

Abstract
The membrane disruption mechanism of pandinin 1 (pin1), an antimicrobial peptide isolated from the venom of the African scorpion, was studied using 31P, 13C, 1H solid-state and multidimensional solution-state NMR spectroscopy. A high-resolution NMR solution structure of pin1 showed that the two distinct α-helical regions move around the central hinge region, which contains Pro19. 31P NMR spectra of lipid membrane in the presence of pin1, at various temperatures, showed that pin1 induces various lipid phase behaviors depending on the acyl chain length and charge of phospholipids. Notably, it was found that pin1 induced formation of the cubic phase in shorter lipid membranes above Tm. Further, the 13C NMR spectra of pin1 labeled at Leu28 under magic angle spinning (MAS) indicated that the motion of pin1 bound to the lipid bilayer was very slow, with a correlation time of the order of 10−3 s. 31P NMR spectra of dispersions of four saturated phosphatidyl-cholines in the presence of three types of pin1 derivatives, [W4A, W6A, W15A]-pin1, pin1(1-18), and pin1(20-44), at various temperatures demonstrated that all three pin1 derivatives have a reduced ability to trigger the cubic phase. 13C chemical shift values for pin1(1-18) labeled at Val3, Ala10, or Ala11 under static or slow MAS conditions indicate that pin1(1-18) rapidly rotates around the average helical axis, and the helical rods are inclined at ∼30° to the lipid long axis. 13C chemical shift values for pin1(20-44) labeled at Gly25, Leu28, or Ala31 under static conditions indicate that pin1(20-44) may be isotropically tumbling. 1H MAS chemical shift measurements suggest that pin1 is located at the membrane-water interface approximately parallel to the bilayer surface. Solid-state NMR results correlated well with the observed biological activity of pin1 in red blood cells and bacteria.
INTRODUCTION
Many pore-forming peptides of relatively small size (15–40 amino acids) have been found in the skin, epithelial cells, and blood of vertebrates, as well as in the hemolymph and venomous secretions of arthropods. Although a number of studies have been conducted to elucidate the action and orientation of antimicrobial peptides, especially linear α-helical peptides, in membrane systems, almost all the peptides studied have been relatively small (<30 amino acids), such as mastoparan X (14 residues) (1–5), ovispirin (18 residues) (6), alamethicin (20 residues) (7,8), PGLa (21 residues) (9), MSI-78 (22 residues) (10), magainin 2 (23 residues) (11), pandinin 2 (24 residues) (12), and melittin (26 residues) (13,14). These disrupt bacterial membranes by carpet-like, toroidal pore, and/or barrel stave mechanisms. On the other hand, relatively longer α-helical peptides, such as pardaxin (33 residues) (15), cecropin A (37 residues) (16), and LL-37 (37 residues) (17), form a helix-bend-helix structure and are known to kill bacteria mainly via a carpet-like mechanism.
Pandinin 1 (pin1) (GKVWDWIKSAAKKIWSSEPVSQLKGQVLNAAKNYVAEKIGATPT) is a 44-amino-acid polypeptide, one of the longest antimicrobial peptides known. It was isolated from the crude venom of the African scorpion Pandinus imperator (18). Pin1 demonstrated high antimicrobial activity against a range of Gram-positive and Gram-negative bacteria, but hemolytic activity against sheep erythrocytes was weak. Although pin1 is predicted to consist of two distinct α-helices separated by a coil region around proline at position 19 (18), there is no further information about the binding mechanism of pin1 in the membrane-mimetic environment.
To elucidate the membrane-binding manner of pin1, we carried out the following steps. First, we solved the structure of pin1 using high-resolution NMR. Second, we prepared three synthetic derivatives of pin1: 1), analogs in which Trp4, Trp6, and Trp15 were substituted with alanine (pin1WA); 2), the N-helix-truncated form of pin1(1-18); and 3), the C-helix truncated form of pin1(20-44), and then carried out some NMR solid-state investigations of pin1 and its derivatives. Third, we investigated the morphological changes of model membranes in the presence of those derivative peptides as a function of lipid chain length using 31P NMR spectroscopy. In addition, we determined the orientation and dynamics of the peptides bound to lipid bilayers using 13C NMR spectroscopy, and we investigated the location of the Trp residue of pin1 and its derivatives in lipid bilayers by measuring the ring current shift of 1H magic angle spinning (MAS) NMR resonances induced by the Trp indole side chain in such peptides. Finally, these solid-state NMR studies of pin1 and its derivatives were compared with their biological activities.
EXPERIMENTAL
Materials
Phospholipids were purchased from Funakoshi (Tokyo, Japan) and used without further purification. The peptides pin1, pin1WA ([W4A, W6A, W15A]-pin1), pin1(1-18), and pin1(20-44) were prepared by solid-phase peptide synthesis using fluoren-9-ylmethoxycarbonyl (Fmoc) methodology on an Applied Biosystems 433A peptide synthesizer. Fmoc-Thr(Ot-butyl)-Wang was used for pin1, pin1WA, and pin1(20-44) and Fmoc-Glu(Ot-butyl)-Wang resin was used for pin1(1-18) to provide free carboxyl at the C-terminus. After synthesis, peptide deprotection, and cleavage from resin, the crude peptides were dissolved in 40% acetonitrile solution and purified by reverse-phase high-performance liquid chromatography (HPLC) on a semipreparative C18 Octa Decyl Silica (ODS) column (10 × 250 mm, Nacalai Tesque, Kyoto, Japan). The mass of the peptides was confirmed by Matrix-assisted laser-desorption ionization-time-of-flight (MALDI-TOF)-MS. Purity of synthetic peptides was higher than 95%.
Structural determination of pin1 in solution
Pin 1 (∼4 mg) was dissolved in 0.5 ml of water/2H 2,2,2-trifluoroethanol (40:60, v/v), pH 3, uncorrected for isotope effects. All 1H NMR experiments were performed on a Bruker (Karlsruhe, Germany) DRX500 spectrometer equipped with a proton/carbon/nitrogen probe and self-shielded triple axis gradients. Two-dimensional spectra were acquired using the states-time proportional phase increment method to achieve F1 quadrature detection (19). A first group of spectra (one COSY, one total correlation spectroscopy (TOCSY), and two nuclear Overhauser enhancement spectroscopy (NOESY)) were recorded at a temperature of 290 K, and the mixing times for both TOCSY and NOESY spectra were 80 ms. Another group of spectra was recorded at 310 K to resolve signal overlap issues.
Spectra were processed using UXNMR on a Silicon Graphic (Mountain View, CA) OCTANE R10000 workstation. The matrices were transformed to a final size of 2048 points in the acquisition dimension and 1024 points in the other dimension. A shifted sine-bell window in both dimensions before Fourier transform multiplied the signal, and thereafter a fifth-order polynomial baseline correction was applied. Proton chemical shifts were calibrated relative to water at 310 K at 4.75 ppm and at 290 K at 4.85 ppm. Spectrum analysis was performed using the XEASY software (20). Identification of the amino-acid spin systems and sequential assignment were performed using the standard strategy described by Wüthrich (21).
Structure calculation
The integration of nuclear Overhauser effect (NOE) data was performed using the manual integration mode of XEASY on NOESY spectra. Distance geometry calculations were performed with the variable function software DIANA 2.8 (22). A preliminary set of 1000 structures was initiated including only intraresidual and sequential upper limit distances converted from peak volumes using the CALIBA routine of the DIANA package. From the first set of structures calculated, the 500 best were kept for a second round, including medium-range upper limit distances, and the remaining 250 best for a third one, with the whole set of upper limit restraints. Finally, three runs of the REDAC (redundant dihedral angle constraints) strategy were performed on the 100 best structures, and the 30 best structures were minimized under NMR restraints using the CNS (crystallography and NMR system) minimization routine (23). The hydrogen bond restraints were obtained using the prediction routine of DIANA and were crossmatched with the NOE input. The values of the torsion angle constraints were obtained via coupling constant measurement on the double quantum filter-correlation spectroscopy (DQF-COSY) spectrum transformed with a final size of 8192/1024 points. The 3JHNα coupling constant was translated into −40°/−70° and −70°/170° angle restraints corresponding respectively to small (<7 Hz) and large (>8 Hz) coupling constants. Visual analysis of the quality of the structures was done using the TURBO software (24). Quantitative analysis was realized with PROCHECK-NMR (25).
Preparation of multilamellar vesicles
The membrane system was made of lipids and cosolubilized with peptide at a peptide/lipid (P/L) molar ratio of 1:20 in chloroform/methanol (2:1). After solvent evaporation under vacuum for one night, the lipid film was hydrated with buffer (20 mM Tris-HCl and 100 mM NaCl (pH = 7.6)) for the 31P and 13C NMR experiments and D2O (100 mM NaCl) for the 1H NMR experiment and vortex mixed. The suspension was freeze thawed for 10 cycles and centrifuged. The supernatant was removed to adjust the water content to ∼80% (w/w) and the suspension transferred to NMR tubes sealed with glue to prevent dehydration.
Solid-state NMR spectroscopy
All solid-state NMR spectra were acquired on a CMX Infinity 300 spectrometer (Chemagnetics, Varian, Palo Alto, CA) operating at a proton resonance frequency of 300 MHz. 31P spectra were acquired using a 5 μs single excitation pulse with 30 kHz continuous wave (CW) 1H decoupling during acquisition. The dwell time was 50 μs, and 256–1024 transients were accumulated for each free induction decay (FID) with a 3 s relaxation delay. The 31P chemical shifts were referenced externally to 85% H3PO4 (0 ppm). For 13C NMR measurements, 1000–16,000 transients were accumulated for each FID with a 53 μs dwell time and a 5 s relaxation delay. For hydrated samples, spectra were acquired using 4 μs excitation pulses with 30 kHz CW 1H decoupling. For lyophilized samples, a cross-polarization (CP) experiment was performed with a contact time of 2 ms, 40 kHz CP, and CW 1H decoupling radio frequency. The 13C chemical shifts were externally referenced to the methine carbon of adamantane (29.5 ppm). 13C spectra were processed using 50 Hz line broadening. 1H MAS NMR spectra were acquired using a 5.3 μs single excitation pulse. The dwell time was 100 μs, and four transients were accumulated for each FID with a 4 s relaxation delay. The 1H shifts were referenced to the proton of HDO (4.5 ppm).
Antimicrobial and hemolytic assays
Growth inhibition curves were obtained using pure peptides at concentrations of 1.6, 3.1, 6.2, 12.5, 25, 50, and 100 μg/ml. Briefly, the inoculum was prepared from fresh bacteria cultures. Serial dilutions of peptides were arranged, and an aliquot of cell suspension was added to each vial. The final volume in each vial was 100 μL, and the cell count was 1.6 × 106 colony forming units/ml for Escherichia coli. The final peptide concentration ranged from 1.6 to 50 μM. After 16–18 h of incubation at 37°C, the optical density (OD) of each vial was measured at 630 nm in an ELISA reader (BioRad, model 450, Hercules, CA). The positive control contained only the bacterial suspension, and the negative control contained only sterile culture medium. Hemolytic activity was determined by incubating suspensions of rabbit red blood cells with serial dilutions of each selected peptide. Red blood cells (10% v/v) were rinsed several times in phosphate-buffered saline (PBS) by centrifugation for 3 min at 3000 g until the OD of the supernatant reached the OD of the control (PBS only). Red blood cells were counted using an hematocytometer and adjusted to ∼7.7 × 106 ± 0.3 × 106 cells/ml. Red blood cells were then incubated at room temperature for 1 h in 10% Triton X-100 (positive control), in PBS (blank), or with pin1 and pin1 derivatives at concentrations of 1.6, 3.1, 6.2, 12.5, 25, 50, and 100 μg/ml. The samples were then centrifuged at 10,000 g for 5 min, the supernatant was separated from the pellet, and its absorbance measured at 570 nm. The relative OD compared to that of the suspension treated with 10% Triton X-100 defined the percentage of hemolysis.
RESULTS
Three-dimensional structure of pin1 in solution
Sequential assignment
The three-dimensional (3D) structure was solved using 1H NMR. The spin systems of pin1 were identified from both COSY and TOCSY spectra. NOESY spectra were used to perform the sequential assignment.
Hydrogen bonds
The exchange rates of amide protons with the solvent D2O were not measurable due to the particular solvent used. All of the hydrogen bonds proposed by DIANA and included in the calculations were fully compatible with the experimental NOE data and occurred in regular secondary structures.
Coupling constants
We manually measured 29 3JHNα meaningful coupling constants on the DQF-COSY spectrum. The missing values were attributed to the N-terminal residue, the prolines, and residues 16–27 and 39–44 either because their Hα-HN signal was quenched by the water resonance or because the value was meaningless.
Secondary structures
The sequential and medium-range NOE correlations and the coupling constant values were summarized in Fig. 1 and used to define the secondary structures. The presence of strong sequential HN-HNi+1 NOEs together with typical Hα-HNi+3 and Hα-HNi+4 connectivities were observed in residues 3–18 and 26–38, in addition to small 3JHNα coupling constants. This corresponds to two distinct α-helices.
FIGURE 1.

Diagram of sequential and short-range NOE connectivities for pin1. The height of the bar indicates the strength of NOE correlation. Coupling constants are shown in the row labeled 3JHNα and indicated by the arrows (>8 Hz).
Structure calculations
The distance geometry calculations were performed with 401 distance constraints that could be clustered into 292 intraresidues and sequential, 105 medium-range and 3 long-range NOEs. Additionally, 34 distance restraints derived from the 17 hydrogen bonds and 25 angle restraints derived from the coupling constant determinations were included in the final calculation, leading to 477 restraints. The 30 calculated structures were in good agreement with the experimental data, since neither distance violation >0.3 Å nor angle violation >10° could be observed. These structures were then energy minimized using the algorithm from the CNS 1.0 software to suppress the undesired nonbonded contacts. This step led to a family of 30 final structures still consistent with experimental restraints, as there was no NOE violation larger than 0.2 Å and no angle violation larger than 5°. The covalent geometry was convenient as indicated by the low root mean-square deviation (RMSD) values on bond lengths and valence angles. The 30 structures form two district bunches of structures with high RMSD values if all the residues are taken into account. The value drops to 1.89 Å (±0.61 Å) for the backbone and 3.18 Å (±0.70 Å) for all the heavy atoms if residues 2–18 are taken into account (Fig. 2 a), and 0.93 Å (±0.38 Å) for the backbone and 1.81 Å (±0.42 Å) for all the heavy atoms if residues 26–38 are taken into account (Fig. 2 b).
FIGURE 2.

Superimposition of the 30 best solution structures of pin1. The α-helix (2-18) is overlaid (left) and the α-helix (26-38) is overlaid (right).
Chain-length dependence of pin1–lipid membrane interactions
To examine the effect of pin1 on lipid membrane, 31P NMR spectra of lipid membrane in the presence of pin1 were measured under static conditions. Fig. 3 shows the 31P NMR spectra of dispersions of four saturated phosphatidyl-cholines, where the acyl chains increase from 12 carbons (1,2-dilauroyl-sn-glycero-3-phosphatidylcholine (DLPC)) to 18 carbons (1,2-distearoyl-sn-glycero-3-phosphatidylcholine (DSPC)) in the absence (Fig. 3 a) and presence (Fig. 3 b) of pin1 (P/L = 1:20) at various temperatures. For the shorter lipids, DLPC and 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC), almost identical phase behavior is obtained below and above Tm (∼−1°C for DLPC and ∼23°C for DMPC) (26). At temperatures above Tm and in the absence of pin1, an axially symmetric, motionally averaged powder pattern was observed. This pattern indicates that the lipids from the multilamellar vesicles (MLV) and lipid bilayers are mainly oriented in the magnet, with their normal perpendicular to the magnetic field (27). In the presence of pin1, however, broad isotropic peaks were observed. Since the external appearance was clear, viscous, and jelly-like, this can be attributed to the presence of a cubic phase (28). For the longer lipids, 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC) and DSPC, similar phase behavior was obtained below and above Tm (∼42°C for DPPC and ∼55°C for DSPC) (26). In the absence of pin1, partially oriented MLVs were observed. On the other hand, in the presence of pin1, at temperatures below Tm, sharp isotropic components were observed, superimposed on a bilayer component. This isotropic component may be ascribable to fast isotopic tumbling of phospholipids, which may be caused by the formation of micelles, small unilamellar vesicles, or small discoidal bilayers (29). At temperatures above Tm, an axially symmetric, motionally averaged powder pattern was observed at 60°C for both membranes. However, at 50°C, the DPPC membrane was shown to be in the cubic phase. For all lipids, spectra showed an intermediate state between multilamellar and cubic phases at temperatures around Tm. In this way, pin1 was shown to induce various lipid phase behaviors depending on acyl chain length and temperature.
FIGURE 3.

31P NMR spectra of dispersions of four kinds of saturated phosphatidyl-cholines, DLPC, DMPC, DPPC, and DSPC, in the (a) absence and (b) presence of pin1 (P/L = 1:20) at various temperatures.
Pin1-acidic phospholipid-containing lipid membrane interactions
Although pin1 demonstrated high antimicrobial activity against a range of Gram-positive and Gram-negative bacteria, it showed relatively weak hemolytic activity against mammalian erythrocytes (30). To compare the interactions of pin1 with bacterial and erythrocyte membranes, we also observed 31P NMR spectra of acidic phospholipid-containing membranes in the presence of pin1 under static conditions, because bacterial membranes contain abundant acidic phospholipids in addition to zwitterionic phospholipids compared to erythrocyte membranes (31,32). Fig. 4 shows the 31P NMR spectra, at various temperatures, of dispersions of four mixtures of phosphatidylcholine and phosphatidylglycerol (PG), where acyl chains increase from 12 to 18 carbons, with a molar ratio of phosphaditylcholine (PC)/PG = 4 in the presence of pin1 (P/L = 1:20). Although DLPC and DMPC membranes showed the formation of a cubic phase above Tm, DLPC/1,2-dilauroyl-sn-glycero-3-phosphatidylglycerol (DLPG) and DMPC/1,2-dimyristoyl-sn-glycero-3-phosphatidylglycerol (DMPG) membranes formed MLVs at these temperatures. In addition, although the formation of micelles was observed in DPPC and DSPC membranes below Tm and an intermediate state between multilamellar and cubic phases at temperatures around Tm, DPPC/1,2-distearoyl-sn-glycero-3-phosphatidylglycerol (DPPG) and DSPC/DSPG membranes showed only multilamellar phase at these temperatures. Comparing Figs. 3 and 4, it can be seen that pin1 forms a three-dimensionally interwoven lipidic cubic phase only in the erythrocyte membrane model (PC membrane) and not in the bacterial membrane model (PC/PG membrane).
FIGURE 4.

31P NMR spectra of dispersions of four kinds of mixed bilayer, DLPC/DLPG, DMPC/DMPG, DPPC/DPPG, DSPC/DSPG in a PC/PG molar ratio of 4:1 in the presence of pin1 (P/L = 1:20) at various temperatures.
Influence of the mutation of tryptophan in pin1 on lipid membrane morphology
To elucidate the cause of the morphological change from lipidic cubic phase to lamellar phase, the mutation of pin1, pin1WA ([W4A, W6A, W15A]-pin1) was prepared. Fig. 5 shows the 31P NMR spectra of dispersions of four saturated phosphatidyl-cholines, DLPC to DSPC, in the presence of pin1WA (P/L = 1:20) at various temperatures. For DLPC lipid, an isotropic peak was observed at 0°C, 20°C, and 40°C. Since the external appearance was milky-white and a little viscous, this can be attributed to the coexistence of cubic and lamellar phases. At −20°C and 60°C, the lamellar phase was observed. For DMPC lipid, membrane lysis occurred below Tm and the lamellar phase appeared above Tm. For the longer lipids DPPC and DSPC, however, 31P NMR spectra showed almost identical phase behaviors in the presence (Fig. 5) and absence (Fig. 3 a) of pin1 at every temperature tested, and the spectra did not show lipidic cubic phases. From these results and those observed by Killian and co-workers (33), we suggest that the three Trp residues in pin1 are a requisite for the formation of the lipidic cubic phase in the presence of pin1 in neutral lipid bilayer.
FIGURE 5.

31P NMR spectra of dispersions of four kinds of saturated phosphatidyl-cholines, DLPC, DMPC, DPPC, and DSPC, in the presence of pin1WA (P/L = 1:20) at various temperatures.
Pin1(1-18) or pin1(20-44)–lipid membrane interactions
The NMR structure of pin1 in solution reveals that the two distinct α-helical regions, which are separated by a hinge region containing Pro19, are moving around the central hinge, as described by the sequential assignment (Fig. 1). It was impossible to fit the 30 structures taking into account all the residues, but the fit was improved if the two helices were taken independently (Fig. 2). To investigate the basis for the formation of the lipidic cubic phase in neutral phospholipid membranes in the presence of pin1, and the method of interaction between the N- or C-terminal parts of pin1 and the membrane, we prepared two peptides, pin1(1-18) and pin1(20-44) (Fig. 6). Secondary structure analysis using circular dichroism spectral data demonstrated that both pin1(1-18) and pin1(20-44) formed an α-helix in a dodecylphosphocholine (DPC) membrane-mimetic environment (10 mM phosphate buffer + 120 mM DPC) and random coil structure in an aqueous environment (10 mM phosphate buffer) (not shown). We observed 31P NMR spectra of dispersions of four saturated phosphatidyl-cholines, DLPC to DSPC, in the presence of pin1(1-18) (Fig. 7 a) or pin1(20-44) (Fig. 7 b) (P/L = 1:20) at various temperatures. These show clearly different properties compared with the spectra obtained in the presence of pin1 (Fig. 3 b). In the presence of pin1(1-18), for the DMPC and DPPC membranes, an isotropic narrow peak was observed below Tm, which may be caused by the membrane lysis, and a bilayer component superimposed on a isotropic component above Tm. When comparing Fig. 3 with Fig. 7 a, it can be seen that pin1(1-18) affects the membrane in a different manner from pin1; it may have a similar effect to melittin (14) and pandinin 2 (pin2) (15) in the DMPC membranes. In the presence of pin1(20-44), however, all lipid membranes showed the lamellar phase at all temperatures (Fig. 7 b). These spectra are almost the same as those in which pin1 is absent (Fig. 3 a). A comparison of the binding mechanisms of pin1(1-18) and pin1(20-44) was later conducted using 13C NMR. Since the molecular weight of pin1(1-18) and pin1(20-44) are ∼½ of that of pin1, they could be more disruptive at a molecular ratio of P/L = 1:10 (34). Finally, to investigate the effect of the division of pin1 on the peptide-lipid membrane interaction, we observed 31P NMR spectra of dispersions of DMPC in the presence of pin1(1-18) and pin1(20-44) (pin1(1-18): pin1(20-44): DMPC = 1:1:20) at various temperatures (Fig. 7 c). Below Tm, the peptides caused membrane lysis. At 20°C and 40°C, the membrane formed an almost magnetically aligned lamellar phase. Although there was some indication of formation of isotropic phases at 60°C, these peptides could not cause complete conversion to the cubic phase. Since these spectra are completely different from the spectra of dispersions of DMPC in the presence of whole pin1 (pin1/DMPC = 1:20) at various temperatures (Fig. 3 b) and are close to the sum of the spectra obtained in the presence of pin1(1-18) (Fig. 7 a) and pin1(20-44) (Fig. 7 b), this naturally indicates that proline-based links between the N- and C-terminal helices are important in causing the formation of the cubic phase in the membrane.
FIGURE 6.

Helical wheel projections of pin1(1-18) and pin1(20-44) (the 13C-labeled residues are indicated by asterisks). Charged, neutral, and hydrophobic residues are shown in black, gray, and white, respectively.
FIGURE 7.

31P NMR spectra of dispersions of four kinds of saturated phosphatidyl-cholines, DLPC, DMPC, DPPC, and DSPC, in the presence of (a) pin1(1-18) (P/L = 1:20) and (b) pin1(20-44) (P/L = 1:20) at various temperatures. (c) 31P NMR spectra of dispersions of DMPC in the presence of pin1(1-18) and pin1(20-44) (pin1(1-18)/pin1(20-44)/lipid = 1:1:20) at various temperatures.
Slow motion of pin1 in neutral membrane
To investigate how pin1 binds to the membrane, we measured the 13C NMR spectra of site-specifically labeled pin1 in DPPC bilayer (P/L = 1:20). Fig. 8, a and b, shows the spectra of [1-13C-Leu28]pin1 in DPPC bilayer, obtained in hydrated state, and under MAS with a spinning speed of 3 and 6 kHz, respectively, at 60°C. Since the signal of pin1 can hardly be seen in Fig. 8 a, and we can see it only with difficulty at the skirt of C=O in the DPPC signal in Fig. 8 b, it may be understood that pin1 moves so slowly in the hydrated state as to cause spectral broadening. Here, when molecules move with correlation time τc, the line width Δ under MAS with the spinning speed ωr is written as follows, if we assume that the line shape is Lorentzian (35):
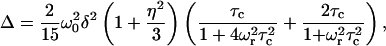 |
(1) |
where ωo shows the Larmor frequency, and the chemical shift anisotropy (CSA) δ and the asymmetry parameter η are given in terms of the principal elements of the chemical shift tensor σ11, σ22, and σ33 as 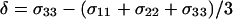 and
and 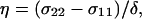 respectively. Depending on the correlation time τc, different line widths can be obtained, showing up as a simulation of different spinning speeds ωr for fixed values of ωo, δ, and η in Fig. 8 c. By comparing this curve with the spectral widths in Fig. 8, a and b, we estimate that the motion of pin1 bound to the lipid bilayer is very slow, with a correlation time of the order of 10−3 s.
respectively. Depending on the correlation time τc, different line widths can be obtained, showing up as a simulation of different spinning speeds ωr for fixed values of ωo, δ, and η in Fig. 8 c. By comparing this curve with the spectral widths in Fig. 8, a and b, we estimate that the motion of pin1 bound to the lipid bilayer is very slow, with a correlation time of the order of 10−3 s.
FIGURE 8.

13C spectra of site-specifically labeled (C=O of Leu28) pin1 in DPPC bilayer (P/L = 1:20), measured at 60°C in hydrated state under MAS. Spinning speeds are (a) 3 kHz and (b) 6 kHz, respectively. The peaks marked by asterisks are due to rotor peak and a first spinning side band. (c) The relationship of line width Δ and correlation time τc under MAS with spinning speed 3 kHz and 6 kHz.
Orientation and dynamics of pin1(1-18) or pin1(20-44) in lipid bilayers
The binding mechanism of pin1(1-18) and pin1(20-44) with lipid bilayers was investigated based on solid-state NMR studies (12,14,36). Fig. 9 a shows the carbonyl region of the 13C NMR spectra of three kinds of 13C=O site-specifically labeled pin1(1-18) (Val3, Ala10, and Ala11) bound to DPPC lipid bilayer (P/L = 1:20), measured at 45°C under static and slow MAS conditions, respectively. Under static conditions, DPPC bilayer showed a well-aligned lamellar phase in the presence of pin1(1-18), as shown in Fig. 7 a. A comparison of the full CSA (Table 1), determined by 13C CP spectra, of lyophilized samples (∼160 ppm) with those obtained under slow MAS conditions (<35 ppm), shows that the CSA under slow MAS conditions was smaller than normal. In addition, the chemical shift powder pattern obtained under slow MAS conditions showed axially symmetrical powder patterns. This indicates that the α-helix rapidly rotates around the average helical axis, assuming the C=O bond direction is nearly parallel to the helical axis (14). Further, the 13C=O chemical shift values δobs obtained under static conditions are close to δ⊥ values under slow MAS conditions, implying that the rotating axis is parallel to the lipid long axis (12,36). We simulated slow MAS spectra in a similar manner using the model shown in previous work (12). Fig. 9 b shows a superimposition of the carbonyl spectra of a variety of samples of 13C-labeled pin1(1-18) bound to DPPC lipid bilayer, obtained under slow MAS conditions, with their best-fitting simulations of spectra obtained using the δ11, δ22, and δ33 values of 13C CP spectra of lyophilized samples (listed in Table 1). They show good agreement, and the best β-orientations are listed in Table 1. These values indicate that pin1(1-18) rapidly rotates around the average helical axis, which is parallel to the lipid long axis, and the helical rods are inclined ∼30° to the lipid long axis. This model is shown in Fig. 9 c. However, Fig. 10 shows the carbonyl region of the 13C NMR spectra of three kinds of site-specifically 13C=O labeled pin1(20-44) (Gly25, Leu28, and Ala31) bound to DMPC lipid bilayer (P/L = 1:20), measured at 40°C. Under static conditions, DMPC bilayer adopts a well-aligned lamellar phase in the presence of pin1(20-44), as shown in Fig. 7 b. The 13C=O chemical shift values, δobs, of [1-13C-Gly25], [1-13C-Leu28], and [1-13C-Ala31]pin1(20-44) were found to be 171.4, 174.2, and 175.1 ppm, respectively. Since these values are relatively consistent with the isotropic values measured by 13C CP spectra (not shown) of lyophilized samples, 171.2, 172.1, and 174.9 ppm, respectively, pin1(20-44) is likely to be isotropically tumbling.
FIGURE 9.

(a) Carbonyl regions of the 13C NMR spectra of a variety of samples of 13C=O-labeled pin1(1-18) bound to DPPC lipid bilayer (P/L = 1:20), measured at 45°C in hydrated state under static and slow MAS conditions (ωr = 100 Hz), respectively. Asterisks denote the phospholipid C=O group. (b) Superimposition of the carbonyl spectra of a variety of samples of 13C-labeled pin1(1-18) bound to DPPC lipid bilayer, obtained under slow MAS condition (solid line, same as Fig. 7 a) with their best-fitting simulations (dotted line). (c) Proposed model for orientation of pin1(1-18) in lipid bilayer.
TABLE 1.
| δobs (ppm)* | δ11 (ppm)† | δ22 (ppm)† | δ33 (ppm)† | β (degree) | |
|---|---|---|---|---|---|
| Val3 | 183.4 | 250.0 | 193.0 | 81.8 | 34° |
| Ala10 | 186.3 | 249.5 | 188.0 | 88.7 | 35° |
| Ala11 | 165.9 | 248.0 | 193.5 | 87.5 | 25° |
Chemical shift values are for hydrated samples under static conditions.
Principal values are for lyophilized powder samples.
FIGURE 10.

(a) Carbonyl regions of the 13C NMR spectra of a variety of samples of 13C=O labeled pin1(20-44) bound to DMPC lipid bilayer (P/L = 1:20), measured at 40°C in hydrated state under static conditions. Asterisks denote the phospholipid C=O group.
Location of Trp residue of pin1 and pin1(1-18) in lipid bilayers
The locations of Trp residues of pin1 in lipid bilayers were investigated by measuring the ring current shift of 1H MAS NMR resonances induced by the Trp indole side chain in pin1 (37). Fig. 11 shows the 1H MAS spectra of dispersions of DPPC in D2O in the absence (a) and presence of pin1 (b), pin1WA (c), pin1(1-18) (d), and pin1(20-44) (e), measured at 60°C with a spinning speed of 3 kHz. The molar ratio of peptides/lipid was 1:20. In the presence of pin1, C2H2 and C3H2 resonances were shifted significantly upfield, indicating that the Trp indole side chains are located near the carbonyl groups of DPPC lipid bilayer. There are three Trp residues (Trp4, Trp6, and Trp15) in pin1, and if we assume that each amino acid has a length of 1.5 Å, the distances Trp4–Trp15 and Trp6–Trp15 are ∼16.5 and 13.5 Å, respectively. The hydrophobic thickness of DPPC is 29.3 Å (38). Although Trp indole side chains can cause upfield shifts in protons located up to 5 Å above and below the indole ring (37,39), insertion of pin1 into the bilayer should result in significant chemical shift changes for the other protons besides C2H2 and C3H2. Therefore, it was concluded that pin1 may extend approximately parallel to the DPPC bilayer surface at the membrane-water interface and that all indole side chains Trp4, Trp6, and Trp15 were located equally near the carbonyl groups of DPPC lipid bilayer. In the presence of pin1(1-18), resonances of C2H2, C3H2, and N(CH3)3 were shifted significantly upfield, indicating that the Trp indole side chains were located near the carbonyl groups of DPPC lipid bilayer. This also indicated that the three Trp residues in pin1(1-18) were located around C2H2, C3H2, and N(CH3)3 and that pin1(1-18) was not deeply inserted in DPPC bilayers. Furthermore, in the presence of pin1WA and pin1(20-44), there were no significant chemical shift changes. Since these contain no Trp residue, the results confirm that the chemical shift changes were due to ring current shift induced by the Trp indole side chain.
FIGURE 11.

1H MAS NMR spectra of dispersions of DPPC in D2O in the absence (a) and presence of (b) pin1, (c) pin1WA, (d) pin1(1-18), and (e) pin1(20-44), measured at 60°C with a spinning speed of 3 kHz. Molar ratios of peptides/DPPC are 1:20. Resonances showing significant chemical shift changes are highlighted with arrows.
Comparison of antimicrobial and hemolytic activity in pin1, pin1WA, pin1(1-18), and pin1(20-44)
To compare the above-mentioned results obtained from solid-state NMR of pin1 and its three derivatives, pin1WA, pin1(1-18), and pin1(20-44), the antimicrobial and hemolytic activities of these peptides were assayed and compared in Fig. 12. The amount of antimicrobial activity in these peptides was in the order pin1 ≫ pin1WA = pin1(1-18) > pin1(20-44) (Fig. 12 a). Hemolytic assays of four kinds of peptides on rabbit erythrocytes showed that pin1 had the highest hemolytic activity in all four types of peptides (Fig. 12 b), pin1(1-18) displayed weaker hemolytic activity, and pin1WA and pin1(20-44) showed hardly any hemolytic activity. Both antimicrobial and hemolytic assay results showed similar tendencies.
FIGURE 12.

Antimicrobial and hemolytic activity of pin1 and pin1 derivatives. (a) E. coli and (b) rabbit red blood cells. Data are the average of three independent experiments for the antimicrobial assays, and the average of two independent experiments for the hemolytic assays. Error bars represent the standard deviation.
DISCUSSION
In this study, the scorpion pore-forming peptide pin1 and its three synthetic derivatives, pin1WA ([W4A, W6A, W15A]-pin1), pin1(1-18), and pin1(20-44), were prepared. Morphological changes in model membranes induced by pin1 and its derivatives, as well as their orientations in lipid bilayers, were investigated. 31P solid-state NMR experiments in the presence of pin1, as shown in Fig. 3, demonstrated the formation of the cubic phase. Up to now, the formation of cubic phase structures in saturated PC bilayers in the presence of peptides has rarely been observed. Although channel-forming peptides gramicidin (40) and WALP-type peptides (33) form cubic structures in saturated PC bilayers, the cubic phase was observed at positions in the phase diagram between the Lα phase and the reversed hexagonal (HII) phases (28); most frequently, the cubic phase was found to be in equilibrium with an HII phase. Notably, in the presence of WALP-type peptides, HII phases were observed when the hydrophobic thickness of the PC lipid bilayer was longer than the peptide length. Further, cubic phases are observed when the hydrophobic thickness of the PC lipid bilayer is about the same length as the peptides. This phase preference may be explained as follows: when the peptide fills only the hydrophobic volume, steric constraints among the lipid headgroups will be relieved, allowing these lipids to organize into structures with highly curved interfaces (33). In the presence of pin1, the HII phase did not appear and the cubic phase formed above Tm instead of the Lα phase for the shorter lipids (DLPC and DMPC). If we assume that each amino acid has a length of 1.5 Å, the length of pin1 will be 66 Å. Since it is much longer than the hydrophobic thickness of the PC lipid bilayer (26.6, 27.9, 29.3, and 30.6 Å for DLPC, DMPC, DPPC, and DSPC, respectively) (38), the cubic phase-forming mechanism of pin1 may be different from that of WALP-type peptides.
Morphological changes in model lipid membranes of erythrocytes (PC) in the absence and presence of pin1 (Fig. 3) were completely suppressed in bacteria model lipid membranes (PC/PG = 4) (Fig. 4). In the presence of pin1, although PC membrane showed various phase behaviors depending on the temperature and the acyl chain length, PC/PG membrane only formed MLVs regardless of the temperature and length of the acyl chain. This difference may be explained by a cation-π interaction between Trp and Tyr in pin1 (Trp4, Trp6, Trp15, and Tyr34) and the lipid headgroups of the lipid bilayer; the cation of a choline group in a PC headgroup might be expected to bind with aromatic side chains Trp and Tyr in pin1 (41). Mixing PG with PC dilutes the cation-π interaction between pin1 and the lipid bilayer. This may be the reason pin1 did not induce the cubic phase in the PC/PG bilayer.
The suppression of the cubic phase was also shown in PC membrane in the presence of pin1WA mutant (Trp4, Trp6, and Trp15 in pin1 were substituted with Ala) (Fig. 5). From this result, we can confirm that Trp residues play a significant role in causing the formation of the cubic phase. Therefore, suppression of the cubic phase could also be described as being caused by a lack of cation-π interaction between the lipid bilayer and Trp in the peptide. In addition, dipole-dipole interaction between the carbonyl groups of PC and the imino group of Trp in the peptide (42) and imino group hydrogen bonding (43) may be responsible. Pin1WA has Tyr34 at the C-terminal; like Trp residue, Tyr residue has an aromatic side chain, but it does not have an imino group. Therefore, only the cation-π interaction between the π-face of the aromatic ring of Tyr of pin1WA and the cation of a choline group in the PC headgroup may be effective, but it is negligible.
Based on the 1H MAS and 13C NMR results for pin1 and pin1(1-18) bound to DPPC lipid bilayer, we can propose a model for the orientation and location of pin1 and pin1(1-18) in DPPC lipid bilayer above Tm (Fig. 13). The hydrocarbon (HC)-core and interface regions of DPPC lipid bilayer are shown. Here, the HC-core region is composed of acyl chains, and the interfacial region is composed of hydrated headgroups and portions of acyl chain methylenes (C2H2 and C3H2) that spill into the interface from the HC core (44). From joint liquid crystallographic refinement of x-ray and neutron diffraction data from Lα-phase lipid bilayers (45,46), the thicknesses of the HC-core and the interfacial region of the 1,2-dioleoyl-sn-glycero-3- phosphatidylcholine (DOPC) bilayer membrane were determined to be ∼29 and 15 Å, respectively. Since the structure of DOPC is the same, comparatively, as DPPC, the thickness of DPPC in Fig. 13 was referred to as that of DOPC. Based on the result from the 1H MAS NMR experiment, we propose that pin1 may be preferentially located within the bilayer interface with a little penetration into the HC core. Further, we estimated that the motion of pin1 bound to lipid bilayer was very slow, with a correlation time on the order of 10−3 s, based on the 13C MAS NMR experiment. Our proposed model for pin1 in Fig. 13 a is consistent with the results of this 13C MAS NMR experiment, because if pin1 is extended approximately parallel to the DPPC bilayer surface at the membrane-water interface, the 44-residue pin1 should disturb lateral diffusion of DPPC lipid bilayer; it is pin1 itself that reduces the motion. Other helix-bend-helix peptides, for example pardaxin (33 residues) (15), cecropin A (37 residues) (16), and LL-37 (37 residues) (17), have mainly shown carpet-type orientation. Their features are very similar to pin1; such relatively long helix-bend-helix-type peptides may have a tendency to adopt a carpet-type orientation. Furthermore, the tryptophan-rich structure of pin1 may be the trigger for the cubic phase. Since cation-π interaction, electric dipolar interaction, and hydrogen bonding between PC headgroups and Trp in pin1 may fix the position of the PC headgroup, the PC acyl chains may be spread out. This could induce negative curvature strain in the lipid bilayer and also in the cubic phase. In contrast, pin1(1-18) is thought to rotate rapidly around the average helical axis with a 30° tilt angle, and most of its parts are located in the interfacial region (Fig. 11 b). This dynamic orientation is similar to that of melittin (14,36) and pin2 (12). Since they have a Pro residue in the middle part of the peptide but pin1(1-18) does not, their dynamic orientations must be unrelated to the presence or absence of Pro. Since tryptophan's interfacial preference has often been reported (44,47), these results are reasonable.
FIGURE 13.

Proposed models for orientation and location of (a) pin1 and (b) pin1(1-18) in DPPC lipid bilayer above Tm.
We used a P/L molar ratio of 1:20 for all solid-state NMR experiments in this work because 13C solid-state NMR experiments require a high P/L molar ratio. Although this molar ratio is relatively higher than the threshold value in which antimicrobial peptides tend to become biologically active (32), this relatively higher molar ratio is within the range used for other solid-state NMR studies of peptide-lipid interactions. As an example, it is well known that antimicrobial peptide PGLa resides on the DMPC membrane surface at a low P/L ratio of 1:200; however, at high peptide concentration (>1:50 molar ratio) the helix axis changes its tilt angle from ∼90° to ∼120°, with the C-terminus pointing toward the bilayer interior (48). Therefore, pin1 and pin1(1-18) could adopt different orientations at a low P/L molar ratio.
The affinity of pin1(20-44) with DMPC lipid bilayer may be the least of the four peptides because in the presence of pin1(20-44) the 13C=O chemical shift values of site-specifically labeled pin1(20-44) bound to DMPC lipid bilayer, measured at above Tm under static conditions, are consistent with the isotropic values of lyophilized samples. This implies that pin1(20-44) has the least biological activity of all these peptides. Although pin1(1-18) and pin1WA exhibit greater biological activities than pin1(20-44), they were much lower than that of pin1. From this result, we can conclude that the N-terminal α-helix is important in membrane binding due to the tryptophan-rich structure of the peptide and that a long helix-bend-helix structure is also required for biological activity.
References
- 1.Matsuzaki, K., O. Murase, H. Tokuda, S. Funakoshi, N. Fujii, and K. Miyajima. 1994. Orientational and aggregational states of magainin 2 in phospholipid bilayers. Biochemistry. 33:3342–3349. [DOI] [PubMed] [Google Scholar]
- 2.Matsuzaki, K., O. Murase, N. Fujii, and K. Miyajima. 1995a. Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry. 34:6521–6526. [DOI] [PubMed] [Google Scholar]
- 3.Matsuzaki, K., O. Murase, and K. Miyajima. 1995b. Kinetics of pore formation by an antimicrobial peptide, magainin 2, in phospholipid bilayers. Biochemistry. 34:12553–12559. [DOI] [PubMed] [Google Scholar]
- 4.Matsuzaki, K., O. Murase, N. Fujii, and K. Miyajima. 1996a. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. 35:11361–11368. [DOI] [PubMed] [Google Scholar]
- 5.Matsuzaki, K., S. Yoneyma, O. Murase, and K. Miyajima. 1996b. Transbilayer transport of ions and lipids coupled with mastoparan X translocation. Biochemistry. 35:8450–8456. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi, S., D. Huster, A. Waring, R. I. Lehrer, W. Kearney, B. F. Tack, and M. Hong. 2001. Orientation and dynamics of an antimicrobial peptides in the lipid bilayer by solid-state NMR spectroscopy. Biophys. J. 81:2203–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.North, C. L., M. Barranger-Mathys, and D. S. Cafiso. 1995. Membrane orientation of the N-terminal segment of alamethicin determined by solid-state 15N NMR. Biophys. J. 69:2392–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bak, M., R. P. Bywater, M. Hohwy, J. K. Thomsen, K. Adelhorst, H. J. Jakobsen, O. W. S. Ørensen, and N. C. Nielsen. 2001. Conformation of alamethicin in oriented phospholipid bilayers determined by 15N solid-state nuclear magnetic resonance. Biophys. J. 81:1684–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bechinger, B., M. Zasloff, and S. J. Opella. 1998. Structure and dynamics of the antibiotic peptides PGLa in membranes by solution and solid-state nuclear magnetic resonance spectroscopy. Biophys. J. 74:981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallock, K. J., D. K. Lee, and A. Ramamoorthy. 2003. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J. 84:3052–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuzaki, K., K. Sgishita, N. Ishibe, M. Ueha, S. Nakata, K. Miyajima, and R. M. Epand. 1998. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry. 37:11856–11863. [DOI] [PubMed] [Google Scholar]
- 12.Nomura, K., G. Corzo, T. Nakajima, and T. Iwashita. 2004. Orientation and pore-forming mechanism of a scorpion pore-forming peptide bound to magnetically oriented lipid bilayers. Biophys. J. 87:2497–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith, R., F. Separovic, T. J. Milne, A. Whittaker, F. M. Bennett, B. A. Cornell, and A. Makriyannis. 1994. Structure and orientation of the pore-forming peptide, melittin, in lipid bilayers. J. Mol. Biol. 241:456–466. [DOI] [PubMed] [Google Scholar]
- 14.Naito, A., T. Nagao, K. Norisada, T. Mizuno, S. Tuzi, and H. Saito. 2000. Conformation and dynamics of melittin bound to magnetically oriented lipid bilayers by solid-state 31P and 13C NMR spectroscopy. Biophys. J. 78:2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hallock, K. J., D. K. Lee, J. Omnaas, H. I. Mosberg, and A. Ramamoorthy. 2002. Membrane composition determines pardaxin's mechanism of lipid bilayer disruption. Biophys. J. 83:1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marassi, F. M., S. J. Opella, P. Juvvadi, and R. B. Merrifield. 1999. Orientation of cecropin A helices in phospholipid bilayers determined by solid-state NMR spectroscopy. Biophys. J. 77:3152–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henzler Wildman, K. A., D. K. Lee, and A. Ramamoorthy. 2003. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry. 42:6545–6558. [DOI] [PubMed] [Google Scholar]
- 18.Corzo, G., P. Escoubas, E. Villegas, K. J. Barnham, W. He, R. S. Norton, and T. Nakajima. 2001. Characterization of unique amphipathic antimicrobial peptides from venom of the scorpion Pandinus imperator. Biochem. J. 359:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marion, D., and K. Wuthrich. 1983. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 113:967–974. [DOI] [PubMed] [Google Scholar]
- 20.Szyperski, T., P. Güntert, G. Otting, and K. Wüthrich. 1992. Impact of protein-protein contacts on the conformation of thrombin-bound hirudin studied by comparison with the nuclear magnetic resonance solution structure of hirudin(1–51). J. Magn. Reson. 99:552–560. [DOI] [PubMed] [Google Scholar]
- 21.Wüthrich, K. 1986. NMR of Proteins and Nucleic Acids. Wiley, New York.
- 22.Güntert, P., W. Braun, and K. Wüthrich. 1991. Efficient computation of three-dimensional protein structures in solution from nuclear magnetic resonance data using the program DIANA and the supporting programs CALIBA, HABAS and GLOMSA. J. Biomol. NMR. 1:111–130. [DOI] [PubMed] [Google Scholar]
- 23.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- 24.Roussel, A., and C. Cambillau. 1989. The TURBO-FRODO graphics package. In Silicon Graphics Geometry Partner Directory. Silicon Graphics, Mountain View, CA. 77–78.
- 25.Laskowski, R. A., J. A. Rullmannn, M. W. MacArthur, R. Kaptein, and J. M. Thornton. 1996. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 8:477–486. [DOI] [PubMed] [Google Scholar]
- 26.Lewis, B. A., and D. M. Engelman. 1983. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 166:211–217. [DOI] [PubMed] [Google Scholar]
- 27.Dempsey, C. E., and A. Watts. 1987. A deuterium and phosphorus-31 nuclear magnetic resonance study of the interaction of melittin with dimyristoylphosphatidylcholine bilayers and the effects of contaminating phospholipase A2. Biochemistry. 26:5803–5811. [DOI] [PubMed] [Google Scholar]
- 28.Lindblom, G., and L. Rilfors. 1989. Cubic phase and isotropic structures formed by membrane lipids—possible biological relevance. Biochim. Biophys. Acta. 988:221–256. [Google Scholar]
- 29.Dufourc, E. J., J. F. Faucon, G. Fourche, J. Dufourcq, T. Gulik-Krzywicki, and M. Le Marire. 1986. Reversible disc-to-vesicle transition of melittin-DPPC complexes triggered by the phospholipid acyl chain melting. FEBS Lett. 201:205–209. [Google Scholar]
- 30.Corzo, G., E. Villegas, F. G.-Lagunas, L. D. Possani, O. S. Belokoneva, and T. Nakajima. 2002. Oxyopinins, large amphipathic peptides isolated from the venom of the wolf spider oxyopes kitabensis with cytolytic properties and positive insecticidal cooperativity with spider neurotoxins. J. Biol. Chem. 277:23627–23637. [DOI] [PubMed] [Google Scholar]
- 31.Ames, G. F. 1968. Lipids of salmonella typhimurium and Escherichia coli: structure and metabolism. J. Bacteriol. 95:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blazyk, J., R. Wiegand, J. Klein, J. Hammer, R. M. Epand, R. F. Epand, W. L. Maloy, and U. P. Kari. 2001. A novel linear amphipathic beta-sheet cationic antimicrobialpeptide with enhanced selectivity for bacterial lipids. J. Biol. Chem. 276:27899–27906. [DOI] [PubMed] [Google Scholar]
- 33.Killian, J. A., I. Salemink, M. R. R. de Planque, G. Lindblom, R. E. Koeppe II, and D. V. Greathouse. 1996. Induction of nonbilayer structures in diacylphosphatidylcholine model membranes by transmembrane α-helical peptides: importance of hydrophobic mismatch and proposed role of tryptophans. Biochemistry. 35:1037–1045. [DOI] [PubMed] [Google Scholar]
- 34.Bonev, B. B., Y.-H. Lam, G. Anderluh, A. Watts, R. S. Norton, and F. Separovic. 2003. Effects of the eukaryotic pore-forming cytolysin equinatoxin II on lipid membranes and the role of sphingomyelin. Biophys. J. 84:2382–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suwelack, D., W. P. Rothwel, and J. S. Waugh. 1980. Slow molecular motion detected in the NMR spectra of rotating solids. J. Chem. Phys. 73:2559–2569. [Google Scholar]
- 36.Toraya, S., K. Nishimura, and A. Naito. 2004. Dynamic structure of vesicle-bound melittin in a variety of lipid chain lengths by solid-state NMR. Biophys. J. 87:3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Case, D. A. 1995. Calibration of ring-current effects in proteins and nucleic acids. J. Biomol. NMR. 6:341–346. [DOI] [PubMed] [Google Scholar]
- 38.Cornell, B. A., and F. Separovic. 1983. Membrane thickness and acyl chain length. Biochim. Biophys. Acta. 733:189–193. [DOI] [PubMed] [Google Scholar]
- 39.Stamm, H., and H. Jackel. 1989. Relative ring-current effects based on a new model for aromatic-solvent-induced shift. J. Am. Chem. Soc. 111:6544–6550. [Google Scholar]
- 40.Van Echteld, C. J. A., B. De Kruijff, A. J. Verkleij, J. Leunissen-Bijvelt, and J. De Gier. 1982. Gramicidin induces the formation of non-bilayer structures in phosphatidylcholine dispersions in a fatty acid chain length dependent way. Biochim. Biophys. Acta. 692:126–138. [Google Scholar]
- 41.Dougherty, D. A. 1996. Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 271:163–168. [DOI] [PubMed] [Google Scholar]
- 42.Wimley, W. C., and S. H. White. 1993. Membrane partitioning: distinguishing bilayer effects from the hydrophobic effect. Biochemistry. 32:6307–6312. [DOI] [PubMed] [Google Scholar]
- 43.Schiffer, M., C. H. Chang, and F. J. Stevens. 1992. The functions of tryptophan residues in membrane proteins. Protein Eng. 5:213–214. [DOI] [PubMed] [Google Scholar]
- 44.Yau, W.-M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of Tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- 45.Wiener, M. C., and S. H. White. 1991. Fluid bilayer structure determination by the combined use of x-ray and neutron diffraction. I. Fluid bilayer models and the limits of resolution. Biophys. J. 59:162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiener, M. C., and S. H. White. 1991. Fluid bilayer structure determination by the combined use of x-ray and neutron diffraction. II. “Composition-space” refinement method. Biophys. J. 59:174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Planque, M. R. R., B. B. Bonev, J. A. A. Demmers, D. V. Greathouse, R. E. Koeppe II, F. Separovic, A. Watts, and J. A. Killian. 2003. Interfacial anchor properties of Tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 42:5341–5348. [DOI] [PubMed] [Google Scholar]
- 48.Glaser, R. W., C. Sachse, U. H. N. Durr, P. Wadhwani, S. Afonin, E. Strandberg, and A. S. Ulrich. 2005. Concentration-dependent realignment of the antimicrobial peptide PGLa in lipid membranes observed by solid-state 19F-NMR. Biophys. J. 88:3392–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]