging effect. Thus we searched for a good fit to the observed data for thin and thick cell walls simultaneously with a single parameter set of D0 and β.
REFERENCES
- 1.Barna, J., and D. Williams. 1984. The structure and mode of action of glycopeptide antibiotics of the vancomycin group. Annu. Rev. Microbiol. 38:339-357. [DOI] [PubMed] [Google Scholar]
- 2.Bischoff, M., M. Roos, J. Putnik, A. Wada, P. Glanzmann, P. Giachino, P. Vaudaux, and B. Berger-Bachi. 2001. Involvement of multiple genetic loci in Staphylococcus aureus teicoplanin resistance. FEMS Microbiol. Lett. 194:77-82. [DOI] [PubMed] [Google Scholar]
- 3.Boyle-Vavra, S., B. L. de Jonge, C. C. Ebert, and R. S. Daum. 1997. Cloning of the Staphylococcus aureus ddh gene encoding NAD+-dependent d-lactate dehydrogenase and insertional inactivation in a glycopeptide-resistant isolate. J. Bacteriol. 179:6756-6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle-Vavra, S., H. Labischinski, C. C. Ebert, K. Ehlert, and R. S. Daum. 2001. A spectrum of changes occurs in peptidoglycan composition of glycopeptide-intermediate clinical Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 45:280-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2002. Staphylococcus aureus resistant to vancomycin—United States. Morb. Mortal. Wkly. Rep. 51:565-567. [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. 2002. Vancomycin-resistant Staphylococcus aureus—Pennsylvania. Morb. Mortal. Wkly. Rep. 51:902-903. [PubMed] [Google Scholar]
- 7.Cetinkaya, Y., P. Falk, and C. G. Mayhall. 2000. Vancomycin-resistant enterococci. Clin. Microbiol. Rev. 13:686-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chesneau, O., A. Morvan, and N. E. Solh. 2000. Retrospective screening for heterogeneous vancomycin resistance in diverse Staphylococcus aureus clones disseminated in French hospitals. J. Antimicrob. Chemother. 45:887-890. [DOI] [PubMed] [Google Scholar]
- 9.Cui, L., and K. Hiramatsu. 2003. Vancomycin-resistant Staphylococcus aureus, p. 187-212. In A. C. Fluit and F. J. Schmitz (ed.), MRSA: current perspectives. Caister Academic Press, Norfolk, England.
- 10.Cui, L., J. Lian, H. Neoh, E. Reyes, and K. Hiramatsu. 2005. DNA microarray-based identification of genes associated with glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 49:3404-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui, L., X. Ma, K. Sato, K. Okuma, F. C. Tenover, E. M. Mamizuka, C. G. Gemmell, M. N. Kim, M. C. Ploy, N. El-Solh, V. Ferraz, and K. Hiramatsu. 2003. Cell wall thickening is a common feature of vancomycin resistance in Staphylococcus aureus. J. Clin. Microbiol. 41:5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui, L., H. Murakami, K. Kuwahara-Arai, H. Hanaki, and K. Hiramatsu. 2000. Contribution of a thickened cell wall and its glutamine nonamidated component to the vancomycin resistance expressed by Staphylococcus aureus Mu50. Antimicrob. Agents Chemother. 44:2276-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferraz, V., A. Duse, M. Kassel, A. Black, T. Ito, and K. Hiramatsu. 2000. Vancomycin-resistant Staphylococcus aureus occurs in South Africa. S. Afr. Med. J. 90:1108-1109. [PubMed] [Google Scholar]
- 14.Finan, J., G. Archer, M. Pucci, and M. Climo. 2001. Role of penicillin-binding protein 4 in expression of vancomycin resistance among clinical isolates of oxacillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 45:3070-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gemmell, C. G. 2004. Glycopeptide resistance in Staphylococcus aureus: is it a real threat? J. Infect. Chemother. 10:69-75. [DOI] [PubMed] [Google Scholar]
- 16.Gustafson, J., A. Strassle, H. Hachler, F. H. Kayser, and B. Berger-Bachi. 1994. The femC locus of Staphylococcus aureus required for methicillin resistance includes the glutamine synthetase operon. J. Bacteriol. 176:1460-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanaki, H., K. Kuwahara-Arai, S. Boyle-Vavra, R. S. Daum, H. Labischinski, and K. Hiramatsu. 1998. Activated cell-wall synthesis is associated with vancomycin resistance in methicillin-resistant Staphylococcus aureus clinical strains Mu3 and Mu50. J. Antimicrob. Chemother. 42:199-209. [DOI] [PubMed] [Google Scholar]
- 18.Hanaki, H., H. Labischinski, Y. Inaba, N. Kondo, H. Murakami, and K. Hiramatsu. 1998. Increase in glutamine-non-amidated muropeptides in the peptidoglycan of vancomycin-resistant Staphylococcus aureus strain Mu50. J. Antimicrob. Chemother. 42:315-320. [DOI] [PubMed] [Google Scholar]
- 19.Hiramatsu, K. 1998. Vancomycin resistance in staphylococci. Drug Resist. Updates 1:135-150. [DOI] [PubMed] [Google Scholar]
- 20.Hiramatsu, K. 2001. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect. Dis. 1:147-155. [DOI] [PubMed] [Google Scholar]
- 21.Hiramatsu, K., L. Cui, M. Kuroda, and T. Ito. 2001. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 9:486-493. [DOI] [PubMed] [Google Scholar]
- 22.Hiramatsu, K., L. Cui, and K. Kuwahara-Arai. 2004. Has vancomycin-resistant Staphylococcus aureus started going it alone? Lancet 364:565-566. [DOI] [PubMed] [Google Scholar]
- 23.Hiramatsu, K., H. Hanaki, T. Ino, K. Yabuta, T. Oguri, and F. C. Tenover. 1997. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 40:135-136. (Letter.) [DOI] [PubMed] [Google Scholar]
- 24.Hiramatsu, K., K. Okuma, X. X. Ma, M. Yamamoto, S. Hori, and M. Kapi. 2002. New trends in Staphylococcus aureus infections: glycopeptide resistance in hospital and methicillin resistance in the community. Curr. Opin. Infect. Dis. 15:407-413. [DOI] [PubMed] [Google Scholar]
- 25.Hood, J., G. F. S. Edwards, B. Cosgrove, E. Curran, D. Morrison, and C. G. Gemmell 2000. Vancomycin-intermediate Staphylococcus aureus at a Scottish hospital. J. Infect. 40:A11. [Google Scholar]
- 26.Kim, M. N., C. H. Pai, J. H. Woo, J. S. Ryu, and K. Hiramatsu. 2000. Vancomycin-intermediate Staphylococcus aureus in Korea. J. Clin. Microbiol. 38:3879-3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsuzawa, H., K. Ohta, S. Yamada, K. Ehlert, H. Labischinski, J. Kajimura, T. Fujiwara, and M. Sugai. 2002. Increased glycan chain length distribution and decreased susceptibility to moenomycin in a vancomycin-resistant Staphylococcus aureus mutant. Antimicrob. Agents Chemother. 46:75-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuroda, M., K. Kuwahara-Arai, and K. Hiramatsu. 2000. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem. Biophys. Res. Commun. 269:485-490. [DOI] [PubMed] [Google Scholar]
- 29.Kuwahara-Arai, K., N. Kondo, S. Hori, E. Tateda-Suzuki, and K. Hiramatsu. 1996. Suppression of methicillin resistance in a mecA-containing pre-methicillin-resistant Staphylococcus aureus strain is caused by the mecI-mediated repression of PBP2′ production. Antimicrob. Agents Chemother. 40:2680-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loll, P. J., A. E. Bevivino, B. D. Korty, and P. H. Axelsen. 1997. Simultaneous recognition of a carboxylate-containing ligand and an intramolecular surrogate ligand in the crystal structure of an asymmetric vancomycin dimer. J. Am. Chem. Soc. 119:1516-1522. [Google Scholar]
- 31.Lu, J., S. Lee, S. Hwa, and A. Yang. 2005. Septic arthritis caused by vancomycin-intermediate Staphylococcus aureus. J. Clin. Microbiol. 43:4156-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maki, H., N. McCallum, M. Bischoff, A. Wada, and B. Berger-Bachi. 2004. tcaA inactivation increases glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 48:1953-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milewski, W. M., S. Boyle-Vavra, B. Moreira, C. C. Ebert, and R. S. Daum. 1996. Overproduction of a 37-kilodalton cytoplasmic protein homologous to NAD+-linked d-lactate dehydrogenase associated with vancomycin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 40:166-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mongodin, E., J. Finan, M. W. Climo, A. Rosato, S. Gill, and G. L. Archer. 2003. Microarray transcription analysis of clinical Staphylococcus aureus isolates resistant to vancomycin. J. Bacteriol. 185:4638-4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakatani, T. 1994. Pharmacokinetic and clinical study of vancomycin. Antibiot. Chemother. 12:123-135. [Google Scholar]
- 36.Oliveira, G. A., A. M. Aquila, R. A. Masiero, C. Levy, S. G. Gomes, L. Cui, K. Hiramatsu, and E. M. Mamizuka. 2001. Isolation in Brazil of nosocomial Staphylococcus aureus with reduced susceptibility to vancomycin. Infect. Control Hosp. Epidemiol. 22:443-448. [DOI] [PubMed] [Google Scholar]
- 37.Paton, R., T. Snell, F. Emmanuel, and R. Miles. 2001. Glycopeptide resistance in an epidemic strain of methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 48:941-942. [DOI] [PubMed] [Google Scholar]
- 38.Ploy, M. C., C. Grelaud, C. Martin, L. de Lumley, and F. Denis. 1998. First clinical isolate of vancomycin-intermediate Staphylococcus aureus in a French hospital. Lancet 351:1212. (Letter.) [DOI] [PubMed] [Google Scholar]
- 39.Reipert, A., K. Ehlert, T. Kast, and G. Bierbaum. 2003. Morphological and genetic differences in two isogenic Staphylococcus aureus strains with decreased susceptibilities to vancomycin. Antimicrob. Agents Chemother. 47:568-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shlaes, D. M., J. H. Shlaes, S. Vincent, L. Etter, P. D. Fey, and R. V. Goering. 1993. Teicoplanin-resistant Staphylococcus aureus expresses a novel membrane protein and increases expression of penicillin-binding protein 2 complex. Antimicrob. Agents Chemother. 37:2432-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shockman, G. D., M. J. Conover, J. J. Kolb, L. S. Riley, and G. Toennies. 1961. Nutritional requirements for bacterial cell wall synthesis. Nutr. Cell Wall 81:44-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sieradzki, K., M. G. Pinho, and A. Tomasz. 1999. Inactivated pbp4 in highly glycopeptide-resistant laboratory mutants of Staphylococcus aureus. J. Biol. Chem. 274:18942-18946. [DOI] [PubMed] [Google Scholar]
- 43.Sieradzki, K., R. B. Roberts, S. W. Haber, and A. Tomasz. 1999. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N. Engl. J. Med. 340:517-523. [DOI] [PubMed] [Google Scholar]
- 44.Sieradzki, K., and A. Tomasz. 1997. Inhibition of cell wall turnover and autolysis by vancomycin in a highly vancomycin-resistant mutant of Staphylococcus aureus. J. Bacteriol. 179:2557-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith, T. L., M. L. Pearson, K. R. Wilcox, C. Cruz, M. V. Lancaster, B. Robinson-Dunn, F. C. Tenover, M. J. Zervos, J. D. Band, E. White, W. R. Jarvis, et al. 1999. Emergence of vancomycin resistance in Staphylococcus aureus. N. Engl. J. Med. 340:493-501. [DOI] [PubMed] [Google Scholar]
- 46.Torres, J., C. Sanders, and A. Lewis. 1979. Vancomycin concentration in human tissues—preliminary report. J. Antimicrob. Chemother. 5:475-477. [DOI] [PubMed] [Google Scholar]
- 47.Tsakris, A., E. Papadimitriou, J. Douboyas, F. Stylianopoulou, and E. Manolis. 2002. Emergence of vancomycin-intermediate Staphylococcus aureus and S. sciuri, Greece. Emerg. Infect. Dis. 8:536-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Utaida, S., P. Dunman, D. Macapagal, E. Murphy, S. Projan, V. Singh, R. Jayaswal, and B. Wilkinson. 2003. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149:2719-2732. [DOI] [PubMed] [Google Scholar]
- 49.Verwer, R., E. Beachey, W. Keck, A. Stoub, and J. Poldermans. 1980. Oriented fragmentation of Escherichia coli sacculi by sonication. J. Bacteriol. 141:327-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weigel, L., D. Clewell, S. Gill, N. Clark, L. McDougal, S. Flannagan, J. Kolonay, J. Shetty, G. Killgore, and F. Tenover. 2003. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302:1569-1571. [DOI] [PubMed] [Google Scholar]
- 51.Williams, D., A. Maguire, W. Tsuzuki, and M. Westwell. 1998. An analysis of the origins of a cooperative binding energy of dimerization. Science 280:711-714. [DOI] [PubMed] [Google Scholar]