

Abstract
The desire to understand tumor complexity has given rise to mathematical models to describe the tumor microenvironment. We present a new mathematical model for avascular tumor growth and development that spans three distinct scales. At the cellular level, a lattice Monte Carlo model describes cellular dynamics (proliferation, adhesion, and viability). At the subcellular level, a Boolean network regulates the expression of proteins that control the cell cycle. At the extracellular level, reaction-diffusion equations describe the chemical dynamics (nutrient, waste, growth promoter, and inhibitor concentrations). Data from experiments with multicellular spheroids were used to determine the parameters of the simulations. Starting with a single tumor cell, this model produces an avascular tumor that quantitatively mimics experimental measurements in multicellular spheroids. Based on the simulations, we predict: 1), the microenvironmental conditions required for tumor cell survival; and 2), growth promoters and inhibitors have diffusion coefficients in the range between 10−6 and 10−7 cm2/h, corresponding to molecules of size 80–90 kDa. Using the same parameters, the model also accurately predicts spheroid growth curves under different external nutrient supply conditions.
INTRODUCTION
Primary malignant tumors are thought to arise from small nodes of cells that have either lost, or ceased to respond to, normal growth regulatory mechanisms, presumably through mutations and/or altered gene expression (1,2). As the tumor develops, this genetic instability causes continued malignant alterations, including invasion, angiogenesis, and metastatic spread, resulting in a large, biologically complex tumor. However, essentially all tumors, both primary and metastatic, go through a relatively simpler, avascular stage of growth, with nutrient supply by diffusion from the surrounding tissue. Before developing a blood supply, these tumors are not capable of acquiring sufficient nutrients to ensure continued exponential growth of the tumor cell mass despite the continuous nutrient supply at the tumor surface. Thus, avascular tumors undergo a quasi-exponential growth phase followed by a saturation phase in which they maintain approximately constant volume. The restricted supply of critical nutrients, such as oxygen and glucose, results in marked gradients within the cell mass. The tumor cells respond through induced alterations in physiology and metabolism as well as through altered gene and protein expression (3,4). In fact, many of the later manifestations of malignancy, including invasion and angiogenesis, are thought to be enhanced, if not precipitated, by the stressful microenvironment that develops in the initial, avascular tumor nodule. Consequently, a better understanding of the regulation of the growth and malignant development of avascular tumors would be beneficial; insights in such systems would also be valuable in understanding the heterogeneous microenvironments found within larger tumors (5).
Multicellular tumor spheroids are a frequently used in vitro model of avascular tumor growth and the microenvironmental and physiological perturbations that occur in tumors (6,7). Spheroids are aggregates of tumor cells that can be grown in precisely controlled external nutrient conditions, and assays of spheroid parameters, such as volume, cell number, viable and necrotic fractions, and saturation size, are relatively easily obtained (8–10). Nutrient supply to spheroids is through diffusion from the surface. Thus, as the aggregate grows, it develops nutrient-deprived inner regions. Spheroids develop many of the hallmark features of avascular tumors, including proliferation arrest, altered metabolism, perturbed gene and protein expression, necrotic death, and therapy resistance (3,6,7). In addition, spheroid growth curves show the same kinetics as those of nodular tumors in vivo, including quasi-exponential growth and saturation in size (8,9,11).
A descriptive model (8) to explain the regulation of growth and viability in spheroids postulates that, at early stages of development, both growth promoters and viability promoters can reach all of the cells in the spheroid. During this early stage, the aggregate is composed of proliferating, viable cells. As the spheroid grows, the concentration of growth promoters decreases in the spheroid center, which eventually falls below a critical value such that cells undergo proliferation arrest and become quiescent. However, since the spheroid continues to grow due to the outer proliferating cells, the central concentration of viability promoters continues to decrease. Once the concentration of viability promoters drops below a critical value, necrotic cell death occurs and the spheroids acquire a necrotic center. Continued cellular metabolism and/or the process of necrosis cause growth inhibitors and viability inhibitors to be secreted and accumulate in the spheroid. When the concentration of growth inhibitors reaches a critical value in the outer spheroid region, cell proliferation is further reduced. When viability inhibitors reach a critical value, they also contribute to the expansion of the necrotic center. Eventually, the thickness of the proliferating layer of cells is reduced to a point at which the number of new cells is equal to the number of cells lost by cell shedding, causing saturation in the spheroid growth. Experimental data supports the idea that simple molecules involved in energy metabolism, such as oxygen and glucose, are the viability promoters in spheroids (8,9). There is also some limited data indicating that growth inhibitors are small protein factors (12). Currently, however, essentially nothing is known about growth promoters or viability inhibitors in this tumor model system, or in avascular tumors in vivo.
Recent molecular research with the spheroid system has suggested that the factors regulating proliferation act through signaling pathways, which are connected to the cyclin-cyclin dependent kinase (CDK) cell-cycle regulatory mechanism. The primary regulatory mechanism in this tumor model seems to be the induction of cyclin-dependent kinase inhibitors (CKIs). LaRue et al. (13) showed that a large upregulation of the CKI p18 occurred in untransformed fibroblasts cultured as spheroids, which accounts for their arrest in the G1-phase and inability to proliferate in aggregate culture. Transformed fibroblasts did not show this upregulation of p18, and spheroids of such cells are able to grow to large sizes. Recently, the same group has demonstrated that the initial induction of G1-phase arrest in large spheroids, presumably in response to some microenvironmental gradient of growth promoters, is due to the upregulation of two CKIs from different families, p18 and p27, with little change in CDKs or other CKIs (14). As spheroids reach sizes near that of growth saturation, with a sizable necrotic center, it was also shown that the innermost cells downregulated all of their cell-cycle regulatory machinery, including cyclins, CDKs, and CKIs (14). The latter result may be due to cell exposure to a growth-inhibitory factor, or may be the result of a prolonged period spent in a nutrient-stressed state. In either case, these results demonstrate that proliferation arrest in this avascular tumor model is controlled by a protein regulatory network operating within the tumor cells.
A predictive model of avascular tumor growth has to account for the complexity of these processes. Important elements that need to be incorporated in such a model include cell proliferation and growth, nutrient consumption and diffusion, waste product production and diffusion, effects of growth promoting and inhibitory factors, intercellular adhesion, and cell-environment interactions, as well as the geometry of the tumor and the cells. Most of the existing models for cellular dynamics in tumors are either simple empirical mathematical expressions (11,15), rate equations of cell populations (16–22), or cellular automaton models of interacting cells, each occupying a single lattice site (23,24). The only previous tumor model that included cell geometry was able to reproduce a layered structure only by introducing an artificial potential (25). A recent model that employs a hybrid of cellular automata for cell representation and continuous equations for chemical and blood flow in a hexagonal grid of blood vessels represents the state-of-the-art in tumor growth modeling (26,27).
In this article, we present a multiscale cellular model to describe the dynamics of avascular tumor growth and development. At the cellular scale, our model considers cell growth and proliferation, intercellular adhesion, and necrotic cell death. At the subcellular scale, we include a protein expression regulatory network for the control of cell-cycle arrest. At the extracellular scale, the model considers diffusion, consumption, and production of nutrients, metabolites, growth promoters, and inhibitors. Data from experiments with multicellular spheroids were used to determine the parameters for the simulations. Starting with a single tumor cell, this model naturally evolves with time to produce an avascular tumor that quantitatively mimics experimental measurements in multicellular spheroids.
METHODS
Multicellular tumor spheroids
Multicellular tumor spheroids of the EMT6/Ro mouse mammary tumor cell line were cultured in suspension as described in detail previously (8,10). Spheroids were initiated from monolayer-cultured cells, which were harvested to generate a uniform-sized population containing 1 × 104 spheroids. These spheroids were then placed into four large spinner flasks fitted with an apparatus for supplying a controlled gas content and humidity, and were replenished with fresh medium every 10–14 h. External concentrations of oxygen and glucose were maintained within 5% of the initial value over a range of concentrations from 0.08 to 0.28 mM oxygen and 1.6 to 16.5 mM glucose (10). As shown in Fig. 1, the spheroids developed a spherically symmetric distribution of proliferating and quiescent cells surrounding a necrotic core.
FIGURE 1.

Illustration of the morphology and proliferative status of cells in multicellular spheroids of the EMT6/Ro cell line. (A) Histological cross-section (optical microscopic image) through the center of a spheroid ∼1200 μm in diameter stained with eosin and hematoxalin, showing the viable rim of cells (red) and the necrotic center (orange). (B) Diagram illustrating the relative distributions of proliferating (green, P) and quiescent (red, Q) cells and central necrosis (gray, N) in a spheroid, relative to the gradients in nutrients and waste products. There is not actually a sharp demarcation between proliferating and quiescent cells as is the case for viable/necrotic boundary; rather, the fraction of proliferating cells decreases continuously across the viable cell rim.
For spheroids <400–500 μm in diameter, sampling was done by removing a volume of the well-mixed spheroid suspension. For measuring the population mean diameter, a 10-ml sample was removed immediately after medium change, assayed, and replaced into the flask. For measuring the number of cells per spheroid, 100 ml of the medium containing the spheroids were removed at the time of medium change every 48 h, dissociated into single cells, and assayed. For larger spheroids, sampling is done by collecting the spheroids that settle in the flask and manually removing the desired number of spheroids. Spheroids were processed for histology after fixation in 10% buffered formalin using standard techniques, and 5-μm-thick sections were stained with eosin and hematoxalin to distinguish the viable and necrotic regions (9,10). Parameters measured in previous studies were used to calibrate and refine the simulation parameters. These parameters include: total spheroid volume, number of cells per spheroid, cell-cycle phase distribution, and the thickness of the viable cell rim (8,9). Previously published data on oxygen and glucose consumption rates (28,29) and oxygen tension distributions within spheroids (30) were also used in our simulations.
Multiscale cellular model
Our multiscale cellular model consists of three levels. At the cellular level, a discrete lattice Monte Carlo model considers cell growth, proliferation, death, and intercellular adhesion. At the subcellular level, a simplified Boolean protein expression regulatory network controls cell-cycle arrest. At the extracellular level, a system of differential equations describes diffusion, consumption, and production of nutrients, metabolites, growth promoters, and inhibitors. The three levels are closely integrated. We use model parameters derived from previous multicellular spheroid experiments.
Cellular model
The cellular model is based on the extended large-Q Potts model (31,32). A simpler version of cellular and extracellular levels has been published previously (32) and a model framework has been developed recently (33). Briefly, the discrete lattice Monte Carlo model partitions the three-dimensional space into domains of cells and cell medium. Every cell is treated as an individual entity with a unique ID number, which occupies all the lattice sites within the cell domain (see, e.g., Fig. 2 in (32)). In this representation, a cell has a finite volume and a deformable shape. A typical cell occupies 27 lattice sites. The extracellular matrix in the spheroids is neglected. Cells have direct contact and interact with each other through surface adhesion and competition for space. The interactions are characterized through a total energy of
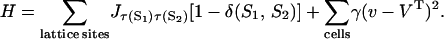 |
(1) |
In the first term, S is the cell ID, Jττ′ corresponds to the adhesive energy between cell types τ and τ′, and δ is the Kroneker δ-function; this term describes the total energy due to cell surface adhesion to each other. Cell-type-dependent adhesion is based on the Differential Adhesion Hypothesis (34,35). Different cell types may express different cell adhesion molecules or a different number of cell adhesion molecules on their surfaces, and thus have different strengths of adhesivity; the differential adhesion hypothesis states that the difference in cell adhesivity drives cell sorting into minimal surface energy configuration. In the second term, v and VT are the current and the target volume of the cell, respectively, and γ is the coefficient corresponding to the elasticity of the cell volume. Any deviation from the target volume gives rise to a volume energy, which keeps the cell volume close to the target volume. Note that our cell type refers to the proliferating status of the cell: proliferating, quiescent, or necrotic and medium, and not the tissue type. Moreover, in our model, the different cell types only differ in their physical properties (cell-cycle duration, metabolic rates, cell adhesion, and maximum volume). External cell culture medium and the necrotic core are treated as special cells. Medium does not have a target volume; thus, proliferating cells can invade the external space when they grow. A necrotic cell, on the other hand, has a target volume set to its current volume, and a large γ-value corresponding to a rigid body. Thus, its space cannot be invaded by the growing mass of viable tumor cells. Every time a cell dies, its volume is added to the target volume of the necrotic core. Our model does not consider apoptosis; cell death means strictly necrosis in this article. This assumption is based on the lack of any experimental data showing apoptosis in EMT6/Ro spheroids, as well as the fact that the majority of cell death in the spheroids occurs by necrosis (6). Spheroid experiments using other cell lines, e.g., Rat 1-T1 and MR1, have shown a low level of single cell death via apoptosis occurring in the viable cell rim despite sufficient nutrient availability (36). This feature can be easily accommodated in our model, although we believe it will have little impact on the overall spheroid dynamics.
FIGURE 2.

Simplified protein regulatory network for the G1-S phase transition. The G1 phase consists of six stages (six levels of grayscale).
This cellular model evolves by a standard Monte Carlo procedure. A random lattice site is selected; the cell ID at this site is changed to the value of one of its unlike neighbors' ID. The probability for accepting such a change is
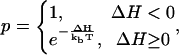 |
(2) |
where ΔH is the total energy difference due to such a change, and T is the effective cell temperature, corresponding to the amplitude of cell membrane fluctuation (37). A Monte Carlo step (MCS) consists of as many trial lattice updates as the total number of lattice sites. The cell system minimizes its total energy. If there is no cell growth, division, or death, this model would reproduce the cell configuration that minimizes the total cell surface adhesion energy (31).
We set the target volume to be twice the initial cell volume, so that the cell grows in time until its volume reaches the target volume. Each cell also carries a cell clock, which ticks to a maximum time corresponding to the duration of a cell cycle. Only when the cell clock reaches the cell-cycle duration and the cell volume reaches the target volume will the cell decide to divide. Cell division is simply reassigning half of the volume to a new cell ID. The daughter cells inherit all properties of their parent.
Extracellular microenvironment
Cells also interact with their microenvironment, which is characterized by local concentrations of biochemicals. The extracellular microenvironment includes nutrients (oxygen and glucose), metabolic waste, growth promoters, and inhibitors. Based on previous measurements with the EMT6 cell line (28), we assume that most of the glucose consumed (75%) flows through the anaerobic glycolysis and produces lactate as waste, while a minority (25%) flows through the Krebbs cycle and respiration; oxygen consumption is connected to glucose consumption through respiratory catabolism to generate CO2, which rapidly diffuses away. Hence, we consider that the main waste in the tumor is lactate, and that the lactate production rate is 1.5 times the glucose consumption rate.
In our model, oxygen and glucose are viability promoters, while lactate is viability inhibitor. Our model considers generic growth and inhibitory factors. Chemical reaction-diffusion is described by
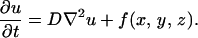 |
(3) |
This is the generic equation for all chemicals in our model (detailed equations are shown in Appendix): the chemical concentration u diffuses with the diffusion coefficient D and is produced (or consumed) at rate f. The metabolic rate f depends on the individual cell's state (proliferating, quiescent, or necrotic); thus, it is a function of location.
We make a few further simplifying assumptions. In reality, a spheroid consists of tumor cells, their extracellular matrix, and the necrotic core; chemicals diffuse in the extracellular matrix, and bind and are internalized (or generated and secreted) by the cells; chemicals also diffuse in the necrotic core with a different diffusion constant. As we do not include the extracellular matrix explicitly in our cellular model, we assume that 1), inside the spheroid the diffusion coefficients are constant, neglecting the differences of diffusion rates in extracellular matrix or cells or necrotic core; and 2), each cell is chemically homogeneous, although different cells might have different chemical concentrations. In the spheroid experiments, oxygen, nutrients, and growth factors are supplied to the surface of the spheroid via convection, and the cell medium is updated frequently such that the chemical concentrations in the medium are kept constant. So we additionally assume that: 3), the cell-culture medium outside the spheroid maintains a constant level of metabolites; and 4), the external medium has no waste or inhibitory factors in it. With these assumptions, we can solve the equations on a much coarser lattice than the lattice for cells. More details are described in the Appendix.
Cell cycle
The passage of a cell through its cell cycle is controlled by cytoplasmic proteins, the main players of which include cyclins, CDKs, CKIs, and the anaphase-promoting complex. Since experiments demonstrate that >85% of the quiescent cells in spheroids are arrested in the G1-phase (9,14), in our model, the cells in their G1-phase have the highest probability of becoming quiescent. To realistically represent this cell-cycle arrest, we include a simplified protein regulatory network to control the transition between the G1- and S-phases. If the cell passes the G1-S transition checkpoint, it will most likely proceed toward mitosis (or division). Arrest of cells in the S- and G2-phases has been documented in spheroids of some cell lines, but not others (14); however, the number of such cells in a spheroid is relatively small.
Our simplified protein regulatory network, shown in Fig. 2, is based on the cell-cycle protein regulatory network for Homo sapiens from the Kyoto Encyclopedia of Genes and Genomes (http://www.kegg.com/). We include the following list of proteins: GSK3β, TGFβ, SMAD3, SMAD4, SCF, CDK inhibitors 4a–d (p15, p16, p18, p19), Kip 1,2 (p27, p57), Cip1 (p21), cyclins D and E, Rb, and E2F. We ignore a few other proteins, such as Cyclin A, from the network because they do not influence the outcome of our network. The proteins we selected come into play at different stages of the G1-phase and their influence differs in duration. For simplicity, we combine the groups of proteins whose expressions have the same effect on the final outcome of the network. Thus, p15 in Fig. 2 stands for p15, p16, p18, and p19 (the whole group of CDK inhibitors 4a–d), and p27 includes p57 expression as well. In our model, these proteins can have only two levels of expression—on and off. If the link pointing to a protein ends with an arrow, it means the link is stimulatory; if the link ends with a bar, it means the link is prohibitory.
This network of proteins is designed to favor the cell transition from G1- to S-phase. However, concentrations of the growth and inhibitory factors directly influence the protein expression, and thus the cell proliferation state. At every time step, we calculate a local factor level of
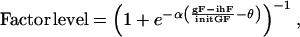 |
(4) |
where gF and ihF are current local concentrations of growth and inhibitory factors, respectively—in which both are outputs of the extracellular chemical equations; initGF is the concentration of growth factors in the medium surrounding the aggregate; θ is a factor level threshold; and α is a free parameter. If the factor level is above the threshold, the protein is turned on under two circumstances: if all the links pointing to it are stimulatory and all the proteins at the beginning of the links are on; or if all the links are prohibitory and the proteins at the beginning of the links are off. All other situations would turn off the protein. If the factor level is below the threshold, this factor level is the probability that a protein will be turned on or off—the higher the factor level (as a result of high growth factor level and low inhibitory factor level), the higher the probability of protein being turned on or off. If the outcome of this Boolean regulatory network is zero, i.e., protein E2F is off, the cell undergoes cell-cycle arrest or turns quiescent. Otherwise, it continues its transit through the cell cycle.
Simulation
The integration of these three parts of the model is illustrated in the flow chart (Fig. 3). The tumor growth starts from a single tumor cell in the center of the lattice with its first set of proteins turned on (top tier in Fig. 2). Cell growth and division follow the cell cycle, which we divide into 16 stages. According to their respective durations, we assign G1-phase to consist of six stages, S-phase of six stages, and G2- and M-phases of four stages combined. Hence the whole cell-cycle duration, which is ∼12 h in an exponentially growing monolayer culture, is equivalent to 16 stages. In our model, cells typically double their volume in four Monte Carlo steps (MCS). So each cell-cycle stage corresponds to 1/4 MCS or ∼3/4 h.
FIGURE 3.

Flow chart of the model showing the integration between the intra-, inter-, and extracellular levels.
During each iteration, we first evolve the cell lattice for 1/4 MCS, then solve chemical reaction diffusion equations for 3/4 h to obtain the concentrations of metabolites for each cell. For cells during G1-phase, the local factor levels change the expressions of proteins in the regulatory network. The cell then reacts to its local environment in the following steps. First, it checks the local chemical environment: a proliferating cell decides whether to proceed to the next stage of the cell cycle or become quiescent, while a quiescent cell decides whether to become necrotic because of hostile environment. Second, it checks the current cell volume: if it has not grown proportional to the time it has lived in this cell cycle, it will become quiescent because of stress (see below for explanation). Finally, a proliferating cell checks whether it has fulfilled the requirements to divide: if yes, it divides into two cells, if not, it continues to progress through the cell cycle. The model then repeats the iterations.
Solid stress (38) and increased interstitial fluid pressure (22,39–41) inside a solid tumor are found to inhibit cell growth in multicellular spheroids and tumors. To account for the effect external pressure may have on the cell cycle, we include check points at the end of each phase of the cell cycle to determine if the cell has increased its volume accordingly. If the cell does not increase its volume proportional to the time it has spent in that and previous phases, it will become quiescent due to pressure exerted by the surrounding tissue. When a cell turns quiescent, it reduces its metabolism and stops its growth. When a cell dies, it becomes part of the necrotic core (special cell with ID 0). For a short period of time (24 h) after the cell dies via necrosis, the cell produces inhibitory factors and some waste.
In spheroid experiments, it has been observed that mitotic cells are shed from the surface of the spheroid at a constant rate per spheroid surface—i.e., ∼218 cells per square millimeter of spheroid surface per hour for the EMT6 cell line (18,42). In our model, if a proliferating cell is at the surface of a spheroid of radius >0.03 cm (18), it can shed away with a 20% shedding probability. Shed cells disappear from further consideration in the model.
Parameters
Our simulations use parameters derived from spheroids using the mouse mammary tumor cells, EMT6/Ro. The experimental data for this particular cell line are abundant (e.g., 8,9,28–30). Although the diffusion coefficients for oxygen, glucose, and lactate are more readily available in literature (listed in Table 1), their metabolic rates are harder to come by. We have derived metabolic rates for oxygen and glucose from different sources (28,29). Stoichiometrically, every glucose molecule that goes through anaerobic catabolism by glycolysis generates two lactate molecules. Since some glucose molecules go through respiratory catabolism instead, we assumed that on average each glucose molecule consumed results in 1.5 lactate molecules, or the production rate of lactate is 1.5 times the consumption rate of glucose (see Table 1). The linkage between waste production and nutrient consumption necessitates that the waste production rate of quiescent cells is also half that of proliferating cells (28,29).
TABLE 1.
Metabolic rates for nutrients, waste, growth, and inhibitory factors for EMT6/Ro
| Oxygen | Glucose | Waste | Growth factors | Inhibitory factors | |
|---|---|---|---|---|---|
| Proliferating cell | 108* | 162* | 240* | 1† | 0† |
| Quiescent cell | 50* | 80* | 110* | 0.5† | 1† |
| Necrotic | 0 | 0 | 0 | 0 | 2† |
| Diffusion constant | 5.94 × 10−2‡ | 1.52 × 10−3‡ | 2.124 × 10−3‡ | 10−6‡ | 10−6‡ |
The unit is [mM/h/cm3].
The unit is [%/h/cm3].
The unit is [cm2/h].
We have two sets of units in the model: lattice size and Monte Carlo steps (MCS) in the cellular model, and centimeters and hours in the extracellular chemical equations. By equating a maximal cell volume in the model to a real cell size (e.g., 4 × 4 × 4 voxels = 1.2 × 103 μm3), we can convert a lattice spacing to centimeters. In addition, by equating the duration of the cell cycle in the model and in real life (e.g., 4 MCS = 12 h), we have the conversion that one MCS is equal to 3 h. With these conversions, all physical parameters can be translated to our model units, and all the model measurements can be translated into real physical units.
When we convert the metabolic and diffusion parameters to model units, we take into account the space occupied by extracellular matrix that is omitted in our cellular model. The lack of data for growth and inhibitory factors allows us to use relative scale for these factors; we assume that the medium supplies 100% of required growth factors, and no inhibitory factors are present outside the spheroid. Consistent with experimental data, we assume that metabolic rates of quiescent EMT6/Ro cells are approximately equal to half of that of proliferating cells (28,29).
The diffusion coefficient for oxygen is derived from extensive microelectrode measurements in spheroids (30,43). The diffusion coefficients for glucose and lactate come from previous experimental determinations on spheroids (44,45). The diffusion coefficients used for the growth and inhibitory factors were determined by an iterative process to determine those that gave the best fits to the experimental data. The diffusion coefficients estimated in this fashion were in the range between 10−6 and 10−7 cm2/h.
As the development of the solid tumor is dominated by cell growth and division, as well as the response to the microenvironment, the simulation results are not sensitive to the differences in cellular adhesion, or the coupling energy coefficients Jττ′ in Eq. 1, at all. The main effect of the coupling energy is to keep cells together, rather than morphogenesis due to differential adhesion. Though no experimental evidence has indicated that quiescent cells have different cell adhesivity than proliferating ones, we keep the differential adhesion capability in the model for further model development (e.g., including endothelial cells). In all the simulations reported below, we use the following set of values for the coupling energy coefficient J: JP,P = JP,Q = 28, JP,N = 24, JP,M = 16, JQ,N = 22, JQ,M = 14, and JN,M = 12, where P, Q, N, and M stand for proliferating, quiescent, necrosis, and medium, respectively. These values could cause cells to “sort,” as in (31). But because cell sorting due to differential adhesion is a slow process, the tumor development is dominated by the growth and division of cells as well as their reaction to the chemical environment. The tumor growth results would not be different if we used one single value for all coupling coefficients.
Volume constraint coefficient γ for proliferating cells usually had value between 1 and 3. To ensure that quiescent cells do not change their volume easily, once a cell turns quiescent, we set the target volume to its current volume and increase its volume constraint to four times its current value. As cells die, their current volumes are added to the necrotic core volume, and the quiescent and proliferating cells cannot grow against the necrotic region.
In our simulations, oxygen concentration below 0.02 mM, glucose concentration below 0.06 mM, and lactate concentration above 8 mM are conditions for cell necrosis. These threshold values are determined in the following process: we start from the lowest oxygen and glucose concentrations used in experiments (e.g., 0.07 mM O2 and 0.8 mM glucose, from (9)), and tune the threshold values to produce tumor growth that best fit to the experimental growth curves. Then we use a fraction of the lowest concentrations, and further tune the values to produce good growth curves.
RESULTS
Starting from one single cell, a tumor in our simulations grows into a spheroidal, layered structure consisting of proliferating and quiescent cells surrounding a necrotic core. Fig. 4 shows the cross sections of the same spheroid at different stages of growth: the initial proliferating cell aggregate; the onset of quiescence; and the appearance of a necrotic core. The last cross section is comparable with the experimental picture in Fig. 1 a.
FIGURE 4.

From simulation, cross-sectional view of a spheroid at different stages of development, with cyan, yellow, and magenta correspond to proliferating, quiescent, and necrotic cells. From left to right, 2 days, 10 days, and 18 days of tumor development, respectively, from a single cell.
Fig. 5 shows the growth curves of a solid tumor in comparison with two sets of experimental data. With 0.08 mM oxygen and 5.5 mM glucose kept constant in the medium, the number of cells (Fig. 5 a) and the tumor volume (Fig. 5 b) first grow exponentially in time for ∼5–7 days. The growth slows down, coinciding with the appearance of quiescent cells. In both the experiments and simulations, spheroid growth saturates after ∼28–30 days. We fit both the experimental and the simulation data to a Gompertz function, y = y0 exp(a(1 − exp(−bt)), to objectively estimate the initial doubling times and the spheroid saturation sizes (8,10). The initial volume and cell number doubling times for the experiments and the simulation differed by less than an hour (8.6–9.5 h). The saturation sizes were more different, with the simulation overestimating the experimental maximal sizes by factors of 2 (cell number) and 2.5 (spheroid volume). Given that the simulation covered a range of spheroid growth of 4–5 orders of magnitude, this agreement with experiment is excellent.
FIGURE 5.

The growth curves of spheroid with 0.08 mM O2 and 5.5 mM glucose in the medium. (a) The number of cells and (b) the volume of spheroid in time. The solid diamonds and squares are experimental data for EMT6/Ro, the circles are simulation results. The solid lines are the best fit with a Gompertz function (see text) for experimental data.
Experimentally, the fraction of cells in the various cell-cycle phases was determined by standard flow cytometric measurement of cellular DNA content as described in detail previously (9). Solid symbols in Fig. 6 are experimental measurements of cell-cycle fraction for G1-, S-, and G2-phases (from (10)). Open symbols are simulation data. We see in both experimental and simulation data that as the spheroid radius increases, the fraction of cells in G1-phase increases, at the same time the fraction of cells in S-phase drops at a comparable rate. Percent of cells in G2-phase remains roughly constant throughout the spheroid growth. The simulation data showed an initially high degree of variability, mostly accounted for by the nonrandom distribution of the initial aggregate and the discrete time sampling involved.
FIGURE 6.

Cell-cycle fraction as a function of time with 0.08 mM O2 and 5.5 mM glucose in the medium. Solid symbols are experimental measurements from the EMT6/Ro cell line; open symbols are the corresponding simulation. (Red lines indicate G1-phase, black lines indicate S-phase, and blue lines indicate the G2-phase.)
During the development of a layered spheroid structure the thickness of the viable rim (proliferating and quiescent cells) remains approximately constant in time (11). In our simulation, we measured thickness of viable rim of spheroids by subtracting the necrotic core radius from the radius of the spheroid. Fig. 7 shows the simulation data for the viable rim thickness as a function of spheroid size for spheroids growing in 0.08 mM of oxygen and 5.5 mM of glucose medium, where open symbols are simulation data and closed symbols experimental measurements. The simulation results agree nicely with experiments in terms of long-term behaviors: the necrotic core increases at almost exactly the same rate as the whole spheroid, while the viable rim size remains roughly constant. However, our data show an initial rapid expansion of necrotic core at the onset of necrosis, which is not present in the EMT6 spheroid data. This rapid initial onset of necrosis in spheroids has been both predicted and demonstrated experimentally in other cell systems (17,46).
FIGURE 7.

Size of viable rim versus spheroid diameter with 0.08 mM O2 and 5.5 mM glucose in the medium. After initial linear growth, viable rim thickness reaches approximately constant value. Solid symbols are experimental data from the EMT6/Ro cell line; open symbols are simulation results.
To test the robustness of our model, we kept all the parameters in the model fixed at the values determined to produce the best fit to the growth of spheroids in 0.08 mM oxygen and 5.5 mM glucose. We then varied only the nutrient concentrations in the medium, as was done in previous spheroid experiments. Fig. 8 shows an example of the good agreement between simulation and experimental growth curves when the external conditions are changed to 0.28 mM O2 and 16.5 mM glucose in the medium. Again, fits of the experimental and simulation data to a Gompertz equation showed good agreement in the initial doubling times and the spheroid saturation sizes.
FIGURE 8.

The growth curves of spheroid with 0.28 mM O2 and 16.5 mM glucose in the medium: (a) the number of cells and (b) the volume of spheroid in time. The solid diamonds and squares are experimental data for EMT6/Ro; open circles are simulation results. The solid lines are the best fit with a Gompertz function to the experimental data.
DISCUSSION
In our simulations, a single tumor cell evolves into a layered structure consisting of concentric spheres of proliferating and quiescent cells at the surface and intermediate layer respectively, and the necrotic core at the center of the spheroid. The spheroid maximum size and the total number of viable cells depend on the chemical environment provided in the medium. Our simulations result in a set of conditions for the cell to undergo necrosis: oxygen concentration below 0.02 mM, glucose concentration below 0.06 mM, and waste (lactate) concentration above 8 mM. Although it has been shown that cells can survive these nutrient/waste concentrations individually, there are currently no experimental data available on the effects of combined exposure to these microenvironmental conditions. These simulation results suggest that cells are able to survive even in a very nutrient-deprived environment. These predictions can be tested experimentally only if chemical concentrations in the spheroid microenvironment can be systematically measured, which is difficult using currently-available techniques such as microelectrodes (30) or bioluminescence (36).
The diffusion coefficients for the growth promoters and inhibitors are found to be in the order of 10−7 and 10−6 cm2/h, respectively. This diffusion constant range is on the order of that for peptide growth and inhibitory factors known to regulate cellular proliferation (e.g., epidermal growth factor, fibroblast growth factor, tumor necrosis factor, and tumor growth factor β) based on extrapolation from previous measurements in spheroids (44). Thus, the model predicts that cellular proliferation in this system is regulated by a combination of limited growth promoters and internally produced growth inhibitors. Interestingly, previous work by Freyer et al. (12) has shown that a peptide inhibitory factor was produced by the necrotic regions of spheroids, and that this inhibitory factor was 80–90 kDa, which would have a predicted diffusion constant of ∼1 × 10−7 cm2/h in spheroids.
Our model produces spheroid volume and cell number growth in remarkable agreement with experimental data (Figs. 5 and 8). Some adjustment of the simulation parameters was performed to optimize this agreement, but the final parameters used in the simulation shown in Fig. 4 are very close to experimentally measured values, when available. Importantly, when all parameters were kept constant but the external concentrations of oxygen and glucose were altered, the simulation produced growth curves very similar to a separate set of experiments done under the altered supply conditions (Fig. 8). This suggests that the underlying model is accurately representing the dynamic development of the tumor mass across a wide range of time and distance scales. The simulation underestimates the number of cells when the tumor grows to a size comparable to the total lattice size (Fig. 8). At this point the numerical artifacts in solving the chemicals result in more accumulation of waste in the tumor, hence more cell death. This artifact also explains the earlier tumor growth saturation for the simulation compared to the experimental data in Fig. 8.
The estimates of cell-cycle fractions as a function of spheroid growth show good agreement with the experimental data, especially at large spheroid sizes (Fig. 6). This suggests that the model, and specifically the protein regulatory network incorporated therein, is able to predict the regulation of cellular proliferation. The major discrepancy between the simulation and the experimental data occurs at small spheroid diameters. This can be explained by the difference in how these two data sets were generated. The experimental data represents an average of many (25–100) individual spheroids, while the simulation shows the values for an individual spheroid. Thus, the early simulation results are greatly affected by the cell-cycle stage of the initial cell or small aggregate. Multiple simulations with different starting conditions or an initial condition with more cells in random stages of cell cycle should average to more accurately match the experimental data.
The simulations of the viable rim thickness indicate that our model is able to reproduce the experimental data, especially at spheroid diameters significantly larger then twice the rim thickness. This strongly suggests that necrotic cell death in spheroids is regulated by a combination of nutrient depletion and waste accumulation, and that the progression of necrosis is uncoupled from the regulation of proliferation (9,10,47). Our simulations, however, show an initial rapid growth of the necrotic core, which later slows down to grow at almost exactly the same rate as the whole spheroid. The data available for the EMT6 cell line do not include measurements in the size range at which this rapid initial expansion is predicted to occur (Fig. 7). However, very careful experiments with human tumor spheroids have demonstrated exactly this rapid initial expansion of necrosis in spheroids (17). Depletion of substrates or accumulation of waste products has been proposed to be the cause (17). Detailed analysis of this rapid initial necrosis is underway.
It is somewhat surprising that the simplified protein regulatory network that controls cell-cycle arrest in our model could produce such a good match to the spheroid data. This result supports the idea that proliferation arrest is regulated by the induction of a few specific proteins, which act primarily in the G1-phase of the cell cycle. The current model is entirely consistent with the recent work showing that G1-specific CKIs are induced, and actively inhibit, their target CDKs relatively close to the spheroid surface (14). Our modeling results also suggest that microenvironmental induction of growth arrest is not caused by restrictions on volumetric expansion of the spheroid. Even though the model incorporates such a mechanism for cell-cycle arrest, the results predict that arrest is actually caused by the induction of G1-phase regulatory proteins. It is important to note that restricted volumetric growth may be an important consideration when spheroids, or nodular tumors, are surrounded by a semirigid matrix (38). Our model can be further refined to include other regulatory pathways, such as S- and G2-phase arrest, as well as to provide a finer degree of protein regulation than the on-off Boolean logic used in the current version. We are investigating whether such refinement can provide a better match of the simulated cell-cycle distributions and growth curves to the experimental data.
More detailed analysis is possible in our model. For example, protein expression levels and chemical concentrations can be relatively easily obtained from the simulations. Unfortunately, other than for oxygen, there are currently no experimental data available for comparison with the chemical composition of the spheroid microenvironment predicted by our model. In the case of oxygen, we have obtained concentration gradient profiles that are consistent with previous measurements using microelectrodes in EMT6 spheroids (30), even though the experimental data were obtained on different spheroids at different times. We are also working on extending the current model past the avascular stage of tumor growth by incorporating angiogenesis through a separate protein regulatory network regulated by the microenvironment.
CONCLUSIONS
We have developed a comprehensive, multiscale cellular model of avascular tumor growth. On the subcellular level, we consider a simple protein network that controls transition between the G1- and S-phases of cell cycle. On the cellular level, our model uses a lattice Monte Carlo model for cell dynamics. On the extracellular level, a set of continuous chemical reaction-diffusion equations describe metabolites, catabolites, growth factors, and inhibitory factors. The environmental parameters we considered were oxygen, glucose, and lactate concentrations, and relative concentrations of growth and inhibitory factors. Simulations under different environmental conditions show both qualitative and quantitative agreement with the experimental data from EMT6/Ro mouse mammary tumor spheroids. The model predicts the survival conditions for cells in the microenvironment, and suggests possible candidates for growth promoters and inhibitors that control cell-cycle arrest.
Acknowledgments
We thank J. Toivanen and V. Dyadechko for providing us with a fast three-dimensional PDE solver, and H. Show for help in finding diffusion coefficients from literature. We also thank the anonymous referees for very constructive comments.
This work was supported by the U.S. Department of Energy under contract No. W-7405-ENG-36 and by grants No. CA-71898, CA-89255, and CA-108853 from the National Cancer Institute.
APPENDIX
The detailed equations for chemicals are
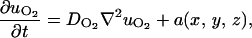 |
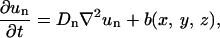 |
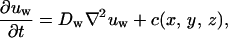 |
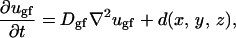 |
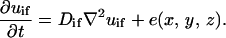 |
In these equations, subscripts O2, n, w, gf, and if stand for oxygen, nutrients, waste, growth, and inhibitory factors, respectively. D is the diffusion coefficient. Variables a, b, c, d, and e are consumption and production rates of these chemicals. Their initial values, ao, b0, and c0 are listed in Table 1.
After we calculate chemical concentrations, we adjust the cells' consumption rate according to the changed concentrations of oxygen and glucose,
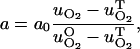 |
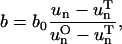 |
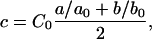 |
where uO is the optimal concentration, and uT is the threshold concentration. The optimal concentration is 0.28 mM for O2 and is 5.5 mM for glucose. The threshold concentrations are determined iteratively as described above near the end of Parameters in Methods (Cellular Model), above. The production rate of waste is directly proportional to the consumption rates for oxygen and glucose; therefore, higher consumption corresponds to higher waste production. As each cell carries its own consumption rates, which vary according to the cell's state as well as the local chemical field, these rates also vary in space.
The boundary conditions at the tumor medium interface are
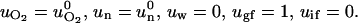 |
At time zero, no chemical is present inside the tumor (single cell).
When cells react to the chemical environment, the quiescent cells produce a small amount of inhibitory factors. During the first 24 h after a cell becomes necrotic, it secretes inhibitory factors at the rate of 0.1 ml/h and waste at the rate of 10 mM/h.
Since each cell is considered to be chemically homogeneous, the chemical reaction diffusion equations need to be solved on an irregular three-dimensional grid with nodes at the center of mass of every cell. We simplify the matter by coarse-graining the cell lattice by a factor of 4, such that the grid is still regular and only a few grid points exist inside each cell, and the concentration within an individual cell is the average of the concentrations on the grid points within that cell.
References
- 1.Osada, H., and T. Takahashi. 2002. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene. 21:7421–7434. [DOI] [PubMed] [Google Scholar]
- 2.Klaunig, J. E., and L. M. Kamendulis. 2004. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 44:239–267. [DOI] [PubMed] [Google Scholar]
- 3.Mueller-Klieser, W. 2000. Tumor biology and experimental therapeutics. Crit. Rev. Oncol. Hematol. 36:123–139. [DOI] [PubMed] [Google Scholar]
- 4.Brown, J. M. 2002. Tumor microenvironment and the response to anticancer therapy. Cancer Biol. Ther. 1:453–458. [DOI] [PubMed] [Google Scholar]
- 5.Vaupel, P., and M. Hockel. 2000. Blood supply, oxygenation status and metabolic micromilieu of breast cancers: characterization and therapeutic relevance (Review). Int. J. Oncol. 17:869–879. [DOI] [PubMed] [Google Scholar]
- 6.Sutherland, R. M. 1988. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 240:177–184. [DOI] [PubMed] [Google Scholar]
- 7.Kunz-Schughart, L. A., M. Kreutz, and R. Knuechel. 1998. Multicellular spheroids: a three-dimensional in vitro culture system to study tumour biology. Int. J. Exp. Pathol. 79:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freyer, J. P., and R. M. Sutherland. 1986. Regulation of growth saturation and development of necrosis in EMT6/Ro multicellular spheroids by the glucose and oxygen supply. Cancer Res. 46:3504–3512. [PubMed] [Google Scholar]
- 9.Freyer, J. P., and R. M. Sutherland. 1986. Proliferative and clonogenic heterogeneity of cells from EMT6/Ro multicellular spheroids induced by the glucose and oxygen supply. Cancer Res. 46:3513–3520. [PubMed] [Google Scholar]
- 10.Freyer, J. P. 1988. Role of necrosis in regulating the growth saturation of multicellular spheroids. Cancer Res. 48:2432–2439. [PubMed] [Google Scholar]
- 11.Marusic, M., Z. Bajzer, J. P. Freyer, and S. Vuk-Pavlovic. 1994. Analysis of growth of multicellular tumor spheroids by mathematical models. Cell Prolif. 27:73–94. [DOI] [PubMed] [Google Scholar]
- 12.Freyer, J. P., P. L. Schor, and A. G. Saponara. 1988. Partial purification of a protein growth inhibitor from multicellular spheroids. Biochem. Biophys. Res. Commun. 152:463–468. [DOI] [PubMed] [Google Scholar]
- 13.LaRue, K. E., M. E. Bradbury, and J. P. Freyer. 1998. Regulation of G1 transit by cyclin kinase inhibitors in multicellular spheroid cultures of rat embryo fibroblast cells transformed to different extents. Cancer Res. 58:1305–1314. [PubMed] [Google Scholar]
- 14.LaRue, K. E., M. Kahlil, and J. P. Freyer. 2004. Microenvironmental regulation of proliferation in EMT6 multicellular spheroids is mediated through differential expression of cyclin-dependent kinase inhibitors. Cancer Res. 64:1621–1631. [DOI] [PubMed] [Google Scholar]
- 15.Marusic, M., Z. Bajzer, S. Vuk-Pavlovic, and J. P. Freyer. 1994. Tumor growth in vivo and as multicellular spheroids compared by mathematical models. Bull. Math. Biol. 56:617–631. [DOI] [PubMed] [Google Scholar]
- 16.Groebe, K., and W. Mueller-Klieser. 1991. Distribution of oxygen, nutrient, and metabolic waste concentrations in multicellular spheroids and their dependence on spheroid parameters. Eur. Biophys. J. 19:169–181. [DOI] [PubMed] [Google Scholar]
- 17.Groebe, K., and W. Mueller-Klieser. 1996. On the relation between size of necrosis and diameter of tumor spheroids. Int. J. Radiat. Oncol. Biol. Phys. 34:395–401. [DOI] [PubMed] [Google Scholar]
- 18.Landry, J., J. P. Freyer, and R. M. Sutherland. 1982. A model for the growth of multicellular tumor spheroids. Cell Tissue Kinet. 15:585–594. [DOI] [PubMed] [Google Scholar]
- 19.Sherrat, J. A., and M. A. J. Chaplain. 2001. A new mathematical model for avascular tumor growth. J. Math. Biol. 43:291–312. [DOI] [PubMed] [Google Scholar]
- 20.Chen, C. Y., H. M. Byrne, and J. R. King. 2001. The influence of growth-induced stress from the surrounding medium on the development of multicell spheroids. J. Math. Biol. 43:191–220. [DOI] [PubMed] [Google Scholar]
- 21.Jackson, T. L., and H. M. Byrne. 2000. A mathematical model to study the effects of drug resistance and vasculature on the response of solid tumors to chemotherapy. Math. Biosci. 164:17–38. [DOI] [PubMed] [Google Scholar]
- 22.Sarntinoranont, M., F. Rooney, and M. Ferrari. 2003. Interstitial stress and fluid pressure within a growing tumor. Ann. Biomed. Eng. 31:327–335. [DOI] [PubMed] [Google Scholar]
- 23.Borkenstein, K., S. Levegruen, and P. Peschke. 2004. Modeling and computer simulations of tumor growth and response to radiotherapy. Radiat. Res. 162:71–83. [DOI] [PubMed] [Google Scholar]
- 24.Mansury, Y., M. Kimura, J. Lobo, and T. S. Deisboeck. 2002. Emerging patterns in tumor systems: simulating the dynamics of multicellular clusters with an agent-based spatial agglomeration model. J. Theor. Biol. 219:343–370. [DOI] [PubMed] [Google Scholar]
- 25.Stott, E., N. F. Britton, J. A. Glazier, and M. Zajac. 1999. Stochastic simulation of benign avascular tumor growth using the Potts model. Math. Comput. Model. 30:183–198. [Google Scholar]
- 26.Alarcon, T., H. M. Byrne, and P. K. Maini. 2004. A multiple scale model for tumor growth. Multiscale Model. Sim. 3:440–475. [Google Scholar]
- 27.Alarcon, T., H. M. Byrne, and P. K. Maini. 2004. Towards whole-organ modelling of tumour growth. Prog. Biophys. Mol. Biol. 85:451–472. [DOI] [PubMed] [Google Scholar]
- 28.Freyer, J. P., and R. M. Sutherland. 1985. A reduction in the in situ rates of oxygen and glucose consumption of cells in EMT6/Ro spheroids during growth. J. Cell. Physiol. 124:516–524. [DOI] [PubMed] [Google Scholar]
- 29.Freyer, J. P. 1994. Rates of oxygen consumption for proliferating and quiescent cells isolated from multicellular tumor spheroids. Adv. Exp. Med. Biol. 345:335–342. [DOI] [PubMed] [Google Scholar]
- 30.Mueller-Klieser, W., J. P. Freyer, and R. M. Sutherland. 1986. Influence of glucose and oxygen supply conditions on the oxygenation of multicellular spheroids. Br. J. Cancer. 53:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glazier, J. A., and F. Garner. 1993. Simulation of the differential adhesion driven rearrangement of biological cells. Phys. Rev. E. 47:2128–2154. [DOI] [PubMed] [Google Scholar]
- 32.Jiang, Y., H. Levine, and J. A. Glazier. 1998. Possible cooperation of differential adhesion and chemotaxis in mound formation of Dictyostelium. Biophys. J. 75:2615–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Izaguirre, J. A., R. Chaturvedi, C. Huang, T. Cickovski, G. Thomas, G. Forgacs, M. Alber, G. Hentschel, S. A. Newman, and J. A. Glazier. 2004. CompuCell, a multi-model framework for simulation of morphogenesis. Bioinformatics. 20:1129–1137. [DOI] [PubMed] [Google Scholar]
- 34.Steinberg, M. S. 1963. Reconstruction of tissues by dissociated cells. Science. 141:401–408. [DOI] [PubMed] [Google Scholar]
- 35.Duguay, D., R. A. Foty, and M. S. Steinberg. 2003. Cadherin-mediated cell adhesion and tissue segregation: qualitative and quantitative determinants. Dev. Biol. 253:309–323. [DOI] [PubMed] [Google Scholar]
- 36.Walenta, S., J. Doetsch, W. Mueller-Klieser, and L. Kunz-Schughart. 2000. Metabolic imaging in multicellular spheroids of oncogene-transfected fibroblasts. J. Histochem. Cytochem. 48:509–522. [DOI] [PubMed] [Google Scholar]
- 37.Beysens, D. A., G. Forgacs, and J. A. Glazier. 2000. Cell sorting is analogous to phase ordering in fluids. Proc. Natl. Acad. Sci. USA. 97:9467–9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helmlinger, G., P. A. Netti, H. C. Lichtenbeld, R. J. Melder, and R. K. Jain. 1997. Solid stress inhibits the growth of multicellular tumor spheroids. Nat. Biotechnol. 15:778–783. [DOI] [PubMed] [Google Scholar]
- 39.Boucher, Y., L. T. Baxter, and R. K. Jain. 1990. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 50:4478–4484. [PubMed] [Google Scholar]
- 40.Boucher, Y., J. Salehi, B. Witwer, G. R. Harsh, and R. K. Jain. 1997. Interstitial fluid pressure in intracranial tumors in patients and in rodents. Br. J. Cancer. 75:829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gutmann, R., M. Leunig, J. Feyh, A. E. Goetz, K. Messmer, E. Kastenbauer, and R. K. Jain. 1992. Interstitial hypertension in head and neck tumors in patients: correlation with tumor size. Cancer Res. 52:1993–1995. [PubMed] [Google Scholar]
- 42.Landry, J., J. P. Freyer, and R. M. Sutherland. 1981. Shedding of mitotic cells from the surface of multicell spheroids during growth. J. Cell. Physiol. 106:23–32. [DOI] [PubMed] [Google Scholar]
- 43.Mueller-Klieser, W. 1984. Method for the determination of oxygen consumption rates and diffusion coefficients in multicellular spheroids. Biophys. J. 46:343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freyer, J. P., and R. M. Sutherland. 1983. Determination of diffusion constants for metabolites in multicell tumor spheroids. Adv. Exp. Med. Biol. 159:463–475. [DOI] [PubMed] [Google Scholar]
- 45.Casciari, J. J., S. V. Sotirchos, and R. M. Sutherland. 1988. Glucose diffusivity in multicellular tumor spheroids. Cancer Res. 48:3905–3909. [PubMed] [Google Scholar]
- 46.Bourrat-Floeck, B., K. Groebe, and W. Mueller-Klieser. 1991. Biological response of multicellular EMT6 spheroids to exogenous lactate. Int. J. Cancer. 47:792–799. [DOI] [PubMed] [Google Scholar]
- 47.Freyer, J. P., P. L. Schor, K. A. Jarrett, M. Neeman, and L. O. Sillerud. 1991. Cellular energetics measured by phosphorous NMR spectroscopy are not correlated with chronic nutrient deficiency in multicellular tumor spheroids. Cancer Res. 51:3831–3837. [PubMed] [Google Scholar]