

Abstract
Human papillomaviruses (HPVs) are small DNA tumor viruses that are the causative agent of warts and are associated with many anogenital cancers. The viral gene encoding the E6 protein has been found to be involved in HPV oncogenesis. E6 is known to inactivate the cellular tumor suppressor, p53. In addition, E6 has been shown to bind to a variety of other cellular proteins. The focus of this study was to determine what role the interactions of E6 with a subset of cellular proteins which contain a common α-helical domain in their E6 binding region (α-helix partners) play in E6-mediated phenotypes. We generated transgenic mice expressing a mutant of E6, E6I128T, which is defective for binding at least a subset of the α-helix partners, including E6AP, the ubiquitin ligase that mediates E6-dependent degradation of the p53 protein, to determine whether binding of α-helix partners plays a role in E6-mediated activities in vivo. Unlike mice expressing the wild-type E6 (strain K14E6WT), the mice expressing E6I128T lacked the ability to alter the radiation-induced block to DNA synthesis and promote the formation of benign skin tumors in conjunction with chemical carcinogens. Additionally, they displayed reduced levels of skin hyperplasia, spontaneous skin tumors, and tumor progression activity compared to those of the K14E6WT mice. From these results, we conclude that a domain in E6 that mediates α-helix partner binding is critical for E6-induced phenotypes in transgenic mice.
Human papillomaviruses (HPVs) are the causative agents of warts. A subset of HPVs, which are classified as high-risk HPVs, induce anogenital cancers such as cervical cancer. The HPV genomes are commonly found integrated into the host genomes in genital cancers. This integration event occurs such that two HPV open reading frames which encode the E6 and E7 proteins almost always remain intact. Further research has established the E6 and E7 open reading frames as oncogenes. E6 has been shown to exhibit oncogenic activities in a variety of cellular transformation assays. Several groups determined that E6 expression was necessary for HPV to transform its natural host cells, human keratinocytes (12, 29). E6 cooperates with Ras to transform baby rat kidney cells (26) and immortalizes primary mouse epithelial cells (41). E6 expression also leads to the immortalization of human mammary epithelial cells (36). Previously, our laboratory generated mice which express the HPV16 E6 protein in their epidermis. These mice of the K14E6WT strain exhibit skin hyperplasia, have an altered radiation response, spontaneously develop malignant skin tumors, and have an increased incidence of skin tumors when treated with chemical carcinogens (39, 40). These results demonstrate that E6 possesses oncogenic properties in vivo.
The first biochemical activity attributed to E6 was its ability to inactivate the tumor suppressor protein p53 (13, 47). The p53 gene is frequently mutated in cancer, and thus, intact p53 is thought to contribute to a general defense mechanism against carcinogenesis. To inactivate p53, E6 binds in a ternary complex with p53 (47) and a cellular ubiquitin ligase, E6AP (14). This interaction results in the ubiquitination and degradation of p53 through the proteosome pathway. In this way E6 abolishes p53 function. However, E6's ability to transform cells does not always correlate with its ability to cause p53 degradation. Mutants of E6 that cannot induce p53 degradation are still able to immortalize mammary epithelial cells (18, 25), transform 3Y1 rat fibroblasts, and confer tumorigenicity onto ψ2 mouse fibroblast cells (19). Conversely, E6 mutations have been identified which retain the ability to induce p53 degradation but are unable to transform cells (6, 18). Furthermore, studies comparing the K14E6WT transgenic mice to p53-null animals identified p53-independent activities of E6. P53-null mice do not develop the epidermal hyperproliferation, nor do they respond to tumor promoters as the K14E6WT mice do, demonstrating the existence of p53-independent activities of E6 in vivo (17, 40). Therefore, E6's oncogenic activities cannot be fully explained by its inactivation of p53.
E6 is known to bind multiple cellular factors. Several E6-binding partners contain similar amino acid motifs in their E6 binding domains (3, 7, 46). This amino acid motif consists of seven amino acids which form part of an α-helical structure that is necessary for interactions with E6 (1). We refer to E6-binding partners that associate with E6 via this motif as α-helix partners. One α-helix partner is E6AP, the cellular ubiquitin ligase necessary for E6-mediated p53 degradation. Other α-helix partners include E6BP (4), a calcium-binding protein of the CREC family, paxillin (44-46), a focal adhesion protein, E6-TP (10), a putative Rap1 GAP protein, and IRF3 (34), a transcriptional regulator involved in interferon response.
Binding of α-helix partners to E6 has been found to be important for the transforming activity of the E6 protein of bovine papillomavirus type 1 (BPV-1). The transforming ability of BPV-1 E6 correlates with its ability to bind paxillin (45). Furthermore, a peptide that competes with the α-helix partners for binding to E6 inhibited the transformation activity of BPV-1 E6 (2). One study provides evidence for a role of interactions with α-helix partners in the transformation activities of HPV-16 E6. In that study, the ability of E6 to immortalize human mammary epithelial cells correlated with its ability to bind and degrade E6TP (9). Another study suggests, however, that α-helix partner binding may not be necessary for HPV E6-induced transformation. In that study, a mutant of E6, E6I128T, that bound the α-helix partners E6AP and E6BP at a level 1 to 5% of that of wild-type E6 was still able to induce the immortalization of mammary epithelial cells (25).
To determine the role of α-helix partner binding in E6's carcinogenic activity in an animal model, we generated mice that express the mutant of E6 (E6I128T) which has been shown to be defective in binding a subset of the α-helix partners. These mice (strain K14E6I128T) lacked alterations in p53-dependent radiation response and the ability to contribute to the induction of papillomas in conjunction with chemical carcinogens. In addition, the K14E6I128T mice exhibited reduced levels of skin hyperplasia, spontaneous skin tumors, and tumor progression activity compared to the K14E6WT mice. These results suggest that a domain of E6 that mediates α-helix partner binding contributes to E6's oncogenic activities in vivo.
K14E6I128T mice express the transgene at a level similar to that of K14E6WT mice.
To assess the importance of α-helix partners in mediating the phenotypes of E6 in vivo, we generated and characterized transgenic mice that express a mutant E6 gene product that has been demonstrated to possess reduced affinity for binding two such partners. This mutant, E6I128T, binds E6AP and E6BP at a level less than 5% of that of wild-type E6. Importantly, it immortalizes human epithelial cells, demonstrating that it retains biological functions. Also, it is expressed at levels equivalent to those of wild-type E6 in vitro. These properties of E6I128T led us to select it for the in vivo function studies. To generate transgenic mice expressing the E6I128T mutant in their epidermis, the E6 open reading frame containing the I128T mutation (E6I128T) was placed between the human keratin 14 (K14) promoter and K14 3′ untranslated region. The recombinant DNA was injected into the male pronuclei of fertilized FVB/N eggs. Founder mice were screened by PCR and Southern analysis. Animals positive for the transgene were bred to generate two independent lines of K14E6I128T mice (lines 6061 and 6072). To monitor transgene expression, we performed in situ hybridization using a probe specific for E6 mRNA (Fig. 1A). The expression pattern of both lines of K14E6I128T mice was similar to the pattern of the K14E6WT mice (40). All lines expressed the transgene primarily in the epidermis and epidermal protrusions (hair follicles-sebaceous glands) in the dermis of the skin.
FIG. 1.

Analysis of transgene expression. (A) In situ hybridization. In situ hybridization was performed as described earlier (40). Transgene mRNA was examined in skin sections from the ears of 9-day-old mice by using an antisense probe. Darkfield images (magnification, ×20) of skin sections from K14E6WT line 5737 mice (panel 1), K14E6WT line 5718 mice (panel 2), nontransgenic mice (panel 3), K14E6I128T line 6061 mice (panel 4), and K14E6I128T line 6072 mice (panel 5) are shown. The epidermal (ep) and dermal (dm) regions of the skin are denoted. Transgene hybridization signals are localized to the epidermis and epidermal protrusions into the dermis. (B) Quantification of transgene expression by real-time PCR. Total RNA from each transgene line was reverse transcribed into cDNA. Real-time PCR analysis was performed with primers 5′-TGGAAGACCTGTTAATGGGCA-3′ and 5′-TGCAGGATCAGCCAATGGTAG-3′ and the Taqman Probe 6-FAM-ACTAGGAATTGTGTGCCCCATCTGTTCTCAG-TAMARA. Shown are RNA equivalents for K14E6WT line 5718 mice, K14E6I128T line 6061 mice, and K14E6I128T line 6072 mice relative to the high-expressing K14E6WT line 5737 mice. The average values ± 1 standard deviation for at least three mice in each group are graphed.
To assess quantitatively the level of transgene expression, a real-time PCR assay of the transgene specific cDNA was designed and performed. Total RNA samples from the skin were reverse transcribed using a poly(dT) primer. Subsequently, the cDNA was amplified with primers specific to the transgene cDNA. Amplification was monitored during the reaction by observing the fluorescence of a 6-carboxyfluorescein (FAM)-labeled Taqman probe specific to the transgene cDNA. A standard curve was generated by using serial dilutions of K14E6WT line 5737 RNA. The threshold (CT) values for the K14E6WT and K14E6I128T reactions were compared to the standard curve values to determine relative abundance (Fig. 1B). The abundance of transgene-specific cDNA in K14E6WT line 5737 was set to 100%. The low-expressing K14E6WT line 5718 expressed the transgene at a level 30% of that of 5737. The signal obtained for K14E6I128T lines 6072 and 6061 expressed the transgene at a level 53 and 100% of that of 5737, respectively.
K14E6I128T expression does not interfere with p53 induction or function following ionizing radiation.
E6I128T has been shown to bind the ubiquitin ligase, E6AP, in vitro with an affinity <5% of that of wild-type E6 (25). Since E6AP binding is required for E6-induced p53 degradation, we wanted to determine whether this mutant could facilitate the degradation of p53 in the epidermis of mice. P53 protein levels were induced by using ionizing irradiation. Cross sections of mouse ears and back skin were subjected to immunohistochemistry for p53. As shown in Fig. 2, p53 was induced in the skin of nontransgenic animals following irradiation. This induction was not present in the K14E6WT mice. However, p53 was induced in the K14E6I128T mice. The failure of the K14E6I128T mice to suppress the induction of p53 protein, as seen in K14E6WT mice, demonstrates that E6I128T is defective in inducing the degradation of p53 in vivo.
FIG. 2.

Levels of p53 protein in nontransgenic mice (panels 1 and 4), K14E6WT line 5737 mice (panels 2 and 5), and K14E6I128T line 6072 mice (panels 3 and 6) after being treated or not treated with irradiation. Skin cross sections from 9-day-old untreated mice (panels 1 to 3) and mice treated with ionizing radiation (4 Gy) 24 h prior to sacrifice (panels 4 to 6) were stained immunohistochemically for p53 protein by using anti-p53 (Novacastra NCL-p53-CM5p) antibody at a 1:500 dilution. After incubation with secondary antibody (30 min) and Vectastain ABC elite reagents (Vector PK-6200) (30 min), slides were exposed to diaminobenzidine substrate for 5 to 20 min. Tissue sections were counterstained by using hematoxylin solution. Examples of p53-positive cells are indicated by arrows.
To determine whether E6I128T inhibits p53 function in the transgenic mice, we looked for the induction of a transcriptional target of p53, namely, the cyclin-dependent kinase inhibitor, p21. Mice were irradiated as described above. Cross sections of mouse skin from K14E6WT, K14E6I128T, and nontransgenic animals were subjected to immunohistochemistry for p21. Figure 3A shows examples of p21-stained cross sections of skin from unirradiated (panels 1 to 3) and irradiated (panels 4 to 6) mice. The percentage of p21-positive cells in the epidermis of unirradiated and irradiated mice from each genotype is graphed in Fig. 3B. The p21 protein was induced from nondetectable (<0.2%) levels to 6.8% of the intrafollicular epithelial cells in the nontransgenic animals following irradiation. Only 1.7% of the cells in the K14EWT mice were p21 positive following irradiation, indicating that wild-type E6 inhibits p53 function in vivo. However, in the K14E6I128T mice, p21 was induced in 6.5% of the cells, which is a finding similar to that for nontransgenic mice. This indicates that E6I128T does not suppress p53 protein function in vivo.
FIG. 3.

Levels of p21 protein in the epidermal cells of nontransgenic mice (panels 1 and 4), K14E6WT line 5737 mice (panels 2 and 5), and K14E6I128T line 6061 mice (panels 3 and 6) after being treated or not treated with irradiation. (A) Cross sections (magnification, ×40) stained immunohistochemically for p21. Nine-day-old mice were either left untreated (panels 1 to 3) or were treated with ionizing radiation (4 Gy) 24 h prior to sacrifice (panels 4 to 6). For p21 detection, tissue sections were exposed to anti-p21 (BD PharMingen 556430) antibody diluted 1:25. After incubation with secondary antibody (30 min) and Vectastain ABC elite reagents (Vector PK-6200) (30 min), slides were exposed to diaminobenzidine substrate for 5 to 20 min. Tissue sections were counterstained using hematoxylin solution. Examples of p21-positive cells are indicated by arrows. (B) Quantification of p21-positive cells in the epidermis of nontransgenic mice, K14E6WT line 5737 mice, and K14E6I128T line 6061 mice. Graphed are the average percentages ± one standard deviation of p21-positive cells in the epidermis of three mice per group. No p21-positive cells were detected in any of the unirradiated nontransgenic mouse skin sections.
K14E6I128T does not abolish radiation response.
In response to ionizing radiation, DNA synthesis is inhibited in epithelial cells of the skin. This inhibition is largely dependent upon the presence of p53. Expression of wild-type E6 abolishes this inhibition (38). To determine whether E6I128T expression alters this radiation response, K14E6I128T mice were irradiated and their DNA synthesis was assessed by monitoring 5′-bromo-2′-deoxyuridine (BrdU) incorporation 24 h postirradiation. Figure 4 shows the percentages of cells in the epidermis of torso skin incorporating BrdU with and without irradiation. The nontransgenic animals displayed an approximately 50% reduction in BrdU incorporation following irradiation. This inhibition of DNA synthesis was completely absent in the K14E6WT mice, which is in agreement with our previous studies (38). The K14E6I128T mice displayed an approximately 50% reduction in DNA synthesis following irradiation, which is similar to the finding for nontransgenic animals.
FIG. 4.

DNA synthesis following ionizing radiation in nontransgenic (NT) mice, K14E6WT line 5737 mice, and K14E6I128T line 6061 mice on a p53-sufficient or p53-deficient background. The graph depicts the percentages of BrdU-positive epidermal cells in untreated mice and mice treated with irradiation (4 Gy) 24 h prior to sacrifice. All mice were injected with BrdU (100 μg/g of body weight) and fluoro-deoxyuridine (83.75 μg/g) 1 h prior to sacrifice. Skin samples were collected, fixed in 10% formalin, paraffin embedded, and cut into 5-μm-thick sections. BrdU was detected by using the BrdU IHC kit (Oncogene HCS24). The total percentage of BrdU-positive cells was calculated by dividing the percentage of BrdU-positive cells by the total number of cells in ten 40× frames per skin section. Error bars indicate the standard deviation for at least three mice per group.
To determine whether E6I128T's lack of alteration of radiation response could be attributed to the lack of p53 inactivation, E6I128T p53-null mice were irradiated. Nontransgenic, p53-null mice displayed a nearly complete elimination of radiation response (Fig. 4). Furthermore, E6I218T p53-null mice displayed a radiation response similar to that of nontransgenic p53-null animals. We interpret these results to mean that the defect in E6I128T's ability to abolish radiation responses is attributable to its inability to inactivate p53.
K14E6I128T mice display reduced levels of epithelial hyperplasia compared to the K14E6WT mice.
The expression of wild-type E6 in the skin of mice has been previously shown to lead to a thickened epidermis. This thickening is due to an increase in the percentage of cells undergoing DNA synthesis (40), especially in the suprabasal layers of the skin. To determine whether E6I128T could also induce epithelial hyperproliferation, we monitored DNA synthesis in the skin of 9-day-old mice. BrdU was injected into mice 1 h prior to sacrifice. BrdU-positive cells were detected by immunohistochemistry. BrdU-positive cells were evident only sporadically in the suprabasal compartment of nontransgenic animals. BrdU-positive suprabasal cells were consistently found in the K14E6WT mice. Fewer BrdU-positive suprabasal cells were found in the K14E6I128T mice than in the K14E6WT mice (Fig. 5). The percentages of BrdU-positive suprabasal cells in nontransgenic, K14E6WT, and K14E6I128T mice are quantified in Table 1. This analysis was performed on 9-day-old and 6-week-old animals. Consistent with previously reported results, the percentage of BrdU-positive cells in the suprabasal layer of the K14E6WT mice (line 5737) was greater than in that of nontransgenic mice. The K14E6I128T mice (line 6061) displayed a lower level of BrdU incorporation in the suprabasal compartment than the K14E6WT mice. This result indicates that E6I128T is less efficient at inducing epithelial hyperproliferation than wild-type E6.
FIG. 5.
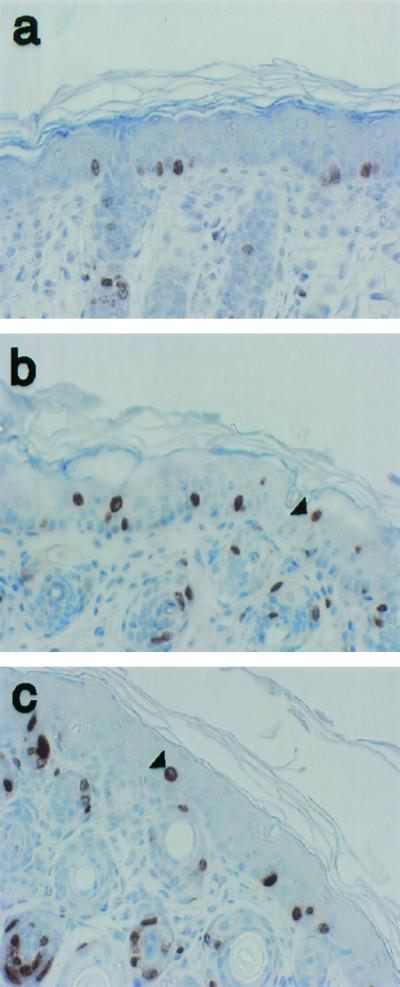
Hyperproliferation in the skin of nontransgenic mice, K14E6WT line 5737 mice, and K14E6I128T line 6061 mice. Shown are cross sections of skin from the ears of 9-day-old nontransgenic mice (a), K14E6WT line 5737 mice (b), and K14E6I128T line 6061 mice (c) stained immunohistochemically for BrdU incorporation. BrdU was injected 1 h prior to sacrifice and detected as described in the Fig. 4 legend. Examples of BrdU-positive suprabasal cells are denoted by arrowheads.
TABLE 1.
Epithelial hyperplasia in nontransgenic, K14E6WT, and K14E6I128T micea
| Mouse group (age) | Suprabasal BrdU-positive cells (% [avg ± SD])
|
||
|---|---|---|---|
| Nontransgenic | K14E6WT | K14E6I128T | |
| 9 days | 1.1 ± 0.1 | 5.0 ± 1.1 | 2.9 ± 1.1 |
| 6 wk | 0.1 ± 0.2 | 6.9 ± 2.1 | 2.0 ± 0.8 |
Nine-day-old or 6-week-old animals were injected with BrdU and fluoro-deoxyuridine 1 h prior to sacrifice, and histology sections of the ears were stained for BrdU as described for Fig. 4. To quantify hyperproliferation, the numbers of BrdU-positive and total suprabasal cells in 10 × 40 microscopic frames per section were counted to calculate the percentage of suprabasal BrdU-positive cells [i.e., (number of BrdU-positive suprabasal cells/total number of suprabasal cells) × 100]. Suprabasal cells were defined as any cells not directly attached to the basement membrane. Values represent the averages for three mice per genotype ± 1 standard deviation.
Inactivation of p53 (i.e., p53-null mice) in and of itself does not lead to epithelial hyperplasia (40). Nevertheless, inactivation of p53 by wild-type E6 could contribute to the induction of hyperplasia by E6. To determine whether E6I128T's reduced ability to induce epithelial hyperplasia was due to its inability to inactivate p53, we assessed the levels of proliferation of K14E6I128T mice on p53-null and p53-sufficient genetic backgrounds. To accomplish this, the homozygous null p53 allele was bred into the K14E6I128T mice. Epidermal hyperproliferation of K14E6I128Tp53+/+ mice was compared to that of K14E6I128Tp53−/− mice (Table 2). The K14E6I128Tp53−/− mice did not display more BrdU incorporation than the K14E6I128Tp53+/+ mice; therefore, the higher level of epithelial hyperproliferation displayed by the K14E6WT mice cannot be due to inactivation of p53.
TABLE 2.
Effect of p53 status on epithelial hyperplasia in K14E6WT and K14E6I128T mice
| Mouse strain | Suprabasal BrdU-positive cellsa (% [avg ± SD])
|
|
|---|---|---|
| p53+/+ | p53−/− | |
| K14E6WT line 5737 | 5.0 ± 0.1 | NDb |
| K14E6I128T line 6072 | 1.3 ± 0.5 | 1.4 ± 0.1 |
| K14E6I128T line 6061 | 2.9 ± 1.1 | 1.7 ± 0.6 |
Percentages of suprabasal cells that were BrdU positive in ear sections of 9-day-old mice. For details, see Table 1. Values represent the averages for three mice per genotype ± 1 standard deviation.
ND, not determined. Previous analysis indicated no differences in the degree of epidermal hyperplasia in the ears of 6-week-old K14E6WT mice of the p53-sufficient versus p53-null background (see reference 40).
K146I128T mice spontaneously develop fewer skin carcinomas than K14E6WT mice.
To determine whether E6 interactions with α-helix partners contribute to E6's carcinogenesis, K14E6I128T mice were monitored for the development of spontaneous skin carcinomas. At between 18 and 21 months of age, two K14E6I128T mice out of 140 developed skin carcinomas (Table 3). This is in contrast to the K14E6WT mouse line 5737, with which 26 out of 180 mice developed spontaneous skin tumors by 15 months of age (P < 0.0002). Therefore, E6I128T induces tumors less efficiently than does wild-type E6.
TABLE 3.
Skin carcinoma development in nontransgenic, K14E6WT, and K14E6I128T mice
| Genotype | Skin carcinoma incidence (no. of mice with tumors/total no.) | Mice with carcinomas (%) | Monitoring period (mo) | Reference(s) or source |
|---|---|---|---|---|
| K14E6WT | 26/180a | 14.4 | 15 | 40 |
| K14E6I128T | 2/140b | 1.4c | 20d | This study |
| FVB/N | 0/150 | <0.7 | 15 | 40 |
| FVB/N | 0/98 | <1 | 24 | 27 |
Tumor incidence in K14E6WT line 5737 mice.
Tumor incidence in K14E6I128T line 6061 and 6072 mice. Note that one additional carcinoma arose in a K14E6I128T founder mouse (line 6086) that was not bred.
The difference between the percentages of K14E6WT and K14E6I128T mice that developed carcinomas was statistically significant (P < 0.0002).
Tumors arose at 12 and 20 months.
E6I128T expression does not increase the yield of chemically induced papillomas.
Wild-type E6 expression has been shown to promote the formation of skin papillomas in mice treated with the initiation agent dimethylbenzanthracene (DMBA) and the promoting agent tetradecanoyl phorbol acetate (TPA). To determine whether E6I128T expression acts as a tumor promoter, we treated K14E6WT, K14E6I128T, and nontransgenic mice with a single dose of DMBA (0.1 μmol) to the skin on the back followed by twice-weekly treatments of TPA for 20 weeks. Mice were monitored during the 20-week TPA treatment for the development of papillomas. This experiment was performed separately for K14E6I128T line 6072 (Fig. 6A) and line 6061 (Fig. 6B). The K14E6WT mice developed more papillomas than did either those of the nontransgenic or K14E6I128T group. The numbers of papillomas that developed in the nontransgenic and K14E6I128T groups were not significantly different. These results indicate that E6I128T fails to possess the tumor promotion activity evident for wild-type E6.
FIG. 6.

Papilloma induction with chemical carcinogens in K14E6I128T mouse lines 6072 (A) and 6061 (B). Groups of 11 to 40 K14E6WT, K14E6I128T, and nontransgenic mice were treated with a single dose of DMBA on day 1, followed by TPA treatments twice weekly for 20 weeks as described previously (39). Papillomas were counted every 2 weeks during TPA treatment. Graphed are the average numbers of papillomas per mouse. Error bars represent 1 standard deviation.
E6I128T expression displays reduced tumor progression activity.
To assess tumor progression, K14E6WT line 5737 mice, K14E6I128T line 6072 mice, and FVB/N nontransgenic mice that had been treated with DMBA and TPA as described above were monitored for the formation of carcinomas (Fig. 7). In agreement with our previous studies, a greater percentage of K14E6WT mice than nontransgenic mice developed skin carcinomas. K14E6I128T mice developed fewer carcinomas than the K14E6WT mice. These data indicate that the ability of E6I128T to contribute to tumor progression is reduced. The experiment is being repeated with the higher-expressing K14E6I128T line (6061), and preliminary results are consistent with those reported for line 6072 (Fig. 7).
FIG. 7.

Carcinoma induction in mice treated with chemical carcinogens. Chemical carcinogens were administered to groups of 11 to 40 K14E6WT, K14E6I128T, and nontransgenic mice as described for Fig. 6. Mice were monitored for 20 weeks following the end of TPA treatment for the development of skin carcinomas. At the time of sacrifice, a portion of the tumor was fixed in 10% buffered formalin for histological analysis. The fixed tissue samples were embedded in paraffin and cut into 5-μm-thick sections for staining with hematoxylin and eosin. The tumor type was determined by histopathological analysis. Graphed are the percentages of mice in each group that developed skin carcinomas.
In this study, we characterized mice expressing a mutant of the HPV16 E6 protein, E6I128T, which is greatly reduced in its binding to all tested α-helix partners (25). Mice of strain K14E6I128T displayed less epithelial hyperproliferation, fewer spontaneous skin tumors, and less tumor progression activity than the K14E6WT mice. We conclude that a domain in E6 that mediates α-helix partner binding contributes to these phenotypes. Furthermore, the K14E6I128T mice lacked an ability to abolish DNA damage response or to promote the formation of papillomas in combination with chemical carcinogens, two properties observed with wild-type E6. This leads us to conclude that the same domain of E6 that mediates α-helix partner binding is necessary for these phenotypes.
Investigations using K14E6I128T mice allow assessment of the importance of α-helix partner binding.
The level of expression of the K14E6I128T transgene mRNA in line 6061 mice was found to be equivalent to the level of expression in K14E6WT line 5737 mice. Although we were unable to measure steady-state protein levels of the transgene in the mice, we think E6I128T was producing equivalent levels of functional protein because similar phenotypes are induced in the lenses of K14E6WT line 5737 and K14E6I128T line 6061 (Minh Nguyen and Anne Griep, personal communication). This observation also demonstrates potential tissue-specific differences in the role of α-helix partner binding in E6's phenotypes. Additionally, in studies previously reported, the E6I128T protein was shown to function in the immortalization of mammary epithelial cells and was expressed at a level similar to that of wild-type E6 in vitro (25). Therefore, direct comparisons of the phenotypes of the K14E6WT and K14E6I128T mice have allowed us to determine the role of α-helix partner binding in the in vivo activities of E6.
The mutant used in this study has been previously shown to bind to the α-helix partners E6AP and E6BP at a level less than 5% of that of wild-type E6 (25). Consistent with this deficiency, we determined that E6I128T expression, unlike that of wild-type E6, is unable to alter p53 levels. This was as expected, as E6AP binding is necessary for E6 to induce the degradation of p53 (15, 35, 42). This in vivo result confirms the prior in vitro data that demonstrated E6I128T to be defective for binding α-helix partners (25). In correlation with the induction in the levels of p53 protein in irradiated K14 E6I128T epidermis, we found p21 to be induced. Song et al. have previously demonstrated that the induction of p21 is largely dependent upon the presence of p53 (38). We conclude that the E6I128T mutant is unable to inhibit the function of p53. This finding demonstrates a correlation between E6's ability to induce the degradation of p53 and its ability to inactivate the function of p53. This is of particular note, given that E6 has been argued to inactivate p53 via an alternative mechanism involving E6's ability to bind CBP (33, 48). We do not know whether E6I128T is impaired in binding CBP.
The K14E6I128T mice do not display radioresistant DNA synthesis.
Although K14E6WT mice fail to block DNA synthesis following irradiation of the skin, the radiation response of the K14E6I128T mice was indistinguishable from that of the nontransgenic animals (Fig. 4). Therefore, E6's binding to α-helix partners seems to be required for this phenotype. Furthermore, the lack of radioresistance in the K14E6I128T mice might be fully compensated by p53 insufficiency. Both nontransgenic and K14E6I128T p53-null mice lacked this radiation response. This result suggests that it is E6I128T's inability to degrade p53 which accounts for its lack of radioresistance. This interpretation is consistent with what is known about p53's role in radiation response. In response to radiation, the p53 protein acts as a transcriptional activator for genes involved in cell cycle arrest, DNA repair, and apoptosis (for a review, see reference 28). Song et al. have shown previously (38) that irradiation leads to blocking of DNA synthesis in the epidermis of the mouse (Fig. 4). In this study, we observed that this block is largely overcome when p53 is inactivated.
Binding of α-helix partners quantitatively contributes to E6-induced hyperproliferation.
E6's interactions with α-helix partners contributes to skin hyperproliferation, as interruption of these interactions led to a less severe phenotype. One potential mediator of these effects is the p53 tumor suppressor protein. However, inactivation of p53 in the presence (Table 2) or absence (40) of E6I128T expression did not increase hyperproliferation of skin. Therefore, p53 inactivation is not responsible for the more efficient proliferative phenotype displayed by K14E6WT mice. Hence, other α-helix partners must be important for the E6-induced hyperproliferation.
Binding of α-helix partners is important for E6 tumor induction.
We observed a decreased spontaneous skin tumor incidence in the K14E6I128T mice compared to the K14E6WT mice. By 20 months of age, only 2 out of 140 (1.4%) of the K14E6I128T mice developed skin tumors. This is in contrast to the 26 out of 180 (14%) K14E6WT mice that developed skin tumors by 15 months of age (this article and reference 40). This result illustrates a reduction in oncogenic activity for the K14E6I128Tmice. Two of the K14E6I128T mice, one of the line 6072 mice, and one of the line 6061 mice did develop spontaneous skin carcinomas. Additionally, a spontaneous carcinoma arose on another K14E6I128T founder mouse (6086) that was never bred. According to the report by Song et al., the researchers never observed skin carcinoma development in nontransgenic animals of the FVB/N inbred strain (37). In addition, others have noted an absence of spontaneous skin carcinoma development in nontransgenic FVB/N mice (27). In the latter study, 98 FVB/N mice were monitored for the development of spontaneous neoplasms for 24 months, which is beyond the time frame during which we observed carcinomas developing in the K14E6I128T mice. No skin carcinomas were detected in the FVB/N animals during the 24-month time period. The only skin tumors observed were neural crest tumors and squamous papillomas, both of which are skin tumor types that are distinct from the squamous cell carcinomas tabulated in our analyses. Therefore, the fact that the K14E6I128T mice did develop some spontaneous skin carcinomas may indicate that E6I128T retains some oncogenic activity. However, due to the low numbers of observed tumors in the K14E6I128T mice, it is not possible at this time for us to determine whether the K14E6I128T mice display a significantly higher spontaneous skin tumor incidence than the nontransgenic FVB/N mice. It is not surprising to us that the K14E6I128T mice, which harbor a mutant E6 protein that is predicted to retain an ability to bind many cellular factors, develop tumors, as we have preliminary data from the analysis of K14E6WT/p53-null mice indicating that the p53-independent activities of E6 contribute to skin carcinogenesis (37). Unfortunately, the rapid and highly penetrant onset of lymphomas and sarcomas in young p53-null FVB/N mice precludes us from characterizing in a reasonable manner the underlying p53-independent mechanisms contributing to E6-mediated carcinogenesis on a p53-null background.
Consistent with the spontaneous tumor data, the E6I128T mutants displayed less synergy with chemical carcinogens. E6I128T lacked the ability of wild-type E6 to induce benign tumors when treated with DMBA and TPA. E6I128T did retain some activity in inducing the progression of benign tumors to malignant carcinomas; however, this activity was reduced compared to that of wild-type E6. In studies similar to ours, treatment of p53-null mice with DMBA and TPA had no effect on the promotion of papillomas (17). Therefore, E6I128T's inability to inactivate p53 does not explain this lack of tumor promotion. More p53-null mice develop malignant skin carcinomas than do p53-sufficent counterparts when treated with DMBA and TPA (17), demonstrating an influence of p53 inactivation on skin tumor progression. We predict that E6I128T's defect in tumor progression is largely due to its inability to inactivate p53.
We have shown that a mutant of E6 whose efficiency in binding α-helix partners is reduced induces a smaller range and reduced magnitude of phenotypes compared to wild-type E6 when expressed in transgenic mice. We interpret this to mean that E6 binding to one or more α-helix partners contributes to these phenotypes. The α-helix partners perform a variety of cellular activities. Current studies are directed at determining which α-helix partner(s) mediates E6's oncogenic activities. One α-helix partner which has the potential to mediate E6's oncogenic activities is E6AP. E6 induces E6AP-dependent degradation of a number of cellular proteins, including not only p53 (15, 35, 42) but also the human homologue of the Drosophila tumor suppressor, hScrib (30), minichromosome maintenance protein 7 (20), and the apoptotic regulator, Bak (43). Thus, the E6:E6AP complex has the potential to modulate a variety of cellular pathways related to cell growth regulation, thereby inducing hyperproliferation and tumors. Alternatively, E6 could induce hyperproliferation and tumors by altering the degradation of E6AP's endogenous substrates. E6 induces the self-ubiquitination of E6AP, which results in a decrease in E6AP half-life (16). E6AP has been shown to be important for regulating the levels of the DNA repair protein, HHR23A (22), and the Src family tyrosine kinase, Blk (31), independently of E6. Perhaps E6 induces carcinogenesis by removing E6AP from its endogenous targets.
Another α-helix partner with the potential to play a role in the oncogenic activities of E6 is E6TP1. This protein is a Rap GTPase-activating protein. Based upon mutational analysis of E6, Gao and colleagues have found a correlation between E6's association with E6TP1 and E6's ability to immortalize mammary epithelial cells (9). The rat homologue of E6TP1, SPAR, binds to Dlg, a PDZ partner of E6 (32). It is intriguing that E6 binds to both Dlg and E6TP1. Perhaps E6, Dlg, and E6TP1 function in concert to confer E6's activities in vivo.
Although the K14E6I128T mutants displayed lower levels of hyperproliferation and tumor progression than the K14E6WT mice, they did not completely lack these phenotypes. This indicates that E6 activities other than α-helix partner binding likely contribute to hyperproliferation and tumor progression. E6 binds to at least nine cellular proteins which do not contain an α-helix domain in their E6 binding region (5, 8, 11, 19, 21, 23, 24, 30, 33, 43, 48). Further analysis is required to determine which of these E6 uses to trigger oncogenesis.
Acknowledgments
We thank Denis Lee for assistance with the in situ hybridization, Henry Pitot for histological analysis of tumors, Kathleen Helmuth for technical assistance with the microinjection, and Jane Weeks and Harlene Edwards for processing tissue sections. We thank Minh Nguyen and Anne Griep for communication of unpublished results. We thank Norman Drinkwater and Bill Sugden for critical reading of the manuscript.
This study was supported by grants from the National Institutes of Health (CA22443, CA09135, and CA07175) and the American Cancer Society.
REFERENCES
- 1.Be, X., Y. Hong, J. Wei, E. J. Androphy, J. J. Chen, and J. D. Baleja. 2001. Solution structure determination and mutational analysis of the papillomavirus E6 interacting peptide of E6AP. Biochemistry 40:1293-1299. [DOI] [PubMed] [Google Scholar]
- 2.Bohl, J., K. Das, B. Dasgupta, and S. B. Vande Pol. 2000. Competitive binding to a charged leucine motif represses transformation by a papillomavirus E6 oncoprotein. Virology 271:163-170. [DOI] [PubMed] [Google Scholar]
- 3.Chen, J. J., Y. Hong, E. Rustamzableh, J. D. Baleja, and E. J. Androphy. 1998. Identification of an alpha helix motif sufficient for association with papillomavirus E6. J. Biol. Chem. 273:13537-13544. [DOI] [PubMed] [Google Scholar]
- 4.Chen, J. J., C. E. Reid, V. Band, and E. J. Androphy. 1995. Interaction of papillomavirus E6 oncoproteins with a putative calcium-binding protein. Science 269:529-531. [DOI] [PubMed] [Google Scholar]
- 5.Degenhardt, Y. Y., and S. J. Silverstein. 2001. Gps2, a protein partner for human papillomavirus E6 proteins. J. Virol. 75:151-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elbel, M., S. Carl, S. Spaderna, and T. Iftner. 1997. A comparative analysis of the interactions of the E6 proteins from cutaneous and genital papillomaviruses with p53 and E6AP in correlation to their transforming potential. Virology 239:132-149. [DOI] [PubMed] [Google Scholar]
- 7.Elston, R. C., S. Napthine, and J. Doorbar. 1998. The identification of a conserved binding motif within human papillomavirus type 16 E6 binding peptides, E6AP and E6BP. J. Gen. Virol. 79:371-374. [DOI] [PubMed] [Google Scholar]
- 8.Gao, Q., A. Kumar, S. Srinivasan, L. Singh, H. Mukai, Y. Ono, D. E. Wazer, and V. Band. 2000. PKN binds and phosphorylates human papillomavirus E6 oncoprotein. J. Biol. Chem. 275:14824-14830. [DOI] [PubMed] [Google Scholar]
- 9.Gao, Q., L. Singh, A. Kumar, S. Srinivasan, D. E. Wazer, and V. Band. 2001. Human papillomavirus type 16 E6-induced degradation of E6TP1 correlates with its ability to immortalize human mammary epithelial cells. J. Virol. 75:4459-4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao, Q., S. Srinivasan, S. N. Boyer, D. E. Wazer, and V. Band. 1999. The E6 oncoproteins of high-risk papillomaviruses bind to a novel putative GAP protein, E6TP1, and target it for degradation. Mol. Cell. Biol. 19:733-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glaunsinger, B. A., S. S. Lee, M. Thomas, L. Banks, and R. Javier. 2000. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 19:5270-5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hawley-Nelson, P., K. H. Vousden, N. L. Hubbert, D. R. Lowy, and J. T. Schiller. 1989. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 8:3905-3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hubbert, N. L., S. A. Sedman, and J. T. Schiller. 1992. Human papillomavirus type 16 E6 increases the degradation rate of p53 in human keratinocytes. J. Virol. 66:6237-6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huibregtse, J. M., M. Scheffner, and P. M. Howley. 1991. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 10:4129-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huibregtse, J. M., M. Scheffner, and P. M. Howley. 1994. E6-AP directs the HPV E6-dependent inactivation of p53 and is representative of a family of structurally and functionally related proteins. Cold Spring Harb. Symp. Quant. Biol. 59: 237-245. [DOI] [PubMed] [Google Scholar]
- 16.Kao, W. H., S. L. Beaudenon, A. L. Talis, J. M. Huibregtse, and P. M. Howley. 2000. Human papillomavirus type 16 E6 induces self-ubiquitination of the E6AP ubiquitin-protein ligase. J. Virol. 74:6408-6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kemp, C. J., L. A. Donehower, A. Bradley, and A. Balmain. 1993. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 74:813-822. [DOI] [PubMed] [Google Scholar]
- 18.Kiyono, T., S. A. Foster, J. I. Koop, J. K. McDougall, D. A. Galloway, and A. J. Klingelhutz. 1998. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396:84-88. [DOI] [PubMed] [Google Scholar]
- 19.Kiyono, T., A. Hiraiwa, M. Fujita, Y. Hayashi, T. Akiyama, and M. Ishibashi. 1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 94:11612-11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhne, C., and L. Banks. 1998. E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J. Biol. Chem. 273:34302-34309. [DOI] [PubMed] [Google Scholar]
- 21.Kukimoto, I., S. Aihara, K. Yoshiike, and T. Kanda. 1998. Human papillomavirus oncoprotein E6 binds to the C-terminal region of human minichromosome maintenance 7 protein. Biochem. Biophys. Res. Commun. 249:258-262. [DOI] [PubMed] [Google Scholar]
- 22.Kumar, S., A. L. Talis, and P. M. Howley. 1999. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem. 274:18785-18792. [DOI] [PubMed] [Google Scholar]
- 23.Lee, S. S., B. Glaunsinger, F. Mantovani, L. Banks, and R. T. Javier. 2000. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J. Virol. 74:9680-9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee, S. S., R. S. Weiss, and R. T. Javier. 1997. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 94:6670-6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, Y., J. Chen, Q. Gao, S. Dalal, Y. Hong, C. Mansur, V. Band, and E. Androphy. 1999. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J. Virol. 73:7297-7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, Z., J. Ghai, R. S. Ostrow, R. C. McGlennen, and A. J. Faras. 1994. The E6 gene of human papillomavirus type 16 is sufficient for transformation of baby rat kidney cells in cotransfection with activated Ha-ras. Virology 201:388-396. [DOI] [PubMed] [Google Scholar]
- 27.Mahler, J. F., W. Stokes, P. C. Mann, M. Takaoka, and R. R. Maronpot. 1996. Spontaneous lesions in aging FVB/N mice. Toxicol. Pathol. 24:710-716. [DOI] [PubMed] [Google Scholar]
- 28.May, P., and E. May. 1999. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene 18:7621-7636. [DOI] [PubMed] [Google Scholar]
- 29.Munger, K., W. C. Phelps, V. Bubb, P. M. Howley, and R. Schlegel. 1989. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 63:4417-4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa, S., and J. M. Huibregtse. 2000. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 20:8244-8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oda, H., S. Kumar, and P. M. Howley. 1999. Regulation of the Src family tyrosine kinase Blk through E6AP-mediated ubiquitination. Proc. Natl. Acad. Sci. USA 96:9557-9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pak, D., S. Yang, S. Rudolph-Correla, E. Kim, and M. Sheng. 2001. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron 31:289-303. [DOI] [PubMed] [Google Scholar]
- 33.Patel, D., S. M. Huang, L. A. Baglia, and D. J. McCance. 1999. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 18:5061-5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ronco, L. V., A. Y. Karpova, M. Vidal, and P. M. Howley. 1998. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 12:2061-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheffner, M., J. M. Huibregtse, R. D. Vierstra, and P. M. Howley. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495-505. [DOI] [PubMed] [Google Scholar]
- 36.Shay, J. W., W. E. Wright, D. Brasiskyte, and B. A. Van der Haegen. 1993. E6 of human papillomavirus type 16 can overcome the M1 stage of immortalization in human mammary epithelial cells but not in human fibroblasts. Oncogene 8:1407-1413. [PubMed] [Google Scholar]
- 37.Song, S. 1999. Ph.D. dissertation. University of Wisconsin, Madison.
- 38.Song, S., G. A. Gulliver, and P. F. Lambert. 1998. Human papillomavirus type 16 E6 and E7 oncogenes abrogate radiation-induced DNA damage responses in vivo through p53-dependent and p53-independent pathways. Proc. Natl. Acad. Sci. USA 95:2290-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song, S., A. Liem, J. A. Miller, and P. F. Lambert. 2000. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology 267:141-150. [DOI] [PubMed] [Google Scholar]
- 40.Song, S., H. C. Pitot, and P. F. Lambert. 1999. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J. Virol. 73:5887-5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Storey, A., and L. Banks. 1993. Human papillomavirus type 16 E6 gene cooperates with EJ-ras to immortalize primary mouse cells. Oncogene 8:919-924. [PubMed] [Google Scholar]
- 42.Talis, A. L., J. M. Huibregtse, and P. M. Howley. 1998. The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)-positive and HPV-negative cells. J. Biol. Chem. 273:6439-6445. [DOI] [PubMed] [Google Scholar]
- 43.Thomas, M., and L. Banks. 1998. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 17:2943-2954. [DOI] [PubMed] [Google Scholar]
- 44.Tong, X., and P. M. Howley. 1997. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 94:4412-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong, X., R. Salgia, J. L. Li, J. D. Griffin, and P. M. Howley. 1997. The bovine papillomavirus E6 protein binds to the LD motif repeats of paxillin and blocks its interaction with vinculin and the focal adhesion kinase. J. Biol. Chem. 272:33373-33376. [DOI] [PubMed] [Google Scholar]
- 46.Vande Pol, S. B., M. C. Brown, and C. E. Turner. 1998. Association of bovine papillomavirus type 1 E6 oncoprotein with the focal adhesion protein paxillin through a conserved protein interaction motif. Oncogene 16:43-52. [DOI] [PubMed] [Google Scholar]
- 47.Werness, B. A., A. J. Levine, and P. M. Howley. 1990. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248:76-79. [DOI] [PubMed] [Google Scholar]
- 48.Zimmermann, H., R. Degenkolbe, H. U. Bernard, and M. J. O'Connor. 1999. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 73:6209-6219. [DOI] [PMC free article] [PubMed] [Google Scholar]