

Abstract
Atomistic molecular dynamics simulations are performed for up to 20 ns to monitor the formation and the stability of complexes composed of single- or double-strand DNA molecules and C60 in aqueous solution. Despite the hydrophobic nature of C60, our results show that fullerenes strongly bind to nucleotides. The binding energies are in the range −27 to −42 kcal/mol; by contrast, the binding energy of two fullerenes in aqueous solution is only −7.5 kcal/mol. We observe the displacement of water molecules from the region between the nucleotides and the fullerenes and we attribute the large favorable interaction energies to hydrophobic interactions. The features of the DNA-C60 complexes depend on the nature of the nucleotides: C60 binds to double-strand DNA, either at the hydrophobic ends or at the minor groove of the nucleotide. C60 binds to single-strand DNA and deforms the nucleotides significantly. Unexpectedly, when the double-strand DNA is in the A-form, fullerenes penetrate into the double helix from the end, form stable hybrids, and frustrate the hydrogen bonds between end-group basepairs in the nucleotide. When the DNA molecule is damaged (specifically, a gap was created by removing a piece of the nucleotide from one helix), fullerenes can stably occupy the damaged site. We speculate that this strong association may negatively impact the self-repairing process of the double-strand DNA. Our results clearly indicate that the association between C60 and DNA is stronger and more favorable than that between two C60 molecules in water. Therefore, our simulation results suggest that C60 molecules have potentially negative impact on the structure, stability, and biological functions of DNA molecules.
INTRODUCTION
Several recent reports have highlighted the potentially hazardous nature of nanomaterials (1–3). For example, it has been shown that carbon nanotubes, arguably the best known nanomaterials, can accumulate in the lungs of rats, and possibly cause the development of granulomas in rats (4,5). The nanomaterials we consider in this work are buckminsterfullerenes (C60) (6,7), because they show several promising potential applications in biology and pharmacology (8–11) and are regarded as one of the building blocks for nanotechnology applications.
The interest in biological applications of C60, as well as that for possible applications in materials science and in other disciplines, has been tempered by concerns that fullerenes may exhibit adverse environmental and health impacts. Early studies showed low toxicity for the C60 molecule itself (12,13). Pharmacokinetic studies with radio-labeled water-soluble C60 showed that after intravenous injection into mice the compound quickly migrates through the body, accumulates in the liver after a few hours (14), and is excreted, either slowly or rapidly depending on the functionalization of the C60 surface (15). These and other studies suggested that fullerene molecules do not exhibit short-term toxicity (16). However, more recent studies suggest that fullerenes may induce oxidative stress in the brain of juvenile largemouth bass (17), and that certain types of C60 derivatives can be quite toxic in cell membrane (18), thus raising the specter of significant negative health impacts from exposure to fullerenes. It is widely believed that severely negative biological outcomes (e.g., replication errors, self-repair mismatches, abnormal functionalities, etc.) can occur when molecules bind to DNA and impact its shape. Such outcomes have the potential of leading to diseased conditions, including cancer. DNA damage of this kind typically requires long times to become detectable and, to the best of our knowledge, no long-term toxicity study related to DNA damage has been reported for fullerenes.
Fullerenes are known to be essentially insoluble in water. However, their solubility can be increased (up to 2 mM) through appropriate coatings (13) or by being suspended in colloidal solution (19). A colloidal solution of uncoated C60 in water can be very stable (up to nine months) at room temperature (20). Hence, it is useful to consider the exposure of biological molecules, such as DNA, to fullerenes in aqueous solutions. In this work, we are interested in investigating the potential long-term toxic effects of fullerenes by understanding how C60 derivatives approach and interact with nucleic acids. To this end, we performed molecular dynamics simulations of aqueous DNA fragments in the presence of C60.
SIMULATION METHODOLOGY
The double-helix DNA molecule used in our simulations consists of 12 basepairs in the sequence of d[AGTCAGTCAGTC]2. The length and sequence of the DNA molecule was chosen as a balance between computational efficiency and biological realism. In particular, a DNA chain of 12 basepairs shows most of the typical properties of a short DNA segment, including a complete 360° rotation in the double-helix in both the A- and B-forms and the ability to spontaneously transition from its A-form to its B-form. Although the simulation of longer chains is clearly desirable, 12 basepairs is approaching the current limit of feasibility for performing multi-nanosecond, atomistically detailed simulations of a DNA segment in an explicit aqueous solution. We chose the sequence d[AGTCAGTCAGTC]2 because it includes and repeats the whole four types of basepairs. The rationale behind this choice consists in our desire to understand if C60 binds preferentially to one specific basepair.
DNA structures were obtained from the AMBER (21) utility software. The head-to-tail length of the DNA is ∼4 nm. Single-strand DNA was obtained by taking one helix from the double-strand B-DNA created. Damaged double-strand DNA was obtained by removing a section of consecutive nucleotides from one helix of the undamaged B-DNA. The section of nucleotide removed contains four bases, together with the backbones connected to them. Resultant gap was of ∼1.4 nm. The dangling bonds in the DNA helix resulting from the removal were saturated by hydrogen atoms. DNA and fullerenes were solvated in 4500–6100 (depending on the system size) TIP3P water molecules (22), within a cubic simulation box of initial size 7 nm. In the simulation box, at least 1.2 nm of water buffer separates the solute (DNA and C60) surface and the box boundaries in any simulation cell directions. Periodic boundary conditions were applied in all three directions. Twenty-two Na+ (11 Na+ for single-strand DNA simulations) counterions were added to electrically neutralize the system. The simulations were performed in the NPT (constant pressure, constant temperature) ensemble; hence, the size of the simulation box is adjusted automatically during the course of the simulation to maintain the pressure at 1 bar.
The potential model for DNA was the all-atom AMBER force field, 1999 version (21). The sp2 carbon atoms in the C60 molecule were modeled as Lennard-Jones (LJ) particle with ɛ/k = 43.2 K and σ = 0.34 nm (23). LJ interaction parameters between different atoms were calculated by the standard Lorentz-Berthelot combing rules. Lennard Jones interactions were computed within a 0.9-nm spherical cutoff without long-range corrections. TIP3P water model was chosen based on previous simulation works (24,25). The potential applied for Na+ was the one built in AMBER. The particle-mesh Ewald method with a fourth-order interpolation was applied to evaluate electrostatic interactions. Initial configurations were obtained by placing one, two, or three C60 molecules near the DNA molecule. The nearest initial distance between the C60 and DNA surfaces was ∼0.7 nm, which is within the LJ cutoff distance. The initial configurations permitted a clear separation between the fullerenes and DNA strands at the beginning of the simulation, while maintaining the simulation box size within a computationally manageable size.
Molecular dynamics simulations were performed within the constant pressure (1 bar) and temperature (300 K) (26) ensemble. The AMBER software package was employed to integrate the equations of motion. Each simulation includes 5000 steps of energy minimization, 40 ps of solvent relaxation, five cycles (each cycle with 1500 steps of energy minimization) of solutes relaxation, 20 ps of equilibration, and 5–20 ns of production. A time step of 2 fs was used and the structural configurations were saved every 2 ps for subsequent analysis.
The binding energy between one DNA and one C60 molecule is defined as
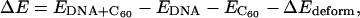 |
where 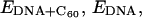 and
and 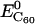 represent the potential energy of the bound DNA-C60 pair, the potential energy of DNA in the bound pair, and the potential energy of the C60 in the bound pair. In our calculations, it is necessary to account for ΔEdeform because significant deformations were observed for DNA segments and thus ΔEdeform is not negligible compared to the total potential energy change. We define ΔEdeform as
represent the potential energy of the bound DNA-C60 pair, the potential energy of DNA in the bound pair, and the potential energy of the C60 in the bound pair. In our calculations, it is necessary to account for ΔEdeform because significant deformations were observed for DNA segments and thus ΔEdeform is not negligible compared to the total potential energy change. We define ΔEdeform as
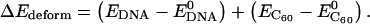 |
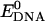 and
and 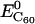 represent the potential energy of DNA and C60 in their native conformations. We use the term “native conformation” to refer to the state that a free DNA or C60 takes when it is in the same solvent and ion environment but with no association interaction force on it. We performed very short simulations (200 ps for C60, B-DNA, ssDNA, and damaged DNA, 10 ps for A-DNA because A-DNA tends to convert into its B-form under the simulation solvent conditions) of free DNA and C60 in water with counterions to calculate
represent the potential energy of DNA and C60 in their native conformations. We use the term “native conformation” to refer to the state that a free DNA or C60 takes when it is in the same solvent and ion environment but with no association interaction force on it. We performed very short simulations (200 ps for C60, B-DNA, ssDNA, and damaged DNA, 10 ps for A-DNA because A-DNA tends to convert into its B-form under the simulation solvent conditions) of free DNA and C60 in water with counterions to calculate 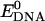 and
and 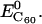 We should point out that the binding energy reported in this work is not free energy. Free-energy calculations are preferable, but the tools available to us for the systems of interest can be highly approximate (such as the Poisson-Boltzmann/surface-area free-energy estimation techniques in MMTSB, The Scripps Research Institute) and we did not attempt to carry out such calculation.
We should point out that the binding energy reported in this work is not free energy. Free-energy calculations are preferable, but the tools available to us for the systems of interest can be highly approximate (such as the Poisson-Boltzmann/surface-area free-energy estimation techniques in MMTSB, The Scripps Research Institute) and we did not attempt to carry out such calculation.
The potential energies were computed from the simulation structures collected, using the software MMTSB. MMTSB operates on the equilibrated structures of DNA and C60 collected during molecular dynamics and invokes an instantaneous potential energy calculation, employing the AMBER package and its utilities. It reports the potential energies of the molecules of interest, or of the associated hybrids.
RESULTS AND DISCUSSION
Our results indicate that C60 forms stable complexes with aqueous DNA (Fig. 1). In our initial simulation configurations C60 and DNA are separated, with solvent molecules in between. The first contact between the C60 molecule and the DNA typically occurs after 1–2 ns. Once the fullerenes diffuse and bind with the nucleotides, C60-DNA complexes are formed and remain stable for up to 20 ns of the simulations. Initially it appeared that the C60 molecules are prone to bind only to the free ends of the double-strand DNA. However, extensive simulations indicate that association of C60 can also occur at the minor groove sites. No association has been observed at the major groove sites in our simulations. These observations are different from a recent experimental observation, according to which a water-soluble (i.e., functionalized) C60 derivative can interact strongly with DNA, the major groove of the double-helix and phosphate backbone acting as the binding sites (27). However, we observe that the C60 considered here is hydrophobic and not hydrophilic.
FIGURE 1.

(a) A typical starting simulation configuration for a C60 molecule interacting with a double-strand DNA molecule in aqueous solution. Water molecules and Na+ ions in the simulation cell are not shown for clarity. During the course of the simulation, C60 approaches either the hydrophobic end (b and c) or the minor groove (d and e) of the double-strand DNA. Once energetically favorable complexes are formed, fullerenes do not leave their docking site for the remainder of our simulations, suggesting that stable complexes are formed. Snapshots of final configurations are reported in c and e. The binding energy, ΔE, for the C60-DNA hybrid is −30.2 or −35.7 kcal/mol for the structures shown in c and in e, respectively. By comparison, the thermal fluctuations of the system is in the order of 0.6 kcal/mol (300 K) and the binding energy between two C60 molecules in the same simulation condition is ΔE = −7.5 kcal/mol. To visualize the progress of the simulation, we report the distance between the center of mass of the C60 molecule and that of one DNA end (defined as the center of mass of the DNA end basepairs C12–G13) as a function of simulation time (b). The same quantity for the C60 molecule and the minor groove site versus simulation time is shown in d. In both cases, it can be seen that after ∼2 ns the fullerene is able to find a docking site in the nucleotide. Once the C60 molecule reaches the docking site, the hybrid remains stable for the remainder of the simulation.
We note that the free ends of the double-strand DNA fragment are the explicit hydrophobic sites exposed in solution, and this may favor the diffusion of hydrophobic fullerenes toward their docking sites. The two free ends of our double-strand DNA segment are constituted by either A-T or G-C basepairs. We performed simulations with C60 in proximity of both ends and did not notice appreciable differences in the binding features. Careful calculation of the binding energy ΔE reveals that the hybrid C60-DNA complexes are energetically favorable compared to the unpaired molecules (the binding energy goes from ΔE = −35.9 kcal/mol, for C60 docked in the minor groove, to −30.4 kcal/mol, for C60 docked at the DNA ends). For comparison, the thermal fluctuations under the simulation condition are in the magnitude of 0.6 kcal/mol (300 K). We also found that the deformation energy of B-DNA is ∼6.5 kcal/mol, which indicates that the C60-DNA association causes an energetically unfavorable deformation in the DNA molecule. However, the deformation energy of C60 observed in simulations is <0.5 kcal/mol and so is negligible.
We also simulated the self-association of C60 molecules in the presence of DNA molecules in the same aqueous solution. The result is summarized in Fig. 2. In this simulation, we placed two C60 molecules and one B-DNA in the simulation box. Self-association between C60 molecules was observed at early stages during the simulation, with a binding energy of ΔE = −7.5 kcal/mol. However, the stable configuration obtained after 5 ns of simulation indicates that the self-association between C60 molecules did not prevent one of the C60 molecules from binding to one end of the DNA. Both C60 molecules migrated to the end of the DNA, with one C60 docked to the DNA end; the other C60 remains associated with the DNA-docked C60.
FIGURE 2.

Self-association of C60 molecules in the presence of a DNA molecule. (a) The starting configuration of two C60 molecules and a B-DNA molecule. (b) The stabilized hybrid of C60-DNA after 5-ns simulation. One C60 binds with an end of the DNA, while the other C60 associates with it. The binding energy between the two C60 molecules is −7.5 kcal/mol. It can be seen from the final configuration that the self-association of the two C60 molecules did not inhibit the binding between one C60 and one hydrophobic end of the DNA.
Despite the large deformation energies discussed earlier, visual observation of the simulation results suggests that the overall shape of the double-strand DNA molecule is not appreciably affected by the association of C60, in agreement with experimental observations of DNA/fullerene derivative hybrid materials (28). However, the impact on the DNA structure is more pronounced when C60 interacts with a double-strand DNA fragment in the A-form (Fig. 3). DNA fragments in their A-form differ from their B-form counterparts in that more hydrophobic contacting surfaces are exposed at the end because of the open channel along the axis. The nucleotide segment modeled in this work is expected to transform into the narrower B-form under the solution conditions considered here (24,25). The simulation started from initial conditions similar to those for C60/B-DNA simulations (see Fig. 1) except the initial DNA structure was replaced by its A-form counterpart. Our results (Fig. 3 a) indicate that a C60 molecule is able to penetrate into one free end of an A-form DNA fragment. A closer look at the basepairs being penetrated indicates that the hydrogen bonds between them are disconnected. The C60 molecule docked in the DNA end is able to come in contact with the face of the basepairs second from the end. The binding energy that corresponds to this docking amounts to −41.9 kcal/mol. The observed deformation energy of the A-DNA is ∼−4.5 kcal/mol. We observe that the deformation energy is negative, while it was positive in our C60/B-DNA simulations. The favorable deformation energy observed for A-DNA is due to the transition of the nucleic acid to its more stable B-form. Although it is known that the hydrogen bonds between the end basepairs of a double-strand DNA may in some cases break during the course of molecular simulation (24,25), such bond breakage is temporary and recovered through thermal fluctuations as the simulation proceeds. In contrast, in our study, we find that the hydrogen-bond breakage encountered when a C60 molecule docks into the end of the A-form DNA, is not restored during the course of the simulation, and appears to be permanent. The docked C60–DNA complex is very stable and the broken hydrogen bonds are not reconnected even after 10 ns of simulation (see Fig. 3 d).
FIGURE 3.

Deformation of an A-form DNA when it interacts with C60 molecules. (a) Two C60 are associated with the DNA molecule. One penetrates into the A1–T24 end of DNA, the other docked by the minor groove. This figure is the snapshot after 10-ns simulation. (b) The stick presentation of a. The docking of one C60 molecule on the minor groove perturbs the stacking of consecutive basepairs, resulting in an angle of ∼20° between the basepair planes that bite the C60 molecule. (c) The C60 molecule penetrates into the first end basepairs (A1–T24) of the DNA and is able to contact with the second basepair (G2–C23) from the end. This is indicated by the distance d between the C60 molecule and center-of-mass of the second basepair as a function of simulation time. (d) The C60 molecule penetrating into one end of the DNA (A1–T24) disrupts the hydrogen bonds of the end basepairs. Here we show the length of one of the hydrogen bonds in the end basepairs A1–T24, with (upper line) and without (lower line) C60 docking into the DNA end. The hydrogen-bond length is defined as the distance between one pair of H-bonding atoms in the end basepairs.
In addition, we observe an appreciable deformation on the basepair stacking angles of the A-form double-strand DNA when the C60 docks on its minor groove sites (Fig. 3 b). Although the A-form DNA under our simulation condition is expected to transform into its B-form with the stacking planes between consecutive basepairs parallel to each other (25), the docking of one C60 into the minor groove induces an angle of 20° between the basepair planes that bite the C60. The deformation of the nucleotide structure results in a slightly greater binding energy (−36.9 kcal/mol) than that of C60 binding to the minor groove of a regular B DNA (−35.7 kcal/mol). Examining the structural details of C60/B-DNA and C60/A-DNA hybrids more carefully, we see that the binding of C60 on the minor groove of A-DNA is somewhat prohibited from fully relaxing into its B-form, as indicated by the nonparallel basepairs in the region near to where C60 docks. This allows the C60 molecule to contact deeper, and with a larger contact area into the A-DNA groove than it does with a B-DNA (see Fig. 4).
FIGURE 4.

Complexes of C60 in the minor groove, starting from the B-DNA (a) and A-DNA (b) conformations. C60 has more contact with DNA starting from A-form than that from B-form, resulting in a slightly greater (more negative) binding energy.
As might be expected on the basis of the double-strand DNA results, the docking of a C60 into a flexible single-strand DNA molecule causes significant deformations in the DNA (Fig. 5 a). We started additional simulations by placing three C60 and one single-strand DNA in the simulation box. After the system reaches equilibrium, we observe that all the C60 molecules in the system are essentially wrapped by nucleotides. One end of the single-strand DNA bends severely because of the two fullerenes docked inside. The other end maintains a relatively native conformation because no C60 molecule binds to it. The C60 docked in the central region of the single-strand DNA induces significant structural deformations on the two basepairs it contacts. The stacking of the base rings is completely deformed and the bases contact the C60 in a face-on pattern. We speculate that a sufficiently long single-strand DNA would completely wrap a C60. However, we did not test this hypothesis, because of computing time limitations. The single-strand DNA and C60 hybrids observed in this work are energetically favorable, with binding energies in the range −27.7 to −39.3 kcal/mol, depending on atomistic docking details. The deformation energy computed for one single-strand DNA (Fig. 5) is ∼5.1 kcal/mol. We noted that a free single-strand DNA is very flexible and therefore can experience large deformation. That helps to explain the relatively small deformation energy of single-strand DNA, even though significant deformation is seen in the ssDNA-C60 hybrids.
FIGURE 5.

(a) Binding of C60 with a single-strand DNA. (b) Radial distribution of water molecules (O atom) around the hydrophobic site (line with no peak) and hydrophilic site (line with peak around r = 4) of DNA after the formation of DNA-C60 hybrid. Here the hydrophobic site is defined as one of the carbon atoms on the base ring of the binding end of the DNA; the hydrophilic site is defined as one of the phosphorus atoms along the sugar frame.
We attribute the binding between the C60 and DNA molecules to the hydrophobic interaction between the C60 and hydrophobic sites on the DNA. As indicated previously, the binding energy being measured is not free energy, but includes contributions from both free energy ΔG and entropic term TΔS. For nonpolar molecules in an aqueous environment, the hydrophobic interaction is dominated by entropy change. This is due to the high entropy decrease during water structural rearrangement for the nonpolar solutes to disperse into the solvent (29). This is supported by our structural analysis of water molecules surrounding the binding sites in the nucleotide compared with a hydrophilic site (Fig. 5 b). Water molecules are completely displaced from the binding site after the docking process stabilizes. Our contention that hydrophobic forces dominate agrees with earlier observations according to which electrostatics are not the driving force for the association between fullerenes and nucleotides (30). Ideally, one could estimate the entropic contribution to the binding process by analyzing the system dynamics using techniques such as those of Smith and co-workers (31,32). However, such analysis is nontrivial and we did not attempt to estimate the entropic interactions.
The above described tendency of C60 to bind and form stable hybrids with the DNA segments led us to investigate the behavior of C60 in proximity of a damaged nucleotide. In this simulation, we removed a piece of nucleotide with the length of four bases from the central part of one DNA strand. The resulting double-strand DNA shows one defect gap in its structure (Fig. 6). The width of the defect gap is ∼1.4 nm. C60 has a diameter of ∼1 nm. We find that when a C60 diffuses near this structure it is rapidly attracted within the defect and forms a stable hybrid with a binding energy of −48.3 kcal/mol. The interaction between the DNA and C60 deformed the stacking of the binding bases for C60 to have a face-on contact with them, as found in C60 and single-strand DNA simulations. The significant deformation of the bases contacting the C60 molecule results in a deformation energy of 11.0 kcal/mol. The hybrid remains very stable during the entire course of simulation. The results suggest that C60 fills in the defect gap, and so thereby may impact the self-repairing process of the damaged nucleotide by blocking the repair sites.
FIGURE 6.

C60 occupies the defect site of the DNA. (a) Starting configuration of defected DNA and C60. (b) The snapshot after 3.8-ns molecular dynamics simulation. The defect site occupied by the C60 molecule corresponds to the vacancy left when a section of the nucleotide was removed. The angles of the basepair planes above and below the buckyball were deformed after the C60 molecule binds to the defect. (c) Distance between the center of mass of the C60 molecule and the center of mass of the bases right above and below the C60 molecule.
CONCLUSIONS
In summary, we have shown, based on molecular dynamics simulations, that C60 can bind to nucleotides and form energetically stable hybrids in aqueous solution. The favorable binding sites in double-strand DNA are the free ends and minor grooves. The binding between C60 and B-form double-strand DNA does not affect the overall shape of the DNA. In contrast, C60 can penetrate into an A-form DNA from the free end and permanently break the hydrogen bonds between the end basepairs. Furthermore, the C60 docked on the minor groove of A-DNA deforms the stacking angles of bases contacting it. C60 binds strongly with single-strand DNA and results in a significant deformation of the nucleotides. In addition, we find that a C60 molecule can readily occupy a defect site in a double-strand DNA and form a stable complex. The set of simulation results presented here suggest the possibility that C60 molecules may interfere with the biological functions performed by DNA, and therefore cause long-term negative side effects in living organisms. However, additional studies are required to determine whether the binding events observed in these simulations have in vivo relevance.
Acknowledgments
The authors thank an anonymous referee for pointing out the role of the deformation energy in the total binding energies.
This research used resources of the Center for Computational Sciences at Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under contract No. DE-AC05-00OR22725. X.C.Z. acknowledges the ORNL-ORAU postdoctoral programs operated by the Oak Ridge Associated Universities. A.S. and P.T.C. additionally acknowledge support of this research from the National Science Foundation through grant No. DMR-0103399 to Vanderbilt University.
Alberto Striolo's present address is School of Chemical, Biological, and Materials Engineering, University of Oklahoma, Norman, OK 73019.
References
- 1.Service, R. F. 2003. Nanomaterials show signs of toxicity. Science. 300:243. [DOI] [PubMed] [Google Scholar]
- 2.Brumfiel, G. 2003. Nanotechnology: a little knowledge…. Nature. 424:246–248. [DOI] [PubMed] [Google Scholar]
- 3.Colvin, V. L. 2003. The potential environmental impact of engineered nanomaterials. Nat. Biotechnol. 21:1166–1170. [DOI] [PubMed] [Google Scholar]
- 4.Lam, C. W., J. T. James, R. McCluskey, and R. L. Hunter. 2004. Pulmonary toxicity of single-walled carbon nanotubes in mice at 7 and 90 days after intratracheal instillation. Toxicol. Sci. 77:126–134. [DOI] [PubMed] [Google Scholar]
- 5.Warheit, D. B., B. R. Laurence, K. L. Reed, D. H. Roach, G. A. M. Reynolds, and T. R. Webb. 2004. Comparative pulmonary toxicity assessment of single-wall carbon nanotubes in rats. Toxicol. Sci. 77:117–125. [DOI] [PubMed] [Google Scholar]
- 6.Kroto, H. W., J. R. Heath, S. C. O'Brien, R. F. Curl, and R. E. Smalley. 1985. C60—Buckminsterfullerene. Nature. 318:162–163. [Google Scholar]
- 7.Krätschmer, W., L. D. Lamb, K. Fostiropoulos, and D. R. Huffman. 1990. Solid C60—a new form of carbon. Nature. 347:354–358. [Google Scholar]
- 8.Tokuyama, H., S. Yamago, E. Nakamura, T. Shiraki, and Y. Sugiura. 1993. Photo-induced biochemical activity of fullerene carboxylic acid. J. Am. Chem. Soc. 115:7918–7919. [Google Scholar]
- 9.Tabata, Y., Y. Murakami, and Y. Ikada. 1997. Photodynamic effect of polyethylene glycol-modified fullerene on tumor. Jpn. J. Cancer Res. 88:11108–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noon, W. H., Y. F. Kong, and J. P. Ma. 2002. Molecular dynamics analysis of a buckyball-antibody complex. Proc. Natl. Acad. Sci. USA. 99:6466–6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao, H. J., and Y. Kong. 2004. Simulation of DNA-nanotube interactions. Annu. Rev. Mater. Res. 34:123–150. [Google Scholar]
- 12.Nelson, M. A., F. E. Domann, G. T. Bowden, S. B. Hooser, Q. Fernando, and D. E. Carter. 1993. Effects of acute and subchronic exposure of topically applied fullerene extracts on the mouse skin. Toxicol. Ind. Health. 9:623–630. [DOI] [PubMed] [Google Scholar]
- 13.Scrivens, W. A., J. M. Tour, K. E. Creek, and L. Pirisi. 1994. Synthesis of C14-labeled C60, its suspension in water, and its uptake by human keratinocytes. J. Am. Chem. Soc. 116:4517–4518. [Google Scholar]
- 14.Yamago, S., H. Tokuyama, E. Nakamura, K. Kikuchi, S. Kananishi, K. Sueki, H. Nakahara, S. Enomoto, and F. Ambe. 1995. In vivo biological behavior of a water-miscible fullerene: 14C labeling, adsorption, distribution, excretion and acute toxicity. Chem. Biol. 2:385–389. [DOI] [PubMed] [Google Scholar]
- 15.Rajagopalan, P., F. Wudl, R. F. Schinazi, and F. D. Boudinot. 1996. Pharmacokinetics of a water-soluble fullerene in rats. Antimicrob. Agents Chemother. 40:2262–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rancan, F., S. Rosan, F. Boehm, A. Cantrell, M. Brellreich, M. Schoenberger, A. Hirsch, and F. Moussa. 2002. Cytotoxicity and phototoxicity of a dendritic C60 mono-adduct and a malonic acid C60 tris-adduct on Jurkat cells. J. Photochem. Photobiol. B Biol. 67:157–162. [DOI] [PubMed] [Google Scholar]
- 17.Oberdörster, E. 2004. Manufactured nanomaterials (Fullerenes, C60) induce oxidative stress in the brain of juvenile largemouth Bass. Environ. Health Perspect. 112:1058–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosi, S., L. Feruglio, T. Da Ros, G. Spalluto, B. Gregoretti, M. Terdoslavich, G. Decorti, S. Passamonti, S. Moro, and M. Prato. 2004. Hemolytic effects of water-soluble fullerene derivatives. J. Med. Chem. 47:6711–6715. [DOI] [PubMed] [Google Scholar]
- 19.Andrievsky, G. V., V. K. Klochkov, A. B. Bordyuh, and G. I. Dovbeshko. 2002. Comparative analysis of two aqueous-colloidal solutions of C-60 fullerene with help of FTIR reflectance and UV-Vis spectroscopy. Chem. Phys. Lett. 364:8–17. [Google Scholar]
- 20.Deguchi, S., R. G. Alargova, and K. Tsujii. 2001. Stable dispersions of fullerenes, C-60 and C-70, in water. Preparation and characterization. Langmuir. 17:6013–6017. [Google Scholar]
- 21.Case, D. A., T. A. Darden, T. E. Cheatham III, C. L. Simmering, J. Wang, R. E. Duke, R. Luo, K. M. Merz, B. Wang, D. A. Pearlman, M. Crowley, S. Brozell, V. Tsui, H. Gohlke, J. Mongan, V. Hornak, G. Cui, and P. Beroza. P., C. Schafmerster, J. W. Caldwell, W. S. Ross, P. A. Kollman. 2004. AMBER 8. University of California, San Francisco.
- 22.Jorgensen, W. L. 1981. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 103:335–340. [Google Scholar]
- 23.Hummer, G., J. C. Rasaiah, and J. P. Noworyta. 2001. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 414:188–190. [DOI] [PubMed] [Google Scholar]
- 24.Kalra, A., S. Garde, and G. Hummer. 2003. Osmotic water transport through carbon nanotube membranes. Proc. Natl. Acad. Sci. USA. 100:10175–10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheatham III, T. E., and P. A. Kollman. 2000. Molecular dynamics simulation of nucleic acids. Annu. Rev. Phys. Chem. 51:435–471. [DOI] [PubMed] [Google Scholar]
- 26.Cheatham III, T. E., and M. A. Young. 2001. Molecular dynamics simulation of nucleic acids: successes, limitations, and promise. Biopolymers. 56:232–256. [DOI] [PubMed] [Google Scholar]
- 27.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. D. Nola, and J. R. Haak. 1984. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 28.Pang, D. W., Y. D. Zhao, P. F. Fang, J. K. Cheng, Y. Y. Chen, Y. P. Qi, and H. D. Abruňa. 2004. Interactions between DNA and a water-soluble C60 derivative studied by surface-based electrochemical methods. J. Electroanal. Chem. 567:339–349. [Google Scholar]
- 29.Israelachvili, J. N. 1985. Intermolecular and Surface Forces: With Applications to Colloidal and Biological Systems. Academic Press, London. 102–107.
- 30.Cassell, A. M., W. A. Scrivens, and J. M. Tour. 1998. Assembly of DNA/fullerene hybrid materials. Angew. Chem. Int. Ed. Engl. 37:1528–1531. [DOI] [PubMed] [Google Scholar]
- 31.Smith, D. E., L. Zhang, and A. D. J. Haymet. 1992. Entropy of association of methane in water—a new molecular dynamics computer simulation. J. Am. Chem. Soc. 114:5675–5676. [Google Scholar]
- 32.Smith, D. E., and A. D. J. Haymet. 1993. Free-energy, entropy, and internal energy of hydrophobic interactions—computer simulations. J. Chem. Phys. 98:6445–6454. [Google Scholar]