

Abstract
Cystic fibrosis transmembrane conductance regulator (CFTR) adenosine triphosphate-dependent chloride channels are expressed in epithelial cells and are associated with a number of genetic disorders, including cystic fibrosis. Venom of the scorpion Leirus quinquestriatus hebraeus reversibly inhibits CFTR when applied to its cytoplasmic surface. To examine the state-dependence of inhibition we recorded wild-type and mutant CFTR channel currents using inside-out membrane patches from Xenopus oocytes. Application of either venom or diphenylamine-2-carboxylate to channels that were either activated (open) or resting (closed) indicate primarily closed state-dependent inhibition of CFTR by venom, whereas diphenylamine-2-carboxylate showed no state-dependence of block. Efficacy of venom-mediated macroscopic current inhibition was inversely related to channel activity. Analysis of single-channel and macropatch data indicated that venom could either inhibit channel opening, if it binds during an interburst closed state or in the absence of cytosolic adenosine triphosphate, or introduce new intraburst closed states, if it binds during an open event. The on-rate of venom binding for intraburst block could be modulated by changing CFTR activity with vanadate or adenylyl-imidodiphosphate, or by introducing the Walker A mutation K1250A. These findings represent the first description of state-dependent inhibition of CFTR and suggest that the active toxin could be used as a tool to study the conformational changes that occur during CFTR gating.
INTRODUCTION
The cystic fibrosis transmembrane conductance regulator (CFTR) forms chloride channels in apical membranes of epithelial cells (1). Loss of function mutations in the gene encoding CFTR cause cystic fibrosis, the most common lethal, autosomal recessive genetic disease in Caucasians, which can result in severe lung disease, pancreatic insufficiency, and infertility (2). However, inappropriate CFTR channel activity is associated with diseases such as secretory diarrhea and polycystic kidney disease (3). Therefore, CFTR is an important medicinal target for therapeutics aimed at correcting channel activity.
CFTR is a member of the adenosine triphosphate (ATP)-binding cassette (ABC) transporter superfamily (4). The channel is a functional monomer with a single polypeptide required to form a single-channel pore (5,6). The polypeptide is composed of two halves, each containing a transmembrane domain and a cytosolic nucleotide binding domain (NBD). The two homologous halves are linked by a regulatory (R) domain. CFTR channel activity requires the presence of hydrolysable nucleoside triphosphates at the NBDs, and the R-domain must be phosphorylated by protein kinase A (PKA) and/or protein kinase C (7,8). Dimerization of NBD1 and NBD2 is an important step during CFTR gating (9). The NBD1/NBD2 dimer configuration promotes the formation of two ATP binding pockets NBD-A and NBD-B, identified according to which portion of the primary sequence contributes the catalytic lysine (9–12); hence, NBD-B includes K1250.
The use of reagents such as 3-isobutyl-1-methylxanthine resulted in a better understanding of the cAMP-dependent regulatory mechanism for CFTR modulation (13,14). These compounds alter channel activity indirectly by inhibiting phosphodiesterase activity causing a decrease in the rate of cAMP degradation, thereby resulting in the potentiation of cAMP-dependent CFTR activity; 3-isobutyl-1-methylxanthine may also alter ATP-dependent gating directly (13,15). Alternatively, non- or poorly-hydrolysable ATP analogs such as adenylyl-imidodiphosphate (AMP-PNP) and adenosine 5′-O-(3-thio)triphosphate (ATPγS), or analogs of ATP hydrolysis products such as vanadate (VO4) or pyrophosphate, have been used to study ATP-dependent gating of CFTR (11,16–18). These compounds directly interact with the channel in the active sites formed by NBD dimerization and alter channel gating. However, since these compounds work by mimicking the activity of ATP or its products, they have not been as useful in answering the following question: Which regions of the CFTR protein change conformation during the gating process subsequent to binding and/or hydrolysis of ATP? The ideal probe for answering this question would be one that interacts with the NBDs during the gating cycle, but can also sense conformational changes that occur outside of the NBDs, which may lead to opening of the channel pore.
We recently reported that a peptide toxin or toxins contained in the venom of the scorpion Leirus quinquestriatus hebraeus (Lqh) reversibly inhibits WT-CFTR only when applied to the cytoplasmic surface of the channels (19). Here we report the complete characterization of the Lqh venom inhibitory activity. In macropatch configuration, the degree of inhibition showed strong dependence upon experimental protocol, particularly with regard to whether the venom and ATP were applied separately or concurrently. In addition, either increasing or decreasing the MgATP concentration used to activate the channels altered the level of inhibition seen with a single concentration of venom. We also found that the potency of venom for intraburst inhibition was reduced in single-channel recordings of WT-CFTR channels with very high open probability or when the venom was applied to K1250A-CFTR channels. In combination with the observation that single channels locked open by treatment with either VO4 or AMP-PNP are less sensitive to inhibition by venom (19), these findings suggest that the inhibition of CFTR is due to interaction of venom with the NBDs in a state-dependent manner. We conclude that the venom alters channel activity by binding during interburst or intraburst closed states, and could be useful for reporting the structural changes that occur subsequent to or during binding and hydrolysis of ATP.
MATERIALS AND METHODS
Preparation of oocytes and cRNA injections
Methods used were similar to those described previously (19,20). Oocytes were harvested from mature Xenopus laevis (Xenopus 1, Ann Arbor, MI). The animals were anesthetized by immersion in tricaine (1.5 mg/mL) and several ovarian lobes were surgically removed under sterile conditions. The follicle cell layer was removed by incubating oocytes for 2 h with 1 mg/mL collagenase in calcium-free OR2 solution: 82.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, and 5 mM HEPES (pH 7.4). The oocytes were washed with calcium-free OR2 and transferred to a modified Liebovitz's L-15 medium with the addition of HEPES (pH 7.5), gentamicin, and penicillin-streptomycin, and incubated at 18°C. For single-channel recordings, cRNA was prepared from a construct (20) carrying the full coding region of CFTR in the pAlter vector (Promega, Madison, WI). The mutant K1250A-CFTR construct was prepared with the QuikChange protocol (Stratagene, La Jolla, CA) using oligonucleotide-mediated mutagenesis. The construct was verified by sequencing across the entire open reading frame before use. For macropatch recordings, cRNAs were prepared from high expression constructs encoding WT-CFTR (pGEMHE-WT) and Flag-cut-ΔR-CFTR (pGEMHE-Flag3-633 and pGEMHE-837-1480), generously provided by Dr. David Gadsby (The Rockefeller University, NY). Oocytes were injected with 5–20 ng of CFTR cRNA along with 0.4 ng of cRNA for the β2-adrenergic receptor for single-channel experiments, allowing activation of CFTR currents by exposure to isoproterenol, or 25–100 ng CFTR cRNA alone for macropatch experiments. Recordings were made 2–5 days after injection.
Electrophysiology
Single-channel and macropatch recordings were obtained using excised, inside-out patches. Oocytes were prepared for study by manually removing the vitelline membrane after shrinking in hypertonic solution (21). Pipettes were pulled from borosilicate glass (Sutter Instruments, Novato, CA). Pipette solution contained 150 mM NMDG-Cl, 5 mM MgCl2, and 10 mM TES (pH 7.4; adjusted with Tris). Intracellular bath solution for excised patches contained 150 mM NMDG-Cl, 1.1 mM MgCl2, 2–10 mM Tris-EGTA, 0.2–5 mM MgATP, and 10 mM TES (pH 7.4; adjusted with Tris). In some experiments 0.1–0.2 mg/mL Lqh venom, 200 μM DPC, 5 mM VO4, and/or 2.75 mM AMP-PNP was added to the intracellular solution. CFTR channels were activated by the catalytic subunit of PKA (50 U/mL) after patch excision into solution containing 1 mM MgATP. Patch-pipette resistances were from 8 to 14 MΩ for single-channel recordings and from 1 to 4 MΩ for macropatch experiments. Typical seal resistances ranged from 100 to ≥ 300 GΩ. All recordings were performed at room temperature (22–25°C).
Single-channel experiments were performed using an Axopatch 200B amplifier (Axon Instruments, Union City, CA) and recorded at 10 kHz to DAT tape (# DTC-EZ700, Sony, San Diego, CA). The membrane potential was held at either −80 mV or −100 mV. In some cases, data were subsequently played back and filtered with a four-pole Bessel filter (Warner Instruments, Hamden, CT) at a corner frequency of 100 Hz and acquired using a Digidata 1322A interface (Axon Instruments) and computer at 400 Hz using the Clampex program of pClamp (Axon Instruments). For intraburst closed-time analysis, experiments were played back and filtered at a corner frequency of 500 Hz and digitized at 2 kHz. Digitized Clampex records were analyzed using Clampfit 9.0 (Axon Instruments) and Igor Pro 4.02 (WaveMetrics, Lake Oswego, OR). In single-channel experiments, recordings of channel activity with venom present began ∼30 s after solution change (19).
Macropatch recordings were also performed with an Axopatch 200B amplifier operated by pClamp software, filtered at 100 Hz, and acquired at 1 kHz with Clampex followed by analysis using Clampfit 9.0. Macropatch currents were obtained by applying one of four voltage protocols. In experiments to measure the on-rate of CFTR inhibition by either DPC or partially fractionated Lqh venom (Lqh-pf venom), the membrane potential was set to 0 mV and then stepped to −80 mV for 150 ms and repeated once per second for 30 s. To determine the maximal inhibitory effect of DPC or venom, the membrane potential was set to 0 mV and then either stepped to various membrane voltages from −100 mV to +100 mV for 150 ms in 20-mV increments, or stepped to −100 mV and held for 50 ms, then ramped to +100 mV over 150 ms. All voltage ramps were run in triplicate and averaged. In the fourth voltage protocol, the membrane potential was stepped from 0 mV to either −80 mV or −100 mV and held throughout the entire course of the experiment. Bath solutions with MgATP, no MgATP, venom, DPC, or MgATP plus venom or DPC were applied to the patches by a fast perfusion system (Warner Instruments, model SF-77B). Solution exchange was complete within <25 ms as judged by the activation of endogenous Cl(Ca) channels in oocytes by exposure to bath solution containing 10 mM Ca2+ (19). WT-CFTR channels were phosphorylated by PKA (50 U/mL) and 1 mM MgATP until the macroscopic current had reached the maximum level for a given patch. After apparent complete CFTR activation, but before employing the fast solution exchange system, the PKA and ATP were flushed from the bath using the ATP-free bath solution until the ATP-dependent current had completely subsided.
Analysis of single-channel and macropatch recordings
Each patch served as its own control before exposure to venom or DPC in all experiments. Transition analysis for single-channel experiments used a 50% cutoff between the open and closed current levels. Open duration analysis was performed on records from patches containing 1–3 active CFTR channels. For initial open duration analysis, records were filtered at 100 Hz and a 100 ms minimum interburst duration cutoff was used to discriminate between interburst gating and brief intraburst closings (22). Subsequent records were filtered at 500 Hz where a 1-s minimum interburst duration cutoff was applied, and a 5-ms minimum intraburst duration cutoff was used to discriminate between brief intraburst closings and transitions to the closed current level due to block by the buffer TES (23). The mean open duration in multichannel patches was determined as previously described (22,24) with the formula
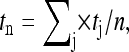 |
(1) |
where ∑ j is the apparent number of active channels, tj is the time that at least j channels are simultaneously open and n is the total number of transitions from an open state to a closed state during the multichannel open event. Thus, a multichannel open burst event is transformed to n single-channel open events with duration t. For the determination of channel open probability (Po), only those patches containing apparently five or fewer open channels were included. Po was calculated as NPo/N, where N is the apparent number of active channels in the patch, determined as the maximum number of channels that were simultaneously open at any point during the control recording of at least 3-min duration; channel number was assumed to be the same upon subsequent exposure to venom. Similar techniques have been used previously to determine CFTR channel number in a given patch (25). Open- and closed-time histograms had bin widths of 2 ms and were constructed from recordings that were a minimum of 360 s in length. Most records used for Po or dwell-time analysis were ∼30 min in duration. Only apparent intraburst closings (or blocked events, in the presence of venom) were included in construction of closed duration histograms; hence, no closings >1000 ms were used in that analysis, because closings of this length were assumed to be due to true channel closure. Since venom-induced inhibition introduced a new population of intraburst closed dwell times not present in control recordings, we were able to estimate the apparent rate constant of venom dissociation (koff) from the mean intraburst blocked intervals (τc) in a given channel record as
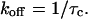 |
(2) |
In macropatch experiments, the mean amplitude of the macroscopic CFTR channel current in all conditions was determined by averaging the current over a 15–20 s stretch immediately before a change in experimental condition.
Venom processing
Lqh venom was dispersed into intracellular bath solution at 2.5 mg dry venom/mL by extensive vortexing followed by brief homogenization using a Potter-Elvenhjem tissue grinder (19). The mucous component was pelleted by centrifugation at 6000 × g for 30 min at 22°C. Assuming that the active component of venom would likely be of low molecular weight, as is the case for most peptide toxins of ion channels, the upper, relatively mucous free solution was recovered and then filtered using a Biomax-10 Micropartition System filter (Millipore, Bedford, MA) with a 10-kDa cutoff, and centrifuged at 2000 × g at 22°C in a fixed angle rotor, to remove the higher molecular-weight components of venom. The resulting mucous-free, filtered venom was stored at −80°C and diluted to given concentrations, based on equivalent dry venom weight, immediately before use. This fraction of venom, which contains several peptide components (19), is referred to as Lqh-pf venom.
Statistics
Results are expressed as mean ± SE for n observations. Comparisons are by paired and unpaired Student's t-test. Differences were considered statistically significant when p < 0.05. Linear regression analysis was performed using SigmaPlot (Jandel Scientific, San Rafael, CA). All statistical tests were performed using SigmaStat 2.03 (Jandel Scientific).
Reagents
Unless otherwise noted, all reagents were obtained from Sigma Chemical (St. Louis, MO). L-15 media was purchased from Gibco/BRL (Gaithersburg, MD). Scorpion venom was purchased from Latoxan (Valence, France). PKA was from Promega. Vanadate stock solutions were prepared by adjusting a 100 mM Na3VO4 solution to pH 10 with NaOH followed by storage at 4°C. Aliquots of stock solution were boiled for 15 min before dilution into buffered bath solution immediately before use (17). DPC was from Aldrich Chemical (Milwaukee, WI), and was dissolved in DMSO at stock concentrations of 0.1–0.5 M and diluted to a 200 μM final concentration immediately before use. The final concentration of DMSO varied up to a maximum of 0.1%, which had no effect on CFTR currents.
RESULTS
Lqh venom preferentially binds to closed CFTR channels
We showed previously that Lqh-pf venom inhibited WT-CFTR currents in excised, inside-out macropatches from Xenopus oocytes (19). In the experiment shown in Fig. 1 A (middle), Lqh-pf venom was applied to phosphorylated CFTR channels that had been allowed to close upon removal of cytosolic ATP. When ATP was returned to the bath, the resulting current density was reduced compared to currents in the absence of venom; application of 0.1 mg/mL Lqh-pf venom to closed CFTR channels in this way led to inhibition of macroscopic current by 24.5 ± 2.5% (n = 9, p ≤ 0.001) at Vm = −80 mV. This result could arise from interaction of the toxin with closed channels before reintroduction of ATP, thus inhibiting their opening, and/or from inhibition of open channels in the presence of ATP. As a control, we examined the effects of DPC when applied to the closed channels in the same way (Fig. 1 A, bottom). DPC is a known CFTR channel pore blocker (20–22), which has not been shown to have any state-dependence of action. Application of 200 μM DPC to closed CFTR channels led to 26.1 ± 2.6% block (n = 5, p ≤ 0.001) upon subsequent activation with ATP, suggesting that, like Lqh-pf venom, DPC can effectively bind to the closed channels. However, since the on-rate of DPC is very high (21), this experimental protocol does not preclude binding of DPC to open channels, as well.
FIGURE 1.

Inhibitory activity of Lqh venom is state-dependent. (A) Representative traces from excised, inside-out macropatch experiments of WT-CFTR are shown before (top) and during application of either 0.1 mg/mL Lqh-pf venom (middle) or 200 μM DPC (bottom); Vm = −80 mV. In each case either Lqh-pf venom or DPC was applied to closed channels for ∼30 s before reopening by MgATP. (B) Representative traces from excised, inside-out macropatch experiments of Flag-cut-ΔR-CFTR are shown with (bottom) and without application of 0.1 mg/mL Lqh-pf venom (top); Vm = −80 mV. (C) Representative traces from excised, inside-out macropatch experiments of WT-CFTR are shown during application of either Lqh-pf venom (top) or DPC (bottom) to activated channels. Lqh-pf venom or DPC were applied to open channels for 30 s; Vm = −80 mV. Bath solution contained 1 mM MgATP, no MgATP, 1 mM MgATP plus either 0.1 mg/mL Lqh-pf venom or 200 μM DPC, or venom or DPC in the absence of ATP. WT-CFTR channels were phosphorylated before the beginning of the experiment. (D) Summary of the effects of either Lqh-pf venom (0.1 mg/mL) or DPC (200 μM) on WT-CFTR steady-state current in the presence of 1 mM MgATP. Mean steady-state current was determined by averaging the current over the final 15–20 s in each condition. (Circles represent individual experiments, some are shown to overlap. Triangles and error bars indicate mean ± SE of n = 5–14 observations for each condition. The asterisks indicate that Lqh venom activity is significantly different when applied to closed versus open CFTR (*p ≤ 0.001).) (E) Effect of application of either 0.1 mg/mL Lqh-pf venom or 200 μM DPC to WT-CFTR while activated by 1 mM MgATP. Arrow indicates the timing of the rapid solution switch from normal bath solution with 1 mM MgATP to bath solution containing MgATP and either venom or DPC. The membrane potential was set to 0 mV and then stepped to −80 mV for 150 ms and repeated once per second for 30 s to measure the on-rate of CFTR inhibition by either DPC or Lqh-pf venom. Mean fractional block ± SE (n = 14 with Lqh-pf venom, n = 5 with DPC) is plotted as a function of time.
Dephosphorylation during recordings did not appear to account for the decreases in CFTR current seen during application of venom or DPC. Evidence for this conclusion can be seen in Fig. 1 A (top), where WT-CFTR activity was recorded using the identical procedures as above without Lqh-pf venom or DPC present; repeated exposure to ATP alone led to the same amount of steady-state current in the second pulse. Furthermore, similar results were seen when Lqh-pf venom was applied to Flag-cut-ΔR-CFTR channels lacking the regulatory R-domain (Fig. 1 B), which does not require PKA-mediated phosphorylation (26). The use of this CFTR variant therefore effectively eliminates the potential for dephosphorylation during the recording, suggesting that the decrease in macroscopic current observed in WT-CFTR is due to venom-induced inhibition or DPC-induced block, respectively. Flag-cut-ΔR-CFTR was inhibited by venom to approximately the same degree as WT-CFTR.
To determine whether venom can inhibit open channels, experiments were performed in which Lqh-pf venom was rapidly applied to CFTR channels that were already fully activated. ATP was rapidly applied to patches containing previously phosphorylated channels for ∼30 s before addition of venom or DPC (Fig. 1 C). Exposure to 200 μM DPC in the continuing presence of 1 mM MgATP led to 31.9 ± 3.1% block of macroscopic current (n = 5, p ≤ 0.001), suggesting that DPC can block CFTR channels in the open state equally well as closed channels (Fig. 1 D). However, application of 0.1 mg/mL Lqh-pf venom onto activated CFTR channels led to only 5.13 ± 1.3% decrease in macroscopic current (n = 14, p = 0.002), indicating that venom is less effective at inhibiting CFTR channels in the open state compared to in the closed state (p ≤ 0.001, Fig. 1 D). These results suggest that Lqh-pf venom inhibits CFTR by preferentially interacting with channels in the closed state. Because the Po of individual CFTR channels in the presence of 1 mM ATP is <0.5 (19,23), venom may interact with open channels that eventually close even in the continued presence of ATP, leading to a slow increase in fractional inhibition through time, as observed (Fig. 1 E). However, the majority of DPC block occurs very rapidly.
Effectiveness of macroscopic inhibition depends on the level of CFTR channel activity
The data presented thus far suggest that Lqh-pf venom may preferentially bind to the closed CFTR channels and then inhibit development of Cl− conductance upon channel activation by ATP. The ATP-dependent gating cycle of WT-CFTR includes several closed states that could be the target of Lqh-pf venom binding (11,18) (Fig. 2 (thick arrow pathway)). Phosphorylated channels reside in the resting (C1) closed state before initial interactions with ATP. Upon binding ATP at NBD-A, channels reside in state C2, which represents the closed state between open bursts (the interburst closed state). The rate of ATP release from NBD-A has been shown to be extremely low (10). Hence, CFTR gating under physiological conditions typically involves transitions from the C2 state through the open states and back to C2, by binding and hydrolysis of ATP at NBD-B, along the path described by the solid rectangle in Fig. 2. In addition, there are two potentially short-lived closed states that occur during the ATP gating cycle. The C3 and C4 closed states occur either immediately before NBD dimerization after ATP binding at NBD-B (C3) or after NBD de-dimerization (C4) as a prerequisite for release of ADP from NBD-B. Three open states are shown, which differ according to what is bound at NBD-B. CFTR activity can be defined by the fraction of time the channel spends in each of these states.
FIGURE 2.

Simplified scheme illustrating proposed CFTR gating steps and channel conformation states that are available for possible interactions with peptide toxin. The thick arrows indicate gating steps for WT-CFTR under standard conditions. C represents closed states before (C1) and after binding of ATP at NBD-A (C2) and NBD-B (C3). The fourth closed state (C4) occurs after ATP hydrolysis at NBD-B and subsequent to de-dimerization of NBDs but before ADP release. Additional non-ATP-dependent closings that occur during open bursts are depicted as “FC” closings. O symbolizes all open states visited during an open burst. AMP-PNP or vanadate treatment alters WT-CFTR gating, causing channel conformations to follow either of the respective pathways depicted by the dashed arrows. Mutations in NBD-B, such as K1250A, result in channels that follow the initial WT-CFTR gating steps; however, rates of ATP binding at NBD-B and hydrolysis of that ATP are greatly reduced (boxes). Results from this study and previous studies suggest that interactions of toxin (B) with CFTR occur while the channel resides in either the C2 or FC states, leading to complete inhibition of Cl− conductance in the C2B or FCB states, respectively.
To determine whether Lqh-pf venom binds to CFTR while the channel is in a given closed state, we first asked whether the venom was a more effective inhibitor of macroscopic currents under conditions that increase the likelihood that the channels were in that closed state. The Po of phosphorylated CFTR channels can be adjusted by changing [ATP] in the intracellular solution (11,18,27). We measured the effect of Lqh-pf venom on phosphorylated WT-CFTR channels in macropatches activated by four concentrations of MgATP ranging from 0.2 mM to 5 mM (Fig. 3 A). In the presence of 1 mM MgATP, treatment with 0.1 mg/mL Lqh-pf venom produced 24.5 ± 2.5% inhibition of macroscopic currents (n = 9). The same concentration of venom had a reduced effect when the channels were activated with 2 mM MgATP (18.4 ± 2.5% inhibition, n = 7). At high [MgATP] (5 mM), 0.1 mg/mL Lqh-pf venom produced only very modest inhibition (14.4 ± 0.6%, n = 3). However, with 0.2 mM MgATP, inhibition of macroscopic currents by Lqh-pf venom was enhanced to 30.8 ± 2.1% (n = 4, p ≤ 0.001 compared to inhibition with 5 mM ATP present). These results suggest that the ability of Lqh-pf venom to inhibit CFTR is dependent on CFTR channel activity, where conditions that promote high channel activity diminish the efficacy of inhibition.
FIGURE 3.

Altering MgATP concentration and channel activity affects the inhibitory potency of venom. (A) Fractional inhibition of mean steady-state current in the presence of 0.1 mg/mL Lqh-pf venom with four concentrations of MgATP. Experiments performed were similar to those shown in Fig. 1 A. Mean WT-CFTR steady-state current was determined by averaging the current over the final 15–20 s at each condition. Columns and error bars indicate mean ± SE of n = 4–10 observations for each condition. The asterisks indicate values that are significantly different from those recorded with 0.2 mM MgATP (*p ≤ 0.01, **p ≤ 0.001). (B) Representative current from voltage ramps in a single excised, inside-out macropatch containing WT-CFTR, at two [MgATP]. Bath solution contained no MgATP, 0.2–2 mM MgATP, 0.1 mg/mL Lqh-pf venom, or 0.2–2 mM MgATP plus 0.1 mg/mL Lqh-pf venom, and was changed in ∼25 ms by a rapid perfusion apparatus. CFTR channels were phosphorylated before the beginning of the experiment. All measurements were made using voltage ramps where the membrane potential was stepped from 0 mV to −100 mV and held for 50 ms before being ramped to +100 mV over 150 ms. Voltage ramps were recorded every 2 min. The peak inward current recorded in each condition is indicated by a dashed line and was used to determine the amount of inhibition. Lowering the [MgATP], leading to decreased CFTR activity, resulted in an increase in inhibitory potency of the venom. (C) Calculation of estimated Po for macropatch experiments as a function of [MgATP]. (Top) The dose-response relationship between [MgATP] and macroscopic CFTR current normalized to the macroscopic current measured at 5 mM MgATP. All measurements were made in excised, inside-out macropatches using voltage ramps where the membrane potential was stepped from 0 mV to −100 mV and held for 50 ms before being ramped to +100 mV over 150 ms. Only currents measured at Vm = −100 mV were used in this analysis. Bath solution contained 0.2–5 mM MgATP, and was changed by a rapid perfusion apparatus. CFTR channels were phosphorylated before the beginning of the experiment. (Middle) CFTR macroscopic currents normalized to current with 1 mM MgATP. (Bottom) Estimated CFTR Po calculated from the values obtained after normalization of macroscopic currents to 1 mM MgATP, where Po under control conditions was 0.339 ± 0.029 (n = 51). (D) Correlation between fractional block by 0.1 mg/mL Lqh-pf venom and CFTR estimated channel Po. The plot includes macropatch recordings of WT-CFTR at four [MgATP]. The line indicates fit of the data by linear regression (p ≤ 0.001, R2 = 0.45).
To confirm within an individual experiment that the inhibitory activity of venom was inversely related to channel activity, and to control for variability between batches of venom, we activated WT-CFTR in macropatches with two concentrations of MgATP in the presence and absence of 0.1 mg/mL Lqh-pf venom that was diluted from a single aliquot of venom stock (Fig. 3 B). After washout of ATP, subsequent exposure to intracellular solution containing Lqh-pf venom and ATP in the same patch led to reduced macroscopic current compared to ATP alone. Importantly, Lqh-pf venom inhibited CFTR more effectively when the channels were activated by low [MgATP] compared to when the same channels were activated with high [MgATP].
The data in Fig. 3, A and B, suggest that ATP may compete with venom at the NBDs, because venom is ineffective at inhibiting macroscopic current in the presence of high [ATP]. However, because CFTR channel activity is also increased as a saturating function of [ATP], the results may also indicate that efficacy of inhibition by venom is dependent upon channel activity more directly, without invoking competition at the ATP binding site. To test this idea, we used the macropatch data to compare fractional inhibition as a function of estimated Po (Fig. 3 C). Because Po in the absence of venom is known for single-channel experiments but not for macropatch experiments, we transformed the macropatch data using true values for Po measured from single channels under defined conditions, making use of the relationship between the Po of phosphorylated CFTR channels and [MgATP] (18). To perform this transformation, we first measured macropatch current as a function of [MgATP]. Studies have shown that 5 mM MgATP can fully activate WT-CFTR macroscopic current (18); thus the relative current at MgATP concentrations <5 mM was determined by normalizing the macropatch current at those concentrations to the current in the presence of 5 mM MgATP (Fig. 3 C, top). Because the mean Po of WT-CFTR with 1 mM MgATP in single-channel patches is known, we then normalized macroscopic currents to those measured with 1 mM MgATP (Fig. 3 C, middle). This transformation allowed us to assign estimated open probabilities to each of the macropatch recordings at various MgATP concentrations (Fig. 3 C, bottom), based on the measured Po of WT-CFTR single channels with 1 mM MgATP (Po = 0.339 ± 0.029, n = 51). A three-parameter Hill equation was used to fit the data, giving p ≤ 0.001, R2 = 0.99. From these calculations we find that CFTR channels in macropatches activated by 0.2 mM MgATP had an estimated Po of 0.170, whereas those recorded with 5 mM MgATP in the bath had an estimated Po of 0.525.
A plot of inhibition of WT-CFTR macroscopic currents by 0.1 mg/mL Lqh-pf venom as a function of estimated Po is shown in Fig. 3 D, where each point represents an individual macropatch experiment at 0.2, 1, 2, or 5 mM MgATP (n = 23). Linear regression analysis suggested that there is a significant relationship between the magnitude of inhibition by 0.1 mg/mL Lqh-pf venom and CFTR estimated channel Po (p ≤ 0.001, R2 = 0.45). It is clear that channels with high activity before application of venom were inhibited poorly by venom, while inhibition of channels with low activity was significantly stronger. This result suggests that the level of channel activity is the determining factor in the ability of the Lqh-pf venom to inhibit, rather than competition at the ATP binding site.
If venom competitively inhibits ATP binding at NBD-B, we also would expect there to be a lengthening of the C2 interburst closings during channel gating. One manifestation of the effect would be a reduction in the macroscopic opening rate of channels in the presence of ATP. However, in our previous study we found no significant change in ATP-induced apparent macroscopic CFTR opening rate upon rapid introduction of ATP (19). A possible explanation for the lack of an effect on apparent macroscopic opening rate is that the Lqh-pf venom remains bound to the channels in the C2 state for very long periods resulting in an extremely low off-rate, so low that the active toxin does not dissociate from the channel within the 30-s wash-on of ATP in the presence of venom during the macropatch protocol. In other words, competitive inhibition at the ATP binding site should only be evident as a change in macroscopic opening rate if the intrinsic rates of binding and unbinding are similar for ATP and the active toxin.
To determine if Lqh-pf venom treatment locked channels into the interburst closed state, we used excised, inside-out multichannel patches. Fig. 4 A shows an example of currents from WT-CFTR channels activated with 50 U/mL PKA and 1 mM MgATP before (left) and during treatment with 0.1 mg/mL Lqh-pf venom (right). Lqh-pf venom treatment resulted in apparent complete knockout of many of the channels that were active in the control recordings; this reduced activity was maintained over several minutes of recording, suggesting that the apparent rate of venom dissociation is very low. Our previous results showed that this effect is reversible (19). Consistent with the lock of channels into the C2 interburst closed state, we often found in patches containing a single WT-CFTR channel at low to moderate Po that exposure to venom rapidly led to disappearance of that channel, despite the continued presence of ATP and PKA (Fig. 4 B). We reasoned that the venom-induced lengthening of the time that the channel spent in the C2 closed state would be more evident if an extremely active variant of CFTR were used in this type of experiment. Removal of Walker A lysines that are important for ATP hydrolysis can greatly increase Po (16). K1250A-CFTR channels exhibit a greatly reduced rate of ATP hydrolysis, resulting in channels that are open for tens of seconds; however, K1250A-CFTR channels also exhibit a reduced opening rate such that they remain in the C2 closed state longer than WT-CFTR channels (18). Studies were performed with K1250A-CFTR in multichannel patches with 50 U/mL PKA and 1 mM MgATP continuously present. Upon treatment with 0.1 mg/mL Lqh-pf venom, similar results were observed in that many of the active channels disappeared from the patch within a few minutes (Fig. 4 C). The very large increase in interburst closed duration as a result of venom binding can be clearly seen since there are no multichannel open events at any point during the recording with venom (∼2 min). These results, along with those from the macropatch recordings, suggest that the venom interacts with CFTR when the channels are in the C2 closed state, shifting them to the C2B state (Fig. 2). Also, the extremely low rate of dissociation of venom from the channel after binding during C2 closings could explain why there is no change in the apparent ATP-dependent macroscopic opening rate in macropatch recordings (19).
FIGURE 4.

Venom-mediated knockout of CFTR channel activity in multichannel patches. (A) Representative recording of several WT-CFTR channels in an excised, inside-out patch before and during treatment with 0.1 mg/mL Lqh-pf venom. (B) Representative single-channel recording of WT-CFTR in an excised, inside-out patch before and during treatment with 0.1 mg/mL Lqh-pf venom. (C) Representative single-channel recording of K1250A-CFTR before and during treatment with 0.1 mg/mL Lqh-pf venom. All measurements were made in the presence of PKA (50 U/mL) and 1 mM MgATP with membrane potential at −80 mV and symmetrical ∼150 mM [Cl−]. CFTR channel activity was filtered at 100 Hz and acquired by computer at 400 Hz. Dashed line represents the closed current level, and downward deflections represent channel openings.
Lqh-pf venom also binds to CFTR during intraburst closings
In our previous study, we showed that exposure to Lqh-pf venom in the continuing presence of 1 mM MgATP and PKA resulted in the introduction of new intraburst closed states lasting hundreds of milliseconds in single-channel recordings of WT-CFTR that were still active during venom treatment (19). Analysis of those recordings showed that 0.1 mg/mL venom caused a 67% decrease in mean burst duration, resulting in a 32% decrease in apparent Po of those channels that were still visible in the patch, in records filtered at 100 Hz. Therefore, a second effect of venom treatment was that the mean duration of CFTR open bursts was apparently shortened because sojourns at the closed current level, representing new blocked states, were introduced into the record. Fig. 5 shows an example of currents from WT-CFTR single channels activated with 50 U/mL PKA and 1 mM MgATP before (top) and during treatment with 0.1 mg/mL Lqh-pf venom (bottom). Upon venom treatment, open bursts appeared to be shortened by the introduction of new closures; the venom-induced closures seemed to be the result of open channel block. However, results from studies with WT-CFTR in macropatch recordings (Fig. 1 C), as well as K1250A-CFTR in recordings of only a few channels (Fig. 4 B), suggested that the venom does not interact with the open state. To clarify this discrepancy, we studied the intraburst behavior of CFTR channels in the presence and absence of venom, in recordings with limited filtering (500 Hz).
FIGURE 5.

Lqh-venom inhibits CFTR intraburst activity. (A) Representative trace of WT-CFTR from an excised inside-out patch with 1 mM MgATP and 50 U/mL PKA continuously present; Vm = −100 mV. Note that brief closings of at least 5 ms in duration occur frequently during an open burst. (B) Representative trace of WT-CFTR with 1 mM MgATP, 50 U/mL PKA, and 0.1 mg/mL Lqh-pf venom continuously present; Vm = −100 mV. Fewer brief closings are evident (lower trace); however, overall burst appearance is altered due to the introduction of venom-induced closings (evident in middle trace). CFTR channel activity was filtered at 500 Hz and acquired by computer at 2 kHz. Dashed line represents the closed current level, and downward deflections represent channel openings.
Inspection of the fine structure of CFTR open bursts in the absence of inhibitors indicates the presence of brief transitions to the closed state, lasting between 5 and 100 ms. These flickery-closure (FC) intraburst closings are not a result of ATP binding or hydrolysis, but instead may reflect alterations of pore architecture during channel openings. Alternatively, they may reflect uncoupling of the transmembrane domain conformation from ATP-mediated gating events at the NBDs. Apparent closings of <5-ms duration are not gating events, but are actually caused by block of the pore by the pH buffer, TES (23,28); such closings are filtered out of most CFTR channel records published. It is the brief FC intraburst closures that give WT-CFTR gating its flickery character. We reasoned that the FC intraburst closed state may represent a target for venom binding. To determine if the Lqh-pf venom does interact with CFTR during the intraburst closings we recorded (at 500 Hz) WT-CFTR in the absence and presence of 0.1 mg/mL Lqh-pf venom (Fig. 5). The expanded records clearly indicate that there are numerous intraburst closings during open bursts of WT-CFTR in control conditions and during venom treatment. For analysis of the CFTR activity under these conditions we used a 5-ms minimum duration cutoff for intraburst closings and a minimum 1-s cutoff for interburst closings; these criteria should help to distinguish FC intraburst closings from TES-induced pore block (closed state transitions <5 ms in duration) and gating-induced channel closures to the C2 state (closed state transitions >1000 ms in duration). If the Lqh-pf venom interacts with the FC intraburst closings under these conditions we would anticipate the following results: 1), there would be no change in the mean open burst duration; and 2), there would be an increase in the mean intraburst closed duration, because the brief intraburst (FC) closed state would be transformed into a longer (i.e., FCB; see Fig. 2) inhibited state. Hence, venom binding during these brief closings would appear to have the effect of prolonging the duration of the intraburst closed state until the toxin dissociates from the channel, which would be evident as an increase in the occurrence of closures with durations of >100 ms and a decrease in the number of brief closings.
We analyzed channel open and closed events in records of WT-CFTR in the absence and presence of 0.1 mg/mL Lqh-pf venom, using records filtered at 500 Hz. Only intraburst closings were used in this analysis; therefore, no closings of >1000 ms duration were included. Treatment of WT-CFTR with 0.1 mg/mL Lqh-pf venom did not result in a significant change in channel open time (overall mean open duration = 670.3 ± 64.7 ms in control versus 582.1 ± 44.6 ms with Lqh-pf venom, p = 0.328, n = 6) (Fig. 6 A). These results suggest that venom does not interact with the open CFTR channel. However, Lqh-pf venom treatment did result in a significant 424% increase in overall mean intraburst closed duration from 36.4 ± 9.4 ms in control conditions to 154.5 ± 28.9 ms during exposure to Lqh-pf venom (p < 0.01, n = 6) (Fig. 6 B). Dwell-time histograms were then constructed from these records (Fig. 6 C). In the absence of venom, the closed-time distribution was fit best by a single-exponential function giving mean τc = 5.2 ms. However, addition of Lqh-pf venom resulted in a closed-time distribution that was best fit by the sum of two exponentials giving mean τc1 = 12.7 ms and mean τc2 = 729.9 ms. This analysis provides an estimated Lqh-pf venom mean block time of 730 ms, giving an estimated koff of ∼1.37 s−1 using Eq. 2. Importantly, the number of brief closed states (5–30 ms in duration) was reduced in the presence of venom. These results suggest that Lqh-pf venom also binds to active CFTR channels while in the FC intraburst closed state, shifting channels to the longer FCB blocked state.
FIGURE 6.

Lqh-pf venom binds to CFTR during brief intraburst closings. (A and B) Effect of 0.1 mg/mL Lqh-pf venom on WT-CFTR overall mean open time duration (A) and overall mean intraburst closed time duration (B) in records filtered at 500 Hz. Overall mean durations in A and B are expressed as the arithmetic averages. Values represent mean open times and mean intraburst closed times recorded during venom application and those measured in control conditions. Columns and error bars indicate mean ± SE of n = 6 observations at each condition. Significant difference from control value: *p < 0.01. (C) Open (left) and closed (right) dwell-time histograms of a single WT-CFTR channel with 1 mM MgATP and 50 U/mL PKA continuously present before (top) and during (bottom) treatment with 0.1 mg/mL Lqh-pf venom; Vm = −80 mV. CFTR channel activity was filtered at 500 Hz and acquired by computer at 2 kHz. Venom had no effect on the mean τo. Note that the majority of intraburst closed times in the control condition are <20 ms. Lqh-pf venom treatment caused a decrease in the number of short closings, but induced the appearance of a new population of closings >100 ms in duration.
Efficacy of intraburst inhibition at the single-channel level depends on open probability
CFTR channel Po typically shows time-dependent variability even under steady-state conditions in the presence of a given cytosolic ATP concentration (18,29). Under standard conditions (50 U/mL PKA and 1 mM MgATP continuously present), Po in our records varied from patch to patch usually in a range between 0.15 and 0.45; the mean Po was 0.339 ± 0.029 (n = 51). The amount of venom-induced inhibition also appeared to vary depending on the level of CFTR channel activity in a given patch before addition of venom. Therefore, to quantify this phenomenon we separated the analysis of venom-induced inhibition in experiments with high activity (Po > 0.35) from that in experiments with low activity (Po < 0.35). In the trace shown in Fig. 5, representative of a low activity experiment, treatment with Lqh-pf venom resulted in 25.5% inhibition (apparent Po changed from 0.231 to 0.172). In a series of experiments of this sort, treatment with 0.1 mg/mL Lqh-pf venom resulted in a 31.4 ± 6.96% decrease in mean apparent Po of WT-CFTR channels with low activity before venom (Po = 0.181 ± 0.031 vs. 0.118 ± 0.018, n = 6, p = 0.029). Channels that displayed elevated activity under control conditions were not as extensively inhibited by the same concentration of venom (Po = 0.584 ± 0.091 vs. 0.554 ± 0.087, n = 2). Visual examination of recordings of highly active WT-CFTR channels indicated that the elevated Po was a result of extremely long open bursts (data not shown). However, inhibition of these highly active channels was improved by exposure to an increased concentration of Lqh-pf venom (0.2 mg/mL). As summarized in Table 1, the amount of inhibition by 0.1 mg/mL venom was significantly different, depending on the level of channel activity under standard conditions with PKA and ATP continuously present. These results suggest that the venom-mediated inhibition of CFTR is dependent on the channel open probability.
TABLE 1.
Lqh-pf venom becomes a more effective blocker at lower CFTR channel activity
| Condition | Venom dose (mg/mL) | Control Po | With venom Po | Fractional inhibition | n |
|---|---|---|---|---|---|
| Wild-type (low Po)* | 0.1 | 0.181 ± 0.031 | 0.118 ± 0.018† | 31.4 ± 6.96† | 6 |
| Wild-type (high Po)‡ | 0.1 | 0.584 ± 0.091 | 0.554 ± 0.087 | 5.08 ± 0.19 | 2 |
| Wild-type (high Po)‡ | 0.2 | 0.419 ± 0.020 | 0.226 ± 0.080 | 46.7 ± 16.4 | 2 |
| Wild-type + VO4§ | 0.2 | 0.569 ± 0.158§ | 0.391 ± 0.121†§ | 31.9 ± 6.32†§ | 3 |
| Wild-type + AMP-PNP§ | 0.2 | 0.618 ± 0.107§ | 0.442 ± 0.095†§ | 29.5 ± 5.22†§ | 3 |
| K1250A | 0.1 | 0.772 ± 0.079 | 0.751 ± 0.074 | 2.66 ± 0.59 | 3 |
Inhibition of CFTR by Lqh-pf venom was determined under several experimental conditions used to control channel-open probability. All measurements are from excised inside-out single-channel patch recordings with 1 mM MgATP and 50 U/ml PKA present; Vm = −80 mV. The cutoff value for distinction between channels with high activity and channels with low activity was identified by measuring the mean open probability of WT-CFTR channels with PKA and 1 mM MgATP present (Po = 0.339 ± 0.029; n = 51). Values are mean ± SE of n experiments.
Control single-channel CFTR open probability < 0.35.
Significantly different from control values, p < 0.05.
Control single-channel CFTR open probability > 0.35.
Data from Fuller et al. (19).
Why would channel Po have an effect on the efficacy of venom-mediated inhibition? Of course, channels with low Po would be expected to exhibit prolonged interburst closed durations (C2), which would increase the likelihood of venom binding to this state, thus inhibiting channel opening. We also hypothesized that the frequency of transitions to the FC intraburst closed state, which represents the other target of venom, is dependent on the level of channel activity. To determine if this were true we measured the time that CFTR channels remained open between intraburst closings under several experimental conditions where we manipulated CFTR channel Po. For this set of experiments, rather than controlling CFTR Po by manipulating cytosolic [MgATP], we increased single-channel activity by applying to the cytoplasmic surface of the patch compounds that have been shown to lock CFTR into the open state for extended periods of time. One such compound is AMP-PNP, a nonhydrolyzable analog of ATP which binds to CFTR's NBD-B in place of ATP and greatly slows channel closing, causing WT-CFTR channels to follow a modified gating scheme and leading to lengthening of burst duration (30) (Fig. 2, upper dashed pathway). A second treatment that can be used to lock CFTR open is application of vanadate, a transition-state analog of the ATP hydrolysis product inorganic phosphate, which binds tightly in place of Pi and interrupts the ATP hydrolysis cycle and channel gating (17) (Fig. 2, lower dashed pathway). In addition, we also employed the Walker A mutant K1250A-CFTR, which gates open much like WT-CFTR, although the rate of ATP binding to NBD-B is somewhat reduced, whereas hydrolysis of ATP is greatly reduced, resulting in open bursts that are many tens of seconds in duration (Fig. 2, boxes).
Single WT-CFTR channels were studied with 50 U/mL PKA, 1 mM MgATP, and either 5 mM vanadate or 2.75 mM AMP-PNP continuously present in the bath solution. All recordings were filtered at 500 Hz to enable examination of the short intraburst FC closings. Fig. 7 A shows a representative trace from one recording where channel activity was stimulated by AMP-PNP. Under these conditions, CFTR Po was elevated as compared to that of channels which are allowed to proceed through the normal gating cycle, as indicated by the long open burst duration (compare Fig. 5, top). In the presence of AMP-PNP (or vanadate, Fig. 7 B), periods of normal channel gating can be seen before channels became locked in the open state. After entry into the locked-open state, there are occasional brief intraburst closures that have durations of ∼5–100 ms. Hence, even in the presence of vanadate or AMP-PNP, CFTR Po remains <1.0. Application of 0.2 mg/mL Lqh-pf venom to the patch in the presence of ATP + AMP-PNP (Fig. 7 A, bottom) or ATP + vanadate (Fig. 7 B, bottom) resulted in the appearance of additional closed states. In control conditions, K1250A-CFTR channels remained almost entirely in the open state (Fig. 7 C, top), with few brief closures. After treatment with Lqh-pf venom, a few intraburst closed/blocked events were identified, although their frequency was low (Fig. 7 C, bottom). Treatment of single K1250A-CFTR channels with 0.1 mg/mL Lqh-pf venom resulted in only 2.66 ± 0.59% inhibition of channel activity (Po = 0.772 ± 0.079 vs. 0.751 ± 0.074, n = 3). Notably, the durations of the venom-induced intraburst blocked states in K1250A-CFTR and WT-CFTR with ATP + AMP-PNP, or WT-CFTR with ATP + vanadate, were similar to those observed in WT-CFTR with ATP alone (400–700 ms). However, the frequency that the blocked events occurred and the total amount of inhibition of Po were decreased, even though the venom concentration was doubled compared to that shown in Fig. 5 (see Table 1).
FIGURE 7.

Venom-mediated intraburst inhibition of single CFTR channels under different experimental conditions, in excised inside-out patches. (A) Representative trace of WT-CFTR with 1 mM MgATP, 50 U/mL PKA, and 5 mM AMP-PNP continuously present before and during application of 0.2 mg/mL Lqh-pf venom. (B) Representative trace of WT-CFTR with 1 mM MgATP, 50 U/mL PKA, and 2.75 mM vanadate continuously present before and during application of 0.2 mg/mL Lqh-pf venom. (C) Representative trace of K1250A-CFTR with 1 mM MgATP and 50 U/mL PKA continuously present before and during application of 0.1 mg/mL Lqh-pf venom. Before binding of AMP-PNP (A top, left) or vanadate (B top, left) closings from normal channel gating are easily discernable. All records at Vm = −100 mV.
The results described above suggested that Lqh-pf venom might be less effective at inhibiting K1250A-CFTR channels or WT-CFTR channels locked open by AMP-PNP or vanadate because those channels occupy the FC state less frequently. To determine if this were true we inspected the intraburst behavior of single WT-CFTR channels with 50 U/mL PKA, 1 mM MgATP, and either 5 mM vanadate or 2.75 mM AMP-PNP continuously present in the absence of venom. Fig. 8 A shows a representative trace of a locked open burst from a recording where WT-CFTR channel activity was stimulated by AMP-PNP. In the expanded segment shown it is evident that FC intraburst closures continued to occur. However, the rate of occurrence appears to be lower than that seen during open bursts of WT-CFTR gating normally with only MgATP and PKA present (compare to Fig. 5 A). Similar results were seen when WT-CFTR channels were locked open with vanadate (Fig. 8 B) or when K1250A-CFTR channels were activated with MgATP (Fig. 8 C).
FIGURE 8.

Frequency of intraburst closings is dependent on experimental condition, in the absence of venom. (A) Representative trace of WT-CFTR from an excised inside-out patch with 1 mM MgATP, 50 U/mL PKA, and 5 mM AMP-PNP continuously present. (B) Representative trace of WT-CFTR from an excised inside-out patch with 1 mM MgATP, 50 U/mL PKA, and 2.75 mM vanadate continuously present. (C) Representative trace of K1250A-CFTR from an excised inside-out patch with 1 mM MgATP and 50 U/mL PKA continuously present. All recordings at Vm = −100 mV. Note that brief closings, ≥5 ms in duration, occur less frequently when CFTR channel activity is manipulated by drug (vanadate or AMP-PNP) or mutation (K1250A), compared to WT-CFTR channels that are activated by MgATP and PKA only. (D) Effect of vanadate, AMP-PNP or K1250A mutation on the frequency of intraburst closings in records filtered at 500 Hz. Columns and error bars indicate mean ± SE of n = 19–40 bursts at each condition. During either AMP-PNP or vanadate treatment locked open events were identified as any open bursts of 5-s minimum duration with a minimum interburst closed duration set at 100 ms. Significant difference from control (WT-CFTR with ATP only) value: *p < 0.001.
To determine the mean open time between FC intraburst closures we analyzed openings that contained a single active channel and at least one intraburst closing between 5 and 100 ms in duration. Because WT-CFTR channels in the presence of AMP-PNP or vanadate cycle between normal gating and locked-open gating (as seen in Fig. 7), only those bursts ≥5 s in duration were used in our analysis of experiments with those reagents. All K1250A-CFTR openings were used. Fig. 8 D shows the observed mean open time between FC intraburst closings, in the absence of venom, which clearly varied depending on experimental conditions. WT-CFTR channels transitioned to FC intraburst closures once every 627.6 ± 25.7 ms under standard control conditions (n = 3 recordings, n = 47 bursts, n = 433 open duration events). It is important to note that, in each of these recordings, the control channel Po was between 0.174 and 0.281, indicating that these channels would have been greatly inhibited by 0.1 mg/mL Lqh-pf venom (Table 1). Channels locked open by AMP-PNP (mean open time between FC closings 1485.2 ± 98.2 ms; n = 2 recordings, n = 27 bursts, n = 176 open duration events) or vanadate (1010.3 ± 58.9 ms; n = 2 recordings, n = 29 bursts, n = 234 open duration events) transitioned to intraburst closures significantly less frequently (p ≤ 0.001 each, compared to WT-CFTR). The largest increase in mean open time between intraburst closings was seen in K1250A-CFTR where the channels remained open for an average of 1580.9 ± 106.8 ms (n = 3 recordings, n = 3 bursts, n = 177 open duration events; p ≤ 0.001 compared to WT-CFTR) between intraburst closings. These results suggest that the frequency at which CFTR moves to the FC state is dependent upon the status of binding and hydrolysis events at NBD-B. Comparing the data in Fig. 8 D with that in Table 1 shows that as these manipulations increased Po and decreased the frequency of transitions to the FC state, venom-mediated inhibition was reduced. Hence, intraburst inhibition of open channels by venom appears to occur upon binding of the active toxin at the FC state.
DISCUSSION
Our previous studies have shown that the venom of the scorpion L. quinquestriatus hebraeus contains a low molecular-weight peptide toxin or toxins that reversibly inhibit(s) WT-CFTR channels exclusively from the cytoplasmic side (19). WT-CFTR channels that exhibited high activity under control conditions were very poorly inhibited by venom. Additionally, WT-CFTR channels that were locked in the open conformation by vanadate or AMP-PNP, and CFTR channels bearing the K1250A mutation, were inhibited by venom only at higher concentrations. These observations suggested that the level of CFTR activity could affect the efficacy of channel inhibition by venom. Data in the present study indicate that the toxin or toxins contained in Lqh venom preferentially bind to closed CFTR channels, suggesting that venom-induced inhibition is state-dependent. Single-channel studies, where CFTR activity was altered by either changing the cytoplasmic [MgATP], treating the channels with vanadate or AMP-PNP, or by eliminating ATP hydrolysis by mutation and thereby slowing channel gating, suggest that the amount of inhibition by Lqh-pf venom is dependent on the channel's Po. Lastly, the data suggest that the toxin or toxins can effectively inhibit CFTR channels only if allowed to interact in either of two closed states.
State-dependent inhibition of cation channels by either a peptide toxin or a small organic compound has been described on several occasions (31–33). N-type Ca2+ channel block by ω-conotoxin GVIA, Shaker K+ channel block by κ-conotoxin PVIIA, and HERG K+ channel block by ergtoxin, are examples of ion channel block by peptide toxins that show state-dependence (34–36). However, few examples of state-dependent inhibition of anion channels have been documented. Accardi and Pusch (37) have shown that p-chlorophenoxy-acetic acid has higher affinity for the closed state of ClC-0 channels than for the open state. Similarly, the buffer 3-(N-morpholino) propanesulfonic acid was shown to block the major subconductance state more readily than the full conductance state of CFTR when applied to the cytoplasmic surface of the channel (28). However, at this time there are no CFTR gating modifier reagents that have been shown to bind in a state-dependent manner.
Allosteric CFTR gating modifiers such as CFTRinh-172 and genistein are believed to bind to the NBDs. However, their usefulness as probes to study CFTR conformational changes during gating is limited due to their complex modes of action. Genistein is a CFTR activator at low μM concentrations, but at high μM concentrations, genistein decreases channel Po due to a prolongation of the closed time (14,24); at high concentrations, genistein also blocks the channel pore (38). CFTRinh-172 is thought to work as a gating modifier by interacting with NBD-1; however, this mechanism has not been rigorously tested (39).
Data presented here show that venom inhibits CFTR channel activity in a closed-state dependent manner by binding to a site that prohibits channel opening from the C2 state and that also inhibits channel reopening from the FC state; the former effect leads to a lengthening of the interburst duration, whereas the latter effect introduces new intraburst closed states. A conservative view would consider that these two behaviors represent binding to the same site, while the kinetics of interaction with that site depend upon the status of ATP binding and hydrolysis at NBD-B. However, as these experiments were performed with only partially fractionated venom we must consider that these two mechanisms may represent the activities of two different toxins, which may bind to two different sites. Individual toxins isolated from venom will be required to address this possibility.
For the following reasons, we propose that the active component or components of Lqh-pf venom interact(s) with CFTR at the NBDs adjacent to but not within the active site of NBD-B:
Recordings from Flag-cut-ΔR-CFTR channels (Fig. 1) suggest that the venom does not inhibit CFTR by interacting with the R-domain.
The results from Figs. 3 and 4 suggest that the venom interacts with channels in the C2 state for a very long time, leading to an apparent reduction in active channel number; these channels simply disappear from the recording, until the venom is washed away (19). This mode of interaction explains the dependence upon [ATP]. Channels that bind toxin in the C2 state are conformationally excluded from opening after binding ATP at NBD-B, and channels that have bound this second ATP and entered into an open burst cannot bind the toxin until they close to the C2 state.
It seems unlikely that toxin binds to the C3 state, since this would lead to a reduction in the apparent macroscopic opening rate upon rapid introduction of ATP, which we did not observe (19), and because binding at the C3 state would be enhanced at high [MgATP], which is in contrast to our findings.
Our data can also exclude the possibility that the interburst duration is lengthened by binding of the toxin in the C4 state, which is the closed state that immediately follows de-dimerization of the NBDs after ATP hydrolysis. Such an effect would require that channels bind ATP, open, hydrolyze the ATP, and then close before entering into the closed-blocked state. This is not consistent with our macropatch recordings upon rapid exposure to ATP in the presence of venom because under this mechanism we would expect to see an increase in macroscopic current due to channel opening followed by a rapid decrease in current as channels close and then become locked into the C4 state. Furthermore, K1250A-CFTR channels and WT-CFTR channels locked open by AMP-PNP basically never reach the C4 state, and yet do show the lengthening of interburst durations (Fig. 4).
Hence, our data are most consistent with the notion that interaction of venom with channels in the C2 closed state underlies the lengthening of interburst closed durations in an ATP concentration-dependent manner.
The apparent dependence of the efficacy of venom-mediated inhibition in the C2 state upon the cytosolic ATP concentration (Fig. 3 A) is suggestive of simple competitive inhibition of ATP binding at NBD-B. However, we favor instead a relationship between efficacy of inhibition and channel activity and therefore the availability of the C2 state, which is partly controlled by ATP binding at NBD-B, for the following reasons:
Our previous experiments indicated no effect of venom on macroscopic opening rate at ATP concentrations near the reported EC50 or >10-fold higher (19).
Although the macropatch data do show a relationship between [ATP] and fractional inhibition (Fig. 3 A), there is also a linear relationship between fractional inhibition and estimated channel Po (Fig. 3 D); the problem here, of course, is that CFTR open probability in the presence of ATP and PKA is dependent upon [ATP].
The low degree of macroscopic inhibition at 5 mM ATP in macropatches is similar to the low degree of inhibition of those single channels that exhibit high control Po (Table 1). Because [ATP] was held constant at 1 mM in these single-channel experiments, the reduced efficacy of venom-mediated inhibition cannot be attributed to competitive inhibition of nucleotide binding at NBD-B.
A combination of mutational analysis and binding studies using labeled ATP will be helpful for clarifying these possible mechanisms.
Venom-mediated inhibition of CFTR channels appears to occur via two mechanisms: one that lengthens interburst durations, by locking channels into the C2 closed state as discussed above, and another that affects intraburst kinetics. The effect on interburst kinetics cannot fully explain the reduced efficacy of inhibition of single K1250A-CFTR channels or single WT-CFTR channels in the presence of AMP-PNP or vanadate. To incorporate those findings, we must include another state amenable to interaction with the toxin. It is well known that brief, flickery closures, with durations between 5 and 100 ms, are seen in WT-CFTR under standard conditions and even in channels that have been locked into an openable state by events in the NBDs such as binding nonhydrolyzable nucleotides. Hence, these closed events likely involve changes in conformation of the pore domain, or in the structures that connect the pore domain and the NBDs, and do not reflect binding and hydrolysis events in the NBDs. To distinguish these closures from those that occur during ATP-dependent gating, we label them FC for flickery closings (Fig. 2). Our previous experiments, analyzing records that were heavily filtered (100 Hz), showed that exposure of WT-CFTR channels to Lqh-pf venom led to a decrease in mean closed durations (τc) as well as a decrease in mean open burst durations (τo) (19). The decrease in τc arose because the population of venom-induced closed states, with durations 400–700 ms, greatly outnumbered the population of long, interburst closed states, with typical durations > 1 sec. These data suggested that the toxin bound to the open state, because the new population of venom-induced closed states appeared to arise from the open current level. This was most apparent in experiments with channels locked open by vanadate, AMP-PNP, or mutation K1250A. However, binding of toxin to the open state is not consistent with the lack of venom-mediated inhibition of current when venom was applied to open channels in macropatch experiments such as that shown in Fig. 1 C. Furthermore, filtering the data at 100 Hz precluded analysis of the brief intraburst closures.
To determine whether venom interacts with CFTR during an intraburst closure, we recorded channel activity with reduced filtering of the data (500 Hz). These records (Figs. 7 and 8) indicate numerous FC closings during open bursts of WT-CFTR under standard conditions and, to a lesser extent, in bursts from WT-CFTR locked open by AMP-PNP or vanadate. Careful examination of the closed time histograms compiled from records with limited filtering indicated that exposure to venom resulted in a decrease in the number of intraburst FC events with durations <20 ms and a concomitant increase in the number of intraburst closed states with duration in the hundreds of milliseconds, which represent the venom-induced intraburst closed states. Hence, the brief FC states appeared to be converted into the much longer FCB states upon binding the toxin (Fig. 2).
As described in Results, the expected Po of individual CFTR channels in the presence of 1 mM ATP is <0.5. One would expect that the Lqh-pf venom would have ample opportunity to achieve significant inhibition of macroscopic currents with this level of channel activity by binding in the C2 state. However, as shown in Fig. 1 C, a 30-s application of Lqh-pf venom onto fully activated CFTR resulted in only minimal channel inhibition. If the C2 interburst and FC intraburst closings are, in effect, the targets for the Lqh-pf venom, then how can we explain these results? We suggest that the level of Lqh-pf venom-mediated inhibition is dependent on the amount of time that CFTR is in the proper conformation during venom application; this is the definition of state-dependence. Multichannel records suggest that the dissociation rate of the Lqh-pf venom from the C2 closed state is extremely slow; however, we have no accurate measure of the venom on-rate to the C2 interburst closed state. It is plausible that the venom on-rate is also relatively slow and that C2 closings in WT-CFTR channels undergoing normal gating are not of sufficient length to warrant venom binding when the channels are fully activated by high concentrations of MgATP; this is likely the case since we continued to see single CFTR channel openings for tens of minutes while Lqh-pf venom was continuously present (for example, see Fig. 5). One would expect that at some point Lqh-pf venom would bind to the channel when it is transiently in the C2 closed state resulting in channel knockout unless the C2 closings are not of sufficient duration when ATP is present in the bath solution.
Which mechanism, interburst inhibition in the C2 state or intraburst inhibition in the FC state, is more important for the overall inhibition of CFTR channel current? The data shown in Fig. 1 suggest that the former accounts for the majority of the inhibitory efficacy of venom at CFTR. The ∼25% inhibition of macroscopic current at 0.1 mg/mL Lqh-pf venom should represent the result of both mechanisms of action. When venom is allowed to interact with channels in the absence of ATP, some fraction of channels bind the active toxin and become locked in the C2 state such that they cannot open upon reintroduction of ATP, but the extent of inhibition by this mechanism cannot be determined without exposure to ATP. However, application of venom to channels that have not bound toxin in the C2 state, and are undergoing normal gating, leads to, at most, ∼5% inhibition (see Fig. 1, C–E). Hence, the remainder, i.e., ∼20% inhibition, must arise from the block of channels in the interburst C2 closed state.
What determines the development of intraburst inhibition? Analysis of single-channel records that were from 200 to 1300 s in duration indicates that WT-CFTR channels reside in a conformation reflecting intraburst closings of 5–20 ms (the FC state) only 0.39 ± 0.09% of the time (n = 7; data not shown). From these results, we can calculate that WT-CFTR channels will reside in the FC state for a total of only 117 ms during the 30-s applications of 1 mM MgATP in the macropatch configuration. These results suggest that a 30-s application of 0.1 mg/mL Lqh-pf venom to open CFTR channels will not likely result in maximum inhibition of channel activity by either mechanism, although if the venom were applied to the activated channels for an extended period then a greater level of inhibition would develop through time; the data in Fig. 1 E are consistent with this.
The previous results could also explain why Lqh-pf venom is significantly less effective when applied to K1250A-CFTR, since the frequency of short intraburst closings in this mutant is much less than that seen with WT-CFTR (Fig. 8 D). Similar results are also seen in recordings of WT-CFTR that were locked open with either vanadate or AMP-PNP, which would explain why an increased concentration of venom was needed to achieve levels of inhibition similar to that seen during venom treatment of WT-CFTR under standard conditions (Table 1).
What determines the frequency of the FC intraburst closings? We hypothesize that the frequency of these events depends on the association between the CFTR NBD dimer and the intracellular loops that connect the transmembrane helices on the cytoplasmic side of the protein. Several studies suggest that the conformation of the CFTR pore changes between the open and closed states, including evidence that changes in anion selectivity and susceptibility to blockade are linked to the ATP hydrolysis cycle (40,41). Where this conformational change takes place is unknown since the regions within the CFTR polypeptide that link binding and hydrolysis of ATP at the NBDs to control of the pore conformation have not yet been identified. However, it seems safe to speculate that the intracellular loops identified in the structures of several ABC transporter proteins, including CFTR, may serve this role (42,43). These intracellular connecting domains (ICDs) also likely contribute to the wide intracellular vestibule of the CFTR pore (44). The tightness of the association between the channel NBDs and ICDs could determine the frequency of intraburst closings since it is believed that FC intraburst closings are the result of pore configuration changes and not due to a step in the ATP-dependent gating cycle (D. Gadsby, personal communication).
The nature of the nucleotide species that occupies NBD-B could be the determining factor in the strength of interaction between the two CFTR domains. Indeed, recent studies have shown that slight structural differences are evident when the ATP-bound NBD dimer is compared to a NBD dimer with ADP and vanadate in place of ATP at NBD-B (43,45,46). Therefore, the ATP/ADP+Pi bound state of the NBD dimer in WT-CFTR could represent the least stable association between the NBDs and the ICDs, resulting in frequent FC intraburst closings (see Fig. 2). The intermediate increase in mean open time between intraburst closings when WT-CFTR channels were treated with vanadate could represent a stabilization of the NBD-ICD interaction due to the modified ATP/ADP+VO4 bound state of the NBD dimer. The ATP/ATP bound state of the NBD dimer could represent a very stable NBD-ICD interaction. The decreases in the frequency of FC closings seen in our single-channel recordings when WT-CFTR channels were treated with poorly hydrolysable ATP analogs such as AMP-PNP attest to this possibility. The K1250A-CFTR results are also consistent with this notion. The lysine-to-alanine mutation causes a large decrease in the hydrolysis rate of the ATP at NBD-B, resulting in long-lived ATP/ATP bound NBD dimers. As a result, the NBD-ICD interaction would be stabilized, resulting in a decrease in the frequency of FC intraburst closings during open bursts in K1250A-CFTR, which is what we see in Fig. 8 C. The structural differences that are dependent on the nucleotide species bound at NBD-B could also have a large effect on the on-rate of venom binding during the FC intraburst closings. This would explain the decrease in intraburst inhibition of K1250A-CFTR channels and of WT-CFTR channels that are locked open with AMP-PNP or vanadate when a low concentration of the venom was applied (Fig. 7). Indeed, the order of susceptibility of the channel to inhibition by venom (WT-CFTR with ATP > WT-CFTR with vanadate > WT-CFTR with AMP-PNP > K1250A-CFTR with ATP) (Table 1) is similar to the predicted order shown in Fig. 8 D.
Small conformational changes in the NBD dimer are needed to signal for channel opening (9). These findings suggest that the NBD dimer, the ICDs, and the channel pore domains lie relatively close to one another. Therefore, the NBDs, the ICDs, or a combination of the two may provide the binding site(s) for the toxin(s) in Lqh-pf venom. In this regard, the state-dependent action of the venom may arise because the structure of the NBDs and ICDs change between the closed and open states. The apparent affinity of the active components of Lqh-pf venom for binding CFTR channels is high, as indicated by the 20-fold slower off-rate from the FC closed state, and potentially 100-fold slower off-rate from the C2 closed state, compared to that of glibenclamide, which represents one of the best-studied small-molecule blockers of CFTR (23). Therefore, the toxins in Lqh venom that target CFTR channels may serve as probes for studying the changes in conformation during gating that occur outside of the NBD dimer interface.
Acknowledgments
We thank Drs. David Dawson and David Gadsby for helpful comments concerning the CFTR gating scheme, Binlin Song for assistance with the preparation of the constructs used in this study, and Christopher Thompson for helpful discussion.
This work was supported by grants from the National Science Foundation (No. MCB-0077575), and the National Institutes of Health (Nos. DK-56481 and DK-066409). During the performance of this study, N.A.M. was an Established Investigator of the American Heart Association.
References
- 1.Quinton, P. M. 1990. Cystic fibrosis: a disease in electrolyte transport. FASEB J. 4:2709–2717. [DOI] [PubMed] [Google Scholar]
- 2.Cheng, S. H., R. J. Gregory, J. Marshall, S. Paul, D. W. Souza, G. A. White, C. R. O'Riordan, and A. E. Smith. 1990. Defective intracellular transport and process of CFTR is the molecular basis of most cystic fibrosis. Cell. 63:827–834. [DOI] [PubMed] [Google Scholar]
- 3.Devuyst, O., and W. B. Guggino. 2002. Chloride channels in the kidney: lessons learned from knockout animals. Am. J. Physiol. 283:F1176–F1191. [DOI] [PubMed] [Google Scholar]
- 4.Riordan, J. R., J. M. Rommens, B. S. Kerem, N. Alon, R. Rozmahel, Z. Grzelczak, J. Zielenski, S. Lok, N. Plavsic, J. L. Chou, M. L. Drumm, M. C. Iannuzzi, F. S. Collins, and L. C. Tsui. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245:1066–1072. [DOI] [PubMed] [Google Scholar]
- 5.Chen, J.-H., X.-B. Chang, A. A. Aleksandrov, and J. R. Riordan. 2002. CFTR is a monomer: biochemical and functional evidence. J. Membr. Biol. 188:55–71. [DOI] [PubMed] [Google Scholar]
- 6.Zhang, Z.-R., G. Cui, X. Liu, B. Song, D. C. Dawson, and N. A. McCarty. 2005. Determination of the functional unit of the Cystic Fibrosis Transmembrane Conductance Regulator chloride channel: one polypeptide forms one pore. J. Biol. Chem. 280:458–468. [DOI] [PubMed] [Google Scholar]
- 7.Gadsby, D. C., and A. C. Nairn. 1999. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 79:S77–S107. [DOI] [PubMed] [Google Scholar]
- 8.Sheppard, D. N., and M. J. Welsh. 1999. Structure and function of the CFTR chloride channel. Physiol. Rev. 79:S23–S40. [DOI] [PubMed] [Google Scholar]
- 9.Vergani, P., S. W. Lockless, A. C. Nairn, and D. C. Gadsby. 2005. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 433:876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basso, C., P. Vergani, A. C. Nairn, and D. C. Gadsby. 2003. Prolonged nonhydrolytic interaction of nucleotide with CFTR's NH2-terminal nucleotide binding domain and its role in channel gating. J. Gen. Physiol. 122:333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vergani, P., A. C. Nairn, and D. C. Gadsby. 2003. On the mechanism of MgATP dependent gating of CFTR Cl− channels. J. Gen. Physiol. 120:17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kidd, J. F., M. Ramjeesingh, F. Stratford, L. J. Huan, and C. E. Bear. 2004. A heteromeric complex of the two nucleotide binding domains of CFTR mediates ATPase activity. J. Biol. Chem. 279:41664–41669. [DOI] [PubMed] [Google Scholar]
- 13.Al-Nakkash, L., and T.-C. Hwang. 1999. Activation of wild-type and ΔF508-CFTR by phosphodiesterase inhibitors through cAMP-dependent and -independent mechanisms. Pflugers Arch. 437:553–561. [DOI] [PubMed] [Google Scholar]
- 14.Schultz, B. D., A. K. Singh, D. C. Devor, and R. J. Bridges. 1999. Pharmacology of CFTR chloride channel activity. Physiol. Rev. 79:S109–S144. [DOI] [PubMed] [Google Scholar]
- 15.Smit, L. S., D. J. Wilkinson, M. K. Mansoura, F. C. Collins, and D. C. Dawson. 1993. Functional roles of the nucleotide-binding folds in the activation of the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA. 90:9963–9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carson, M. R., S. M. Travis, and M. J. Welsh. 1995. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J. Biol. Chem. 270:1711–1717. [DOI] [PubMed] [Google Scholar]
- 17.Baukrowitz, T., T.-C. Hwang, A. C. Nairn, and D. C. Gadsby. 1994. Coupling of CFTR Cl− channel gating to an ATP hydrolysis cycle. Neuron. 12:473–482. [DOI] [PubMed] [Google Scholar]
- 18.Zeltwanger, S., F. Wang, G.-T. Wang, K. D. Gillis, and T.-C. Hwang. 1999. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. J. Gen. Physiol. 113:541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuller, M. D., Z.-R. Zhang, G. Cui, and N. A. McCarty. 2004. Inhibition of CFTR channels by a peptide toxin of scorpion venom. Am. J. Physiol. Cell. 287:C1328–C1341. [DOI] [PubMed] [Google Scholar]
- 20.McDonough, S., N. Davidson, H. A. Lester, and N. A. McCarty. 1994. Novel pore lining residues in CFTR that govern permeation and open-channel block. Neuron. 13:623–634. [DOI] [PubMed] [Google Scholar]
- 21.McCarty, N. A., S. McDonough, B. N. Cohen, J. R. Riordan, N. Davidson, and H. A. Lester. 1993. Voltage-dependent block of the cystic fibrosis transmembrane conductance regulator Cl− channel by two closely related acrylaminobenzoates. J. Gen. Physiol. 102:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang, Z.-R., S. Zeltwanger, and N. A. McCarty. 2000. Direct comparison of NPPB and DPC as probes of CFTR expressed in Xenopus oocytes. J. Membr. Biol. 175:35–52. [DOI] [PubMed] [Google Scholar]
- 23.Zhang, Z.-R., S. Zeltwanger, and N. A. McCarty. 2004. Steady-state interactions of glibenclamide with CFTR: evidence for multiple sites in the pore. J. Membr. Biol. 199:15–28. [DOI] [PubMed] [Google Scholar]
- 24.Wang, F., S. Zeltwanger, I. C. Yang, A. C. Nairn, and T. C. Hwang. 1998. Actions of genistein on cystic fibrosis transmembrane conductance regulator channel gating. Evidence for two binding sites with opposite effects. J. Gen. Physiol. 111:477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muanprasat, C., N. D. Sonawane, D. Sailinas, A. Taddei, L. J. V. Galietta, and A. S. Verkman. 2004. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J. Gen. Physiol. 124:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Csanády, L., K. W. Chan, D. Seto-Young, D. C. Kopsco, A. C. Nairn, and D. C. Gadsby. 2000. Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J. Gen. Physiol. 116:477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winter, M. C., D. N. Sheppard, M. R. Carson, and M. J. Welsh. 1994. Effect of ATP concentration on CFTR Cl− channels: a kinetic analysis of channel regulation. Biophys. J. 66:1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishihara, H., and M. J. Welsh. 1997. Block by MOPS reveals a conformational change in the CFTR pore produced by ATP hydrolysis. Am. J. Physiol. Cell. 273:C1278–C1289. [DOI] [PubMed] [Google Scholar]
- 29.Ikuma, M., and M. J. Welsh. 2000. Regulation of CFTR Cl− channel gating by ATP binding and hydrolysis. Proc. Natl. Acad. Sci. USA. 97:8675–8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang, T.-C., G. Nagel, A. C. Nairn, and D. C. Gadsby. 1994. Regulation of the gating of cystic fibrosis transmembrane conductance regulator Cl channels by phosphorylation and ATP hydrolysis. Proc. Natl. Acad. Sci. USA. 91:4698–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terlau, H., A. Boccaccio, M. O. Baldomero, and F. Conti. 1999. The block of Shaker K+ channels by κ-conotoxin PVIIA is state-dependent. J. Gen. Physiol. 114:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang, Y.-C., and C.-C. Kuo. 2002. Inhibition of Na+ current by imipramine and related compounds: different binding kinetics as an inactivation stabilizer and as an open channel blocker. Mol. Pharm. 62:1228–1237. [DOI] [PubMed] [Google Scholar]
- 33.Chen, H., F. Sesti, and S. A. N. Goldstein. 2003. Pore- and state-dependent cadmium block of Iks channels formed with MinK-55C and wild-type KCNQ1 subunits. Biophys. J. 84:3679–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stocker, J. W., L. Nadasdi, R. W. Aldrich, and R. W. Tsien. 1997. Preferential interaction of ω-conotoxins with inactivated N-type Ca2+ channels. J. Neurosci. 17:3002–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naranjo, D. 2002. Inhibition of single Shaker K channels by κ-conotoxin-PVIIA. Biophys. J. 82:3003–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milnes, J. T., C. E. Dempsey, J. M. Ridley, O. Crociani, A. Arcangeli, J. C. Hancox, and H. J. Witchel. 2003. Preferential closed channel blockade of HERG potassium currents by chemically synthesized BeKm-1 scorpion toxin. FEBS. Lett. 547:20–26. [DOI] [PubMed] [Google Scholar]
- 37.Accardi, A., and M. Pusch. 2003. Conformational changes in the pore of ClC-0. J. Gen. Physiol. 122:277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lansdell, K. A., Z. Cai, J. F. Kidd, and D. N. Sheppard. 2000. Two mechanisms of genistein inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in murine cell line. J. Physiol. 524:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taddei, A., C. Folli, O. Zegarra-Moran, P. Fanen, A. S. Verkman, and L. J. Galietta. 2004. Altered channel gating mechanism for CFTR inhibition by a high affinity thiazolidinone blocker. FEBS Lett. 558:52–56. [DOI] [PubMed] [Google Scholar]
- 40.Dawson, D. C., X. Liu, Z.-R. Zhang, and N. A. McCarty. 2003. Anion conduction by CFTR mechanisms and models. In The CFTR Chloride Channel. K. Kirk, and D. C. Dawson, editors. Landes Publishing, Georgetown, Texas. 1–34.
- 41.McCarty, N. A. 2000. Permeation through the CFTR chloride channel. J. Exp. Biol. 203:1947–1962. [DOI] [PubMed] [Google Scholar]
- 42.Rosenberg, M. F., A. B. Kamis, L. A. Aleksandrov, R. C. Ford, and J. R. Riordan. 2004. Purification and crystallization of the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 279:39051–39057. [DOI] [PubMed] [Google Scholar]
- 43.Reyes, C. L., and G. Chang. 2005. Structure of the ABC transporter MsbA in complex with ADP·vanadate and lipopolysaccharide. Science. 308:1028–1031. [DOI] [PubMed] [Google Scholar]
- 44.Seibert, F. S., P. Linsdell, T. W. Loo, J. W. Hanrahan, J. R. Riordan, and D. M. Clarke. 1996. Cytoplasmic loop three of cystic fibrosis transmembrane conductance regulator contributes to regulation of chloride channel activity. J. Biol. Chem. 271:27493–27499. [DOI] [PubMed] [Google Scholar]
- 45.Hung, L. W., I. X. Wang, K. Nikaido, P. Q. Liu, G. F. Ames, and S. H. Kim. 1998. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature. 396:703–707. [DOI] [PubMed] [Google Scholar]
- 46.Change, G., and C. B. Roth. 2001. Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science. 293:1793–1800. [DOI] [PubMed] [Google Scholar]