

Abstract
Membrane-peptide interactions are involved in many crucial biological and pharmacological activities. To clarify the interaction mode of membrane-peptide complexes, it is important to analyze both the dynamic properties and the contact residues of the membrane-bound peptide. In this study, we investigated the dynamic properties of a peptide bound to a lipid bilayer, using relaxation and amide-water exchange analyses, and directly determined the membrane-peptide interface, using the cross-saturation method. For the models of a lipid bilayer and a peptide, isotropic bicelles and mastoparan were used, respectively. The results indicate that mastoparan had a heterogeneous distribution of motion over various timescales and interacted with the lipid bilayer by using its hydrophobic side; the molecule was located within the lipid bilayer rather than on the surface, as thought previously. This study shows that the cross-saturation method is useful for determining the interface of not only protein-protein but also membrane-peptide complexes.
INTRODUCTION
Many peptides exhibit interesting biological and pharmacological activities. For example, animals and plants defend themselves against microorganisms by using antimicrobial peptides, and peptide hormones elicit physiological effects by binding to G-protein coupled receptors. Some peptides function by direct absorption to and translocation across cell membranes (1), whereas other peptides function by binding to receptors. In the latter case, the two-step ligand transportation model suggests that the peptide initially binds to the cell membrane, undergoes two-dimensional diffusion through the membrane, and interacts with its receptor (2). Thus, membrane-peptide interactions may be an essential requirement for expression of peptide activities.
To clarify the interaction mode of membrane-peptide complexes, it is important to analyze not only contact residues but also the dynamic properties of the peptide, because the membrane itself exhibits a rather fluid and dynamic character. Such information would be especially useful for elucidating the action mechanisms of membrane-acting peptides (1). Various methods have been used to study the contact residues of membrane-peptide complexes, including fluorescence probes (3), spin-labeled lipids (4), and secondary chemical shift calculation (5). Several methods require the preparation of the sample with some modification, however, and cannot precisely detect the interaction because of its chemical derivatization. Other methods can examine only the hydrophobic environment of the peptide in the membrane. Thus, these methods indirectly detect the interaction between the membrane and the peptide. Therefore, another approach that can directly determine the interface is needed. The cross-saturation (CS) method, which we developed several years ago, is able to accurately identify the molecular interface of protein-protein complexes in solution (6). In this study, we applied this method to directly detect the interaction between a membrane and a peptide.
In this study, to model the membrane-peptide interaction, isotropic bicelles were used as the membrane, and mastoparan was used as the peptide. Isotropic bicelles are suitable as a mimic of lipid bilayers for high-resolution NMR studies of the membrane-peptide interaction (5). Mastoparan is a 14-amino acid peptide and characteristic of an amphiphilic α-helix in a phospholipid environment. The dynamic properties of mastoparan bound to an isotropic bicelle were investigated using the relaxation and amide-water chemical exchange analyses (7). In addition, the interface between the isotropic bicelle and mastoparan was determined using the CS method. We discuss the interaction mechanism of the lipid bilayer-peptide complex with regard to the dynamic properties and contact residues. The results show that the CS method may be used to analyze the interactions of not only protein-protein but also membrane-peptide complexes.
MATERIALS AND METHODS
Sample preparation
For the relaxation and amide-water chemical exchange analyses, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) (Avanti Polar Lipids) were dissolved in a buffer solution containing 10 mM acetate-d4 (pH 6.0) and 10% 2H2O. For the CS experiment, DMPC and DHPC-d22 (Avanti Polar Lipids, Alabaster, AL) were dissolved in a buffer solution containing 10 mM acetate-d4 (pH 6.0) and 95% 2H2O. These solutions were mixed and vortexed to yield a molar ratio [DMPC]/[DHPC] of 0.5 and a total phospholipid concentration of 10% w/v. The mixture was cooled to 4°C for 15 min and was then heated to 37°C for 30 min. This cycle was repeated several times to ensure sample homogeneity.
Mastoparan was expressed in Escherichia coli cells as a recombinant fusion protein, His10-ubiquitin-mastoparan-Gly (8). The cells were grown to an A600 of 0.5 at 37°C, induced with 1 mM isopropyl-β-D-thiogalactopyranoside, and incubated for an additional 3 h. Cell pellets were lysed by sonication, and the supernatant was applied to a HisTrap HP column (Amersham Biosciences, Piscataway, NJ). The ubiquitin-tag was cleaved specifically at the C-terminus of ubiquitin with yeast ubiquitin hydrolase, which was separately overexpressed in E. coli and isolated (8). The obtained mastoparan-Gly was purified by a COSMOSIL 5-C8-AR-300 column (Nacalai Tesque, Kyoto, Japan) and α-amidated by peptidylglycine α-amidating enzyme (Wako, Osaka, Japan). The resulting mastoparan was purified again by a COSMOSIL 5-C8-AR-300 column and lyophilized. Mastoparan uniformly labeled with 13C and 15N for the spectral assignments was prepared by growing cells in minimal media with 13C6-glucose and 15N-anmonium chloride. Mastoparan uniformly labeled with 2H and 15N for the dynamic analyses and CS experiment was prepared in 2H2O minimal media with 2H7-glucose and 15N-anmonium chloride.
NMR spectroscopy
All NMR experiments were performed at 37°C on a Bruker Avance 600 equipped with a cryo-cooled probe, Avance 700, or Avance 800 spectrometer (Bruker Biospin, Karlsruhe, Germany). The sequential assignments of the amide group resonances of mastoparan with and without isotropic bicelles were achieved by a set of three-dimensional triple-resonance experiments (HNCACB and CBCA(CO)NH) (9). All spectra were processed by NMRPipe (10), and data analyses were assisted by the software Sparky (11). All amide group resonances were assigned, except for the residue Ile1.
For the relaxation and amide-water exchange analyses, samples were dissolved in a buffer containing 10 mM acetate-d4 (pH 6.0) and 10% 2H2O. The final concentrations of mastoparan and isotropic bicelles were ∼0.1 mM and 5% w/v, respectively. Under this condition, the lipids assume isotropic bicelles (23). A series of inverse-detected two-dimensional 1H-15N NMR experiments were performed to determine backbone 15N R1 and R1ρ relaxation rates and heteronuclear 1H-15N steady-state nuclear Overhauser effect (NOE) values at a 1H resonance frequency of 700 MHz (12,18). In the R1ρ experiment, continuous wave spin-lock at 2.5 kHz B1-field was used and 1H 180° pulses were applied during the spin-lock period according to the pulse scheme of Massi et al. (pulse scheme 2 of Fig. 1 in Massi et al. (18)). Since all of the main chain amide resonances of mastoparan which bound to isotropic bicelle were within the spectral range of 700 Hz for 15N dimension, a 15N spin-lock power of 2.5 kHz resulted in an effective field that was tipped between 82° and 90° from the laboratory z axis. Furthermore, B1 field strength is negligible compared to the steady magnetic field B0. Under such conditions, we could approximate R1ρ as R2 (19–21). Since R1ρ experiments can use much higher effective field strength (2.5 kHz in this experiment) than R2 (CPMG) experiments, the exchange contribution in R1ρ experiment was considered to be further suppressed compared to the R2 experiment. The R1 experiment was recorded with seven delays of 50, 100, 200, 500, 800, 1100, and 1500 ms, whereas the six delays in the R1ρ experiment were 32, 48, 64, 80, 100, and 120 ms. Recycle delays of 2 s were employed in the measurement of R1 or R1ρ values. Peak picking and measuring peak height were done by using NMRPipe software (10), and the intensities of the crosspeaks corresponding to individual residues were then fitted to a single-exponential decay function to obtain the values of R1 or R1ρ by using procedures implemented in NMRPipe (10). 1H-15N NOE values were determined from two spectra, recorded with and without 1H saturation. A steady state was reached after 3 s of 120° 1H saturation pulses spaced every 5 ms, and a total recycle delay time of 5 s was used. The spectral density functions J(0), J(ωN), and J(0.87ωH) were calculated by reduced spectral density mapping as described by Farrow et al (13). During the calculation of these spectral density functions, R1ρ was treated as R2, as discussed above (20,21). Amide-water chemical exchange analysis was performed according to the procedures of Spera et al. (7). Two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) spectra were recorded with and without presaturation of the H2O resonance for 1.2 s, and a total recycle delay time of 3 s was employed. An attenuation factor, Mps/Mo, was calculated for each HSQC resonance. Because mastoparan was perdeuterated, cross-relaxation from Hα protons close to the water resonance was safely neglected.
FIGURE 1.

15N relaxation parameters for mastoparan in the presence of isotropic bicelles: (a) R1 relaxation rates; (b) R1ρ relaxation rates; and (c) heteronuclear 1H-15N NOEs.
For the CS experiment, samples were dissolved in a buffer containing 10 mM acetate-d4 (pH 6.0) and 95% 2H2O. The concentrations of mastoparan and isotropic bicelles were ∼0.1 mM and 5% w/v, respectively. The low proton content of the solvent was designed to suppress the spin diffusion caused by the exchangeable amide proton in mastoparan. The CS experiment was performed as described by us elsewhere (6). Saturation for the protons of the isotropic bicelles was achieved using the WURST-2 decoupling scheme (14). The saturation frequency was set at 0.9 ppm, and the maximum radio frequency amplitude was 0.17 kHz for WURST-2 (adiabatic factor Q0 = 1). Saturation time was 3.0 s, and adjusting delay was 1.0 s. All NMR spectra were processed and analyzed with NMRPipe (10), and the intensity reduction ratios were calculated, such that,
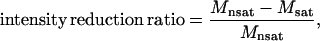 |
in which Mnsat is the signal intensity without irradiation, and Msat is the signal intensity with irradiation (22).
RESULTS AND DISCUSSION
Secondary structure of mastoparan bound to the lipid bilayer
1H-15N HSQC spectra of mastoparan were recorded in the absence and presence of isotropic bicelles. The amide group chemical shifts of 1H-15N HSQC spectra differed depending on the absence or presence of isotropic bicelles (data not shown). Resonance assignments were performed by a set of three-dimensional triple-resonance experiments (HNCACB and CBCA(CO)NH) (9). The obtained assignments for the proton resonances (Hα and Hβ) were consistent with those established by Vold et al. (5), indicating that mastoparan bound to isotropic bicelles under the experimental condition here.
The predominant secondary structure of mastoparan bound to the lipid bilayer is a helical conformation between Leu3 and Lys14 (5). The results from the chemical shift index analyses using Hα (5), Cα, and Cβ and the 15N NOESY-HSQC spectrum of mastoparan bound to the lipid bilayer confirmed this helical structure.
Dynamic properties of mastoparan bound to the lipid bilayer
For relaxation analyses of mastoparan bound to the lipid bilayer, longitudinal (R1) and transverse (R1ρ) 15N NMR relaxation rates and 1H-15N heteronuclear NOEs were measured (Fig. 1) (12). The timescales of motions are represented by the calculated reduced spectral density functions. The reduced spectral density functions J(0), J(ωN), and J(0.87ωH) that describe the motion of the H-N bonds derived from the relaxation parameters R1, R1ρ, and 1H-15N NOE are shown in Fig. 2 (13). Whereas the spectral density function at zero frequency, J(0), is sensitive to motions on all timescales, the high-frequency spectral density functions J(ωN) and J(0.87ωH) are sensitive to fast internal motions on the timescales of 1/ωN and 1/ωH (15,16). The residues Leu6–Leu11 exhibit J(0.87ωH) values consistently lower than 5 ps/rad, indicating a lack of internal motion as generally shown in the core region of a protein (15). The residues Lys4–Ala5 and Lys12–Leu14 exhibit J(0.87ωH) values consistently lower than 7 ps/rad, indicating moderate internal motion. The residues Asn2 and Leu3 exhibit higher J(0.87ωH) values than those of the other residues, indicating that a higher degree of flexibility exists in these regions. J(0) values approximately follow the opposite trend of J(0.87ωH) values.
FIGURE 2.

Reduced spectral density of mastoparan in the presence of isotropic bicelles: (a) reduced spectral density J(0); (b) reduced spectral density J(ωN); and (c) reduced spectral density J(0.87ωH).
The result of the amide-water chemical exchange experiment shows that the residues Asn2–Ala5 exhibited lower intensity ratios than those of the other residues (Fig. 3) (7). This result can usually be interpreted as the fraying of the helix (7). The result from the exchange contribution analysis between R1ρ and R2 experiments, which is sensitive to motions on the scale of milliseconds, approximately shows the same trend and supports the hypothesis regarding the fraying of the N-terminal region of the helix (data not shown).
FIGURE 3.

Amide-water chemical exchange analysis of mastoparan interacting with the lipid bilayer. Ratios of amide intensities with and without presaturation of H2O resonance are plotted as a function of residue number in the peptide sequence.
Although mastoparan bound to the lipid bilayer basically held the helical structure, starting from residue Leu3 and ending with Lys14, the residues Leu3–Ala5 and Lys12–Leu14 had fast motion (on the scale of picoseconds to nanoseconds) and the residues Leu3–Ala5 also had slower motion (on a scale greater than milliseconds). Thus, the helical peptide bound to the lipid bilayer did not possess uniform distribution of motion but showed a heterogeneous distribution of motion with various timescales.
Contact residues of mastoparan bound to the lipid bilayer
Mastoparan was uniformly labeled with 2H and 15N. Protonated DMPC and deuterated DHPC were used for the preparation of isotropic bicelles to eliminate a possible unusual saturation transfer effect, which originates from the peptide bound to the edge region of isotropic bicelles. Uniformly labeled mastoparan bound to partially labeled isotropic bicelles. The CS experiment was performed with an excess amount of isotropic bicelles relative to mastoparan to detect the resonances of the peptide in the bound state. The complex was irradiated at a frequency corresponding to DMPC acyl proton resonances exclusively affecting isotropic bicelles, because no aliphatic protons exist in deuterated mastoparan. The saturation was not limited within isotropic bicelles but was transferred via CS phenomena to the residues at the interface of the peptide. Thus, the irradiation applied to the complex resulted in selective intensity losses for the bound mastoparan resonances. The contact residues on the peptide can be identified by comparing the peak intensities on 1H-15N HSQC spectra with and without the saturation.
In this experiment, it turned out that the saturation effect in the lipid bilayer was not efficiently transferred to the residues at the interface of mastoparan, even though acyl protons were confirmed to be fully saturated in the irradiation scheme. For example, in the FB-Fc complex (64 kDa) the saturation period for 1.2 s led to intensity reduction ratios of >0.5 in the contact residues (6). In this study, the necessity of a long saturation period (3.0 s) reflected the dynamic aspect of the lipid bilayer environment. In that respect, we confirmed that the saturation of acyl chain protons of DMPC by the irradiation scheme used in this study was not transferred to the choline and glycerol protons and the saturation caused by selective irradiation of each choline or glycerol proton was not transferred to the peptide (data not shown).
Fig. 4 showed the results of the CS experiment. The irradiation of the lipids resulted in the different intensity losses for each resonance. On the basis of the spectra in the presence of the lipid bilayer with and without irradiation, we calculated the reduction ratios of the peak intensities (Fig. 5 a). The residues with intensity reduction ratios of >0.28 were Leu6, Ala7, Leu9, Ala10, Lys12, Ile13, and Leu14. The residues affected by the irradiation were distributed on one side of the helical structure (Fig. 5 b). Although the residues Ala7, Lys12, and Leu14 were profoundly affected by the irradiation, the residues Leu3 and Ala5, located in the depth of the lipid bilayer in the helix model (Fig. 5 b), were less affected. This observation indicates that the saturation effect in the lipid bilayer was not efficiently transferred to residues Leu3 and Ala5 because of their dynamic properties, as discussed in the previous section.
FIGURE 4.

Results of the CS experiment. (a, b) 1H-15N HSQC spectra observed for uniformly 2H,15N-labeled mastoparan in the presence of isotropic bicelles without (a) and with (b) the irradiation of the lipid; (c and d) F2 CSs through L3, L9, and A10 (the corresponding F1 frequencies were indicated with arrows in the spectra) without (c) and with (d) irradiation. The assignments are indicated with the one-letter amino acid code and residue number in a.
FIGURE 5.

Overview and results of the CS experiment. (a) Plots of the intensity reduction ratios of the signal intensities with and without the irradiation of the lipid. The residues with signal intensity reduction ratios of >0.28 are shown in black. (b) The helical wheel representation of mastoparan (Leu3–Leu14). The residues with signal intensity reduction ratios of >0.28 are shown in black circles. (c) Solution structure of mastoparan with detergent micelles (Protein Data Bank entry 1D7N; (24)). The residues that were affected by the irradiation are shown in black.
The downfield amide-proton shifts, commonly observed for amide protons on the more hydrophobic side of α-helices in proteins and associated with Leu3, Ala7, and Ala10, and the smaller upfield shift of the Leu14 amide resonance most likely resulted from this side of the helix being in the hydrophobic part of the lipid bilayer (5). The CS experiment showed here that mastoparan contacted the lipid bilayer with its hydrophobic side and then interacted in a deeper position of the lipid bilayer. We suggest that only the residues Lys4, Ala8, and Lys11 are located at the surface of the lipid bilayer, possibly near the choline groups of DMPC, and the other residues are buried in the depth of the acyl chains of the lipid bilayer (Fig. 5, b and c). The possibility exists that mastoparan is located on and rolls over the lipid bilayer surface between residues Ala7 and Lys12 in the helix model. However, the interpretation that mastoparan is buried within the lipid bilayer is supported by the differential quenching in transferred NOE experiments of mastoparan X, using spin-labeled phosphatidylcholine (K. Hosoda, S. Noguchi, T. Kohno, and K. Wakamatsu, unpublished results). Moreover, a previous fluorescence study indicated that the membrane-bound peptide is located a few angstroms below the headgroup of the lipid bilayer (17).
CONCLUSIONS
In this work, we investigated the dynamic properties of a peptide bound to a lipid bilayer and determined the interface between the lipid bilayer and the peptide. The CS phenomenon is driven by cross-relaxation, which is sensitive to the dynamic properties of the molecule. Therefore, in the CS experiment, the residues of the peptide with fast motion indicated lower intensity reduction ratios, even though these residues were positioned toward the molecular interface. In other words, the region in motion could not contribute to the tight interaction with the target molecule, the lipid bilayer, because of its motion. Thus, the CS method is useful for determining the molecular interface of a complex as well as discriminating the tight interaction in the molecular interface. Moreover, by using transverse relaxation-optimized spectroscopy (TROSY) techniques, this approach could be extended to the interaction between lipid bilayers and membrane proteins, which is reconstituted in the bicelles.
Acknowledgments
We thank R. Scott Prosser for helpful discussions.
This work was supported by grants from New Energy and the Industrial Technology Development Organization. T. Kohno acknowledges support through a grant from the National Project on Protein Structural and Functional Analyses. K. Wakamatsu is grateful for support by grants-in-aid for scientific research from the Japan Society for the Promotion of Science.
Takefumi Nakamura's present address is Institute of Life Sciences, Ajinomoto Co., 1-1 Suzuki-cho, Kawasaki-ku, Kawasaki, Kanagawa 210-8681, Japan.
References
- 1.Matsuzaki, K. 1999. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta. 1462:1–10. [DOI] [PubMed] [Google Scholar]
- 2.Inooka, H., T. Ohtaki, O. Kitahara, T. Ikegami, S. Endo, C. Kitada, K. Ogi, H. Onda, M. Fujino, and M. Shirakawa. 2001. Conformation of a peptide ligand bound to its G-protein coupled receptor. Nat. Struct. Biol. 8:161–165. [DOI] [PubMed] [Google Scholar]
- 3.Fujita, K., S. Kimura, and Y. Imanishi. 1994. Self-assembly of mastoparan X derivative having fluorescence probe in lipid bilayer membrane. Biochim. Biophys. Acta. 1195:157–163. [DOI] [PubMed] [Google Scholar]
- 4.Vogel, A., H. A. Scheidt, and D. Huster. 2003. The distribution of lipid attached spin probes in bilayers: application to membrane protein topology. Biophys. J. 85:1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vold, R. R., R. S. Prosser, and A. J. Deese. 1997. Isotropic solutions of phospholipid bicelles: a new membrane mimetic for high-resolution NMR studies of polypeptides. J. Biomol. NMR. 9:329–335. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi, H., T. Nakanishi, K. Kami, Y. Arata, and I. Shimada. 2000. A novel NMR method for determining the interfaces of large protein-protein complexes. Nat. Struct. Biol. 7:220–223. [DOI] [PubMed] [Google Scholar]
- 7.Spera, S., M. Ikura, and A. Bax. 1991. Measurement of the exchange rates of rapidly exchanging amide protons: application to the study of calmodulin and its complex with a myosin light chain kinase fragment. J. Biomol. NMR. 1:155–165. [DOI] [PubMed] [Google Scholar]
- 8.Kohno, T., H. Kusunoki, K. Sato, and K. Wakamatsu. 1998. A new general method for the biosynthesis of stable isotope-enriched peptides using a decahistidine-tagged ubiquitin fusion system: an application to the production of mastoparan-X uniformly enriched with 15N and 15N/13C. J. Biomol. NMR. 12:109–121. [DOI] [PubMed] [Google Scholar]
- 9.Clore, G. M., and A. M. Gronenborn. 1994. Multidimensional heteronuclear nuclear magnetic resonance of proteins. Methods Enzymol. 239:349–363. [DOI] [PubMed] [Google Scholar]
- 10.Delaglio, F., S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer, and A. Bax. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6:277–293. [DOI] [PubMed] [Google Scholar]
- 11.Goddard, T. D., and D. G. Kneller. SPARKY 3. University of California, San Francisco.
- 12.Farrow, N. A., R. Muhandiram, A. U. Singer, S. M. Pascal, C. M. Kay, G. Gish, S. E. Shoelson, T. Pawson, J. D. Forman-Kay, and L. E. Kay. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 33:5984–6003. [DOI] [PubMed] [Google Scholar]
- 13.Farrow, N. A., O. Zhang, A. Szabo, D. A. Torchia, and L. E. Kay. 1995. Spectral density function mapping using 15N relaxation data exclusively. J. Biomol. NMR. 6:153–162. [DOI] [PubMed] [Google Scholar]
- 14.Kupce, E., and G. Wagner. 1995. Wideband homonuclear decoupling in protein spectra. J. Magn. Reson. B. 109:329–333. [Google Scholar]
- 15.Raussens, V., C. M. Slupsky, B. D. Sykes, and R. O. Ryan. 2003. Lipid-bound structure of an apolipoprotein E-derived peptide. J. Biol. Chem. 278:25998–26006. [DOI] [PubMed] [Google Scholar]
- 16.Viles, J. H., D. Donne, G. Kroon, S. B. Prusiner, F. E. Cohen, H. J. Dyson, and P. E. Wright. 2001. Local structural plasticity of the prion protein. Analysis of NMR relaxation dynamics. Biochemistry. 40:2743–2753. [DOI] [PubMed] [Google Scholar]
- 17.Chung, L. A., J. D. Lear, and W. F. DeGrado. 1992. Fluorescence studies of the secondary structure and orientation of a model ion channel peptide in phospholipid vesicles. Biochemistry. 31:6608–6616. [DOI] [PubMed] [Google Scholar]
- 18.Massi, F., E. Johnson, C. Wang, M. Rance, and A. G. Palmer. 2004. NMR R1ρ rotating-frame relaxation with weak radio frequency fields. J. Am. Chem. Soc. 126:2247–2256. [DOI] [PubMed] [Google Scholar]
- 19.Peng, J. W., and G. Wagner. 1994. Investigation of protein motions via relaxation measurements. Methods Enzymol. 239:563–596. [DOI] [PubMed] [Google Scholar]
- 20.Habazettl, J., L. C. Myers, F. Yuan, G. L. Verdine, and G. Wagner. 1996. Backbone dynamics, amide hydrogen exchange, and resonance assignments of the DNA methylphosphotriester repair domain of Escherichia coli Ada using NMR. Biochemistry. 35:9335–9348. [DOI] [PubMed] [Google Scholar]
- 21.Xu, Q., and C. A. Bush. 1996. Dynamics of uniformly 13C-enriched cell wall polysaccharide of Streptococcus mitis J22 studied by 13C relaxation rates. Biochemistry. 35:14512–14520. [DOI] [PubMed] [Google Scholar]
- 22.Nishida, N., H. Sumikawa, M. Sakakura, N. Shimba, H. Takahashi, H. Terasawa, E. Suzuki, and I. Shimada. 2003. Collagen-binding mode of vWF-A3 domain determined by a transferred cross-saturation experiment. Nat. Struct. Biol. 10:53–58. [DOI] [PubMed] [Google Scholar]
- 23.Glover, K. J., J. A. Whiles, G. Wu, N. Yu, R. Deems, J. O. Struppe, R. E. Stark, E. A. Komives, and R. R. Vold. 2001. Structural evaluation of phospholipids bicelles for solution-state studies of membrane-associated biomolecules. Biophys. J. 81:2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hori, Y., M. Demura, M. Iwadate, A. S. Ulrich, T. Niidome, H. Aoyagi, and T. Asakura. 2001. Interaction of mastoparan with membranes studied by 1H-NMR spectroscopy in detergent micelles and by solid-state 2H-NMR and 15N-NMR spectroscopy in oriented lipid bilayers. Eur. J. Biochem. 268:302–309. [DOI] [PubMed] [Google Scholar]