

Abstract
Cardiac excitation-contraction coupling is initialized by the release of Ca from the sarcoplasmic reticulum (SR) in response to a sudden increase in local cytosolic [Ca] ([Ca]i) within the junctional cleft. We have tested the hypothesis that functional ryanodine receptor (RyR) regulation plays a major role in the regulation of myocyte Ca. A mathematical model with unique characteristics was used to simulate Ca homeostasis. Specifically, the model was designed to accurately represent the SR [Ca]-dependence of release from a variety of experimentally produced data sets. The simulated data for altered RyR Ca sensitivity demonstrated a regulatory feedback loop that resulted in the same release at lower [Ca]SR. This suggests that the primary role of myocyte RyR regulation may be to decrease SR [Ca] without decreasing the size of the [Ca]i transient. The model results suggest that this action moderates the increased SR [Ca] observed with adrenergic stimulation and may keep the [Ca]SR below the threshold for delayed afterdepolarizations and arrhythmia. However, increased Ca affinity of the RyR increased the probability of delayed afterdepolarizations when heart failure was simulated. We conclude that RyR regulation may play a role in preventing arrhythmias in healthy myocytes but that the same regulation may have the opposite effect in chronic heart failure.
INTRODUCTION
Cardiac excitation-contraction coupling (ECC) works by local Ca-induced SR Ca-release (CICR), where Ca current (ICa) is the trigger. It has been suggested that CICR is altered by changes in free sarcoplasmic reticulum (SR) [Ca] (([Ca]SR) (1–3)). Most Ca within the SR is bound, primarily to calsequestrin, but the effect of higher [Ca]SR upon release may be via increased [Ca]i sensitivity of the ryanodine receptor (RyR) as demonstrated in bilayers (4,3). The precise mechanism by which this regulation may take place is unclear but may involve junctin, triadin, and calsequestrin (5).
Whatever the mechanism, increasing [Ca]SR causes a nonlinear increase in SR Ca release rate, which is increasingly steep at normal to high SR Ca load (6–12,2,13,14). Such a steep relationship could have a dramatic effect upon the overall physiology of the myocyte leading to release instabilities (15). The role of luminal Ca control of SR Ca release is not completely clear, though recent evidence points toward a role in terminating SR Ca release (16,17).
Normal rabbit myocytes exhibit low diastolic SR Ca release (Jleak) at low [Ca]SR, but this increases steeply with increasing [Ca]SR (18,14). This SR Ca leak must be balanced by net Ca uptake via the SR Ca pump (JSRCaP) at steady state. At 100 nM [Ca]i the unidirectional forward flux through the SR Ca pump (JSRCaPF) is an estimated 25 μmol/l cytosol/s. If the leak out of the SR is 4–15 μM/s (18,14), the residual efflux from the SR will be accounted for by the unidirectional reverse Ca flux through the SR Ca pump (JSRCaPR, i.e., ≥50% of the counterflux). This means that up to 50% of the diastolic SR Ca-pump flux is in a futile cycle at the cost of ATP. Such a system appears counterintuitive and raises the question of what role this diastolic release may play in ECC.
Complicating this issue further, data indicate that Jleak is increased in isolated cardiac myocytes from animals in chronic heart failure (19,20). This effect appears to be caused by RyR dysregulation, possibly due to hyperphosphorylation (21–24) and mediated by an increased RyR Ca affinity. The precise role this increased Jleak plays in the pathophysiology of heart failure (HF) is unknown, but it is likely complicated by other Ca handling defects during HF (e.g., increased Na-Ca exchange, decreased JSRCaP, and decreased K currents (IK1 (25)).
Finally, because phosphorylation of the RyR may occur under physiological conditions such as exercise (23) this raises questions about how altered RyR gating is involved in altered cardiac contractility. Eisner's group has elegantly shown that when CICR is altered by increasing (26) or decreasing (27) flux through the RyR, the effect on Ca transients is only transient. The [Ca]i transient amplitude rapidly returns to near the pretreatment level, whereas SR Ca content ([Ca]SRT) changes to a new steady-state level (i.e., a sort of autoregulation of Ca transients). These studies raise the question: why would the cell phosphorylate the RyR normally in response to physiological stimuli if Ca transients are unaltered?
Here we examine how luminal SR Ca regulation can be expected to modulate ECC under conditions where RyR is altered. We use mathematical modeling that allows examination of individual processes in the overall function. This analysis is in some cases impractical in real cells where simultaneous effects on other interacting systems occur. We examine the questions: 1), Why is the ability to alter release at the RyR necessary for obtaining a normal physiological response? and 2), What role does dysregulation of SR Ca release at the RyR play in the pathophysiology of the cardiac myocyte in chronic heart failure?
We find that: 1), increased Jleak has more profound effects on SR [Ca] and Δ[Ca]i at low frequencies of stimulation, 2), luminal SR Ca regulation of RyR gating hastens autoregulation of [Ca]i transients, thus fine tuning the response, and 3), increased cytosolic Ca affinity of the RyR decreases [Ca]SRT and could limit Ca overload and spontaneous SR Ca release under conditions that normally result in higher [Ca]SRT (e.g., high adrenergic tone); however, 4), the increased diastolic SR Ca release may result in delayed afterdepolarizations (DAD) in HF.
METHODS
The mathematical Ca handling model used is described in detail by Shannon et al. (28) and briefly in the Appendix. The code for the model can be downloaded from http://www.luhs.org/depts/physio/personal_pages/bers_d/index.html. The parameters used are found in Table 1. All values for the parameters below can be found in Table 2 and in the Appendix on a figure-by-figure basis.
TABLE 1.
Channels and transporters in the model
| Channel/transporter | Description |
|---|---|
| INa | Fast Na current |
| INaBk | Na background current |
| Na-K pump | ATP-dependent Na-K transporter |
| IKr | Rapid K current |
| IKs | Slow K current |
| Ito,s | Slow transient outward current |
| Ito,f | Fast transient outward current |
| IK1 | Delayed rectifier current |
| ICl(Ca) | Ca-dependent Cl current |
| IClbk | Cl background current |
| ICa | L-type Ca current |
| ICabk | Ca background current |
| INCX | Na-Ca exchange current |
| Jleak | SR Ca leak flux (JSRCaLeak + diastolic JSRCaRel) |
| ISLCaP | SL Ca pump current |
| JSRCaP | SR Ca pump flux |
| JSRCaPF | Forward unidirectional SR Ca pump flux |
| JSRCaPR | Reverse unidirectional SR Ca pump flux |
| JSRCaRel | SR Ca release flux |
| JSRCaLeak | Passive SR Ca leak flux |
TABLE 2.
Parameter values
| Parameters | Values | Description |
|---|---|---|
| Bmax-SR | 140.0 μmol/L cytosol | Bmax for Ca binding within the SR |
| EC50-SR | 0.45 mM | EC50 for [Ca]SR-dependent activation of SR Ca release |
| H | 1.787 | Hill coefficient of SR Ca pump |
| HSR | 2.5 | Hill coefficient for [Ca]SR-dependent activation of SR Ca release |
| Kmf | 0.246 μM | Km for Ca of SR Ca pump forward mode |
| Kmr | 1.7 mM | Km for Ca of SR Ca pump reverse mode |
| Kd-SR | 0.65 mmol/L SR | Kd for Ca binding within the SR |
| kleak | 5.348 × 10−6 ms−1 | Passive SR Ca leak rate constant |
| kiCa | 0.5 mM−1 ms−1 | Baseline, non-SR-dependent transition rate constant for the RyR |
| Kim | 0.005 ms−1 | Rate transition constant for the RyR (Fig. 1, text) |
| koCa | 10 mM−2ms−1 | Baseline, non-SR-dependent transition rate constant for the RyR |
| kom | 0.06 ms−1 | Rate transition constant for the RyR (Fig. 1, text) |
| Ks | 25 ms−1 | SR Ca release rate constant |
| MaxSR | 15.0 | Maximum parameter [Ca]SR-dependent activation of SR Ca release |
| MinSR | 1.0 | Minimum parameter [Ca]SR-dependent activation of SR Ca release |
| Vmax | 286 μmol/L cytosol | Vmax of the SR Ca pump |
Key structural features include four compartments (in series): a junctional cleft, a subsarcolemmal compartment, a bulk myoplasmic compartment, and the SR (28). Ca released into the junctional cleft diffuses to the subsarcolemmal compartment then to the bulk cytosol where SR Ca uptake occurs.
The action potential was constructed from individual currents found in the normal rabbit ventricular myocyte. The channels and transporters that were included are listed in Table 1. Na and K currents were simulated as in Shannon et al. (28). Ca is exported from the cell by the sarcolemmal (SL) Ca pump and the Na-Ca exchanger. Both were dependent upon the free Ca concentration just under the SL membrane in the subsarcolemmal compartment ([Ca]SL). The Na-Ca exchanger included modulation of activity by external Na and Ca and by [Ca]SL, including an allosteric Ca-dependent activation site represented as described by Weber et al. (29).
The SR Ca pump was considered to be reversible with both a forward (JSRCaPF) and reverse (JSRCaPR) unidirectional flux. The net flux (JSRCaP) was the difference between these two unidirectional fluxes:
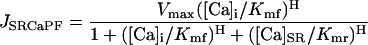 |
(1) |
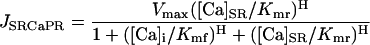 |
(2) |
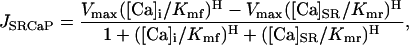 |
(3) |
where Vmax is the maximum velocity of the transporter, Kmf and Kmr are the Km for Ca of the forward and reverse modes, respectively, H is the Hill coefficient, [Ca]i is the free bulk cytosolic concentration, and [Ca]SR is the free SR Ca concentration. Ca is only taken up from the bulk cytosolic compartment into the SR compartment where it is buffered by luminal proteins such that the total SR [Ca] ([Ca]SRT) is equal to the sum of the bound SR [Ca] and [Ca]SR:
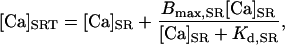 |
(4) |
where Bmax,SR is the Bmax for Ca binding and the Kd,SR is the Kd for Ca binding to the SR luminal buffering proteins.
SR Ca release through the RyR was modeled as a modified version of that described by Stern et al. (30). Regulation by [Ca]SR was added by modifying the Ca activation and inactivation binding constants with a standard Michaelis relationship:
 |
(5) |
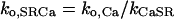 |
(6) |
 |
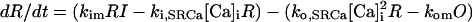 |
(7) |
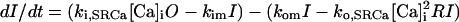 |
 |
 |
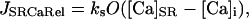 |
(8) |
where JSRCaRel is the RyR-dependent Ca release flux and the rate constants ko,SRCa and ki,SRCa are not diffusion limited. MaxSR and MinSR are the values of kSR where the maximum and minimum response to this substrate are achieved. Values are in Table 1 where the resulting flux is in mmol/l SR/ms.
In addition to the SR Ca release a passive SR Ca leak flux (JSRCaLeak) was added:
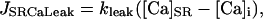 |
(9) |
where kleak is a simple rate constant. The total SR Ca leak rate (Jleak) during diastole is therefore the sum of this passive leak and the residual diastolic Ca release through the RyR. Both of these fluxes are responsible for transfer of Ca from the SR lumen to the junctional cleft space. The net flux across the SR membrane (JSR) is therefore:
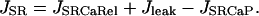 |
(10) |
Characteristics of excitation-contraction coupling
The basic characteristics of ECC in our mathematical model are described in Shannon et al. (28). When square pulses of voltage are applied from a holding potential of −80 mV, both the peak [Ca]i transient and the integrated JSRCaRel are graded as a function of peak ICa. The release is also dependent upon SR Ca load in the manner measured physiologically. That is both the fractional release and the gain of ECC (∫JSRCaRel/∫Ca) rise as a nonlinear function of [Ca]SRT that gets steeper at higher values (2,28).
RESULTS
Effect of diastolic SR Ca leak in normal myocytes
Fig. 1 shows the influence of SR Ca leak upon the frequency dependence of Δ[Ca]i and [Ca]SR. Peak [Ca]i and [Ca]SR increased with frequency, as expected from rabbit myocyte experiments (31,28), reflecting the steep relationship between SR Ca release and [Ca]SR (Fig. 3 (2,14,28)). When Jleak was reduced to <1 μM/s, the positive Δ[Ca]i-frequency relationship is lost, particularly at lower frequencies where both Δ[Ca]i and [Ca]SR decline with increasing stimulation rate. This appears to be because the SR Ca uptake continues to take place during the diastolic period in the presence of lower SR Ca leak, ultimately until the SR [Ca] nears the thermodynamically limiting [Ca]SR/[Ca]i gradient (i.e., where the free energy in the form of ATP can no longer pump Ca uphill (32)). At shorter cycle lengths, the diastolic period is reduced such that less uptake into the SR is allowed and less Ca is accumulated.
FIGURE 1.

Δ[Ca]i-frequency relationship. Dependence of Δ[Ca]i (A) and [Ca]SR (B) on frequency with different levels of Jleak.
FIGURE 3.


Altered RyR activation parameters. (A) Simulation of 100 μM tetracaine (reducing maximal release flux by 70%), with (left) and without (right) [Ca]SR-dependent RyR regulation. Also shown are the total cellular Ca influx (ICa + SL Ca leak) and total cellular Ca efflux (INCX + SL Ca pump). The difference is the Ca gained or lost by the cell at each beat. The bottom panel diagramically demonstrates the feedback loop that is operative during the adjustment to the new steady state (27). (B) Simulation of 100 μM caffeine (increasing RyR leak by ∼2×; Ko,SRCa increased by fivefold), with (left) and without (right) [Ca]SR-dependent RyR regulation. As above, the total cellular Ca influx and efflux and the difference at each beat is shown. The diagram (bottom) shows the regulatory feedback loop whereby altered release results in autoregulation of Ca transients with altered [Ca] (26).
When Jleak was doubled, as may occur in heart failure (20), by both increasing kleak and the Cai affinity of the RyR, both Δ[Ca]i and [Ca]SR were lower, particularly at lower frequencies (with little difference at 3 Hz). This can be understood because at higher frequency there is both less time for diastolic fluxes to have impact and the much greater systolic fluxes dominate the behavior. The SR Ca leak might serve to allow for cardiac reserve that the myocyte may draw upon as beating frequency increases. Notably, experimentally in HF the force-frequency relationship tends to be less positive or even negative (33). Because enhanced Jleak by itself produces the opposite effect, increased Jleak cannot explain this hallmark of HF.
Role of the [Ca]SR dependence of release in normal ECC
Luminal SR Ca release regulation was a novel aspect in our mathematical model. Fig. 2 A illustrates what happens when luminal regulation is abruptly turned off (by freezing RyR regulation by [Ca]SR at the end-diastolic value). Thus, declining [Ca]SR no longer contributes to closure of RyRs. The first [Ca]i transient after this change is much larger than its predecessor and the time-to-peak [Ca]i is prolonged. The Ca transient subsequently returns to nearly same steady-state amplitude, but the time-to-peak [Ca]i remains prolonged.
FIGURE 2.

[Ca]SR-dependent RyR regulation. (A) At 10 s the regulation of RyR by [Ca]SR was frozen at the diastolic level, resulting in prolonged SR Ca release. (B) Sudden increase in intra-SR Ca buffering (doubling [calsequestrin], CSQN). Detailed model parameters are located in the online supplement.
Terentyev et al. (17) showed that calsequestrin overexpression (which should slow [Ca]SR decline) delayed the turnoff of release. Fig. 2 B simulates this with an abrupt increase in intra-SR Ca buffering, while holding [Ca]SR initially constant. Once again, the next [Ca]i transient is increased in both amplitude and time-to-peak, but the amplitude then recovers to nearly the same steady state while the time to peak is still prolonged. Thus, the model is further verified by effectively reproducing this important experimental data. The data further suggest that the regulation of SR Ca release by luminal Ca plays a role in terminating release.
The role of SR Ca on SR Ca release was also tested in the model by simulating experiments where intrinsic RyR properties are altered. Fig. 3 A (left) demonstrates the effect of partial RyR block, e.g., simulating experiments with low tetracaine concentrations and matching results of Overend et al. (27). Upon “addition of tetracaine” (we reduced RyR conductance to 30% of normal, as ks in Eq. 8), [Ca]i transient magnitude and SR Ca release declines, but then recovers to close to the same steady-state magnitude as [Ca]SR increases over several beats. Tetracaine washout results in an overshoot of Ca transients, followed by a decline. The data demonstrate the effects of the feedback loop illustrated at the bottom of Fig. 3 A. The decreased release results in less [Ca]i-dependent inactivation of the RyR (on the cytosolic side) and of ICa (increasing Ca influx). The decreased peak [Ca]i also decreases Ca extrusion by Na-Ca exchange (INCX) and, indirectly, an increase in SR Ca uptake. As [Ca]SRT increases, release increases causing the recovery of the peak [Ca]i in the steady-state response. The resting Ca also rises slightly. This is due to the higher and lower total [Ca] within the cell in the presence of tetracaine and caffeine, respectively. Though most of this difference is exhibited by a change in total SR [Ca], some of it also comes from the cytosol because the two compartments communicate through the SR Ca transport mechanisms (RyR and the SR Ca pump). Although the peak [Ca]i appears to be slightly higher, the Δ[Ca]i is, in fact, slightly lower once the resting [Ca]i is subtracted. This is the expected regulatory behavior from a feedback loop.
Fig. 3 B (left) shows the opposite reaction in response to an increased RyR affinity for cytosolic [Ca], e.g., in response to low caffeine as in Trafford et al. (26). Once again, the model parallels the data as caffeine increases the Ca release (causing increased [Ca]i-dependent ICa inactivation and decreased Ca influx) and increases the [Ca]i transient magnitude (causing increased INCX and loss of Ca from the cell) eventually nearly the same steady-state Ca transient is reached in the presence of caffeine as existed before its addition (but at lower [Ca]SRT).
Once again, the replication of these actual experimental results in Fig. 3 further verifies the model and confirms its appropriateness for use in exploring the effects of SR Ca upon release and the effects of release upon the other Ca regulatory processes. Having established this, we used the model to further investigate the importance of luminal regulation in the feedback loop responsible for the autoregulation in a way that cannot currently be done experimentally. The right panels of Fig. 3, A and B, show the response to “tetracaine” and “caffeine”, respectively, when the luminal regulation of the RyR is removed from the model (as in Fig. 3 A). Four things are evident: 1), the luminal regulation is not necessary for the response or for the completion of the feedback loop. 2), The speed of the response is slower, i.e., more beats are required to respond to the perturbation and bring about a new steady state. 3), The digital cell does not recover to a level as near to the steady state before treatment (i.e., the efficacy of the feedback response is reduced). 4), The extent of the change in [Ca]SR that is required to bring about the new steady state is considerably larger. The results demonstrate intrinsic autoregulation that may keep the [Ca]i transient magnitude relatively constant (34) and suggest the possibility that the luminal regulation may play a role in keeping the changes in [Ca]SR to a minimum in response to changes in the cellular environment.
The demonstration of this intact feedback loop within the model is significant. The data demonstrate the role of changing release through alteration of [Ca]SRT in stabilizing the physiology of the myocyte and it was important that this same stabilizing influence be present in the model. It is also important to note that when the release-load relationship becomes too steep, this same effect can cause instability, specifically alternans (15). This instability is also demonstrable within the model when the Hill coefficient for the SR luminal Ca release effect is increased to a higher level (data not shown).
The role of altered RyR Ca affinity in mediating adrenergic effects on cardiomyocytes
When the Ca affinity of the RyR is doubled (as may occur upon RyR phosphorylation), there is very little effect upon the steady-state Ca transient. The effect is functionally analogous to the effects of low [caffeine] in Fig. 3 B. The myocyte adjusts itself to generate a similar Ca transient to those present before the altered affinity and this Ca transient will be the result of higher fractional release at a smaller [Ca]SRT.
The data in Fig. 4 A represent the relationship between SR Ca release and [Ca]SR (demonstrated indirectly by showing the relationship to [Ca]SRT as in Shannon et al. (2)). SR Ca release in this case is defined as the “gain” of the system or the integrated SR Ca released, divided by the amount of stimulus Ca entry (i.e., integrated ICa). Note that when the Ca affinity of the RyR is increased, a leftward shift in the curve results such that the same gain is achieved at a lower [Ca]SR (dotted line). Thus, the only major effect of increasing the affinity of the RyR for cytosolic Ca is the decrease in the resulting steady-state SR Ca content at the same gain. In other words, the same stimulus Ca causes the same release at a lower SR Ca content.
FIGURE 4.

Altered RyR Ca affinity and β-adrenergic agonists. (A) Gain of ECC from model as a function of [Ca]SR with and without increased RyR [Ca]i affinity. The leftward shift shows the same gain is achieved at a decreased [Ca]SR. (B) Model diastolic SR Ca buffering relationship with the normal myocyte (•), with adrenergic effects (○), and with adrenergic effects minus the altered RyR Ca affinity (♦). (C) [Ca]SR is lower during the isoproterenol response with high RyR Ca affinity.
We propose that this decrease in [Ca]SRT is exactly the reason why RyR regulation may be important, and the SR Ca buffering curve in Fig. 4 B demonstrates why this may be advantageous. Under control conditions (1 Hz), [Ca]SR is near the Kd for Ca binding (i.e., by calsequestrin, the major SR Ca buffering protein (32,35). Thus the system is situated in a position where SR Ca is well buffered and small variations in SR [Ca] don't cause large variations in [Ca]SR, allowing controlled regulation of release on a beat-to-beat basis.
However, with adrenergic stimulation (e.g., twofold increase in both ICa and SR Ca pump affinity but no change in RyR Ca affinity), the [Ca]SRT rises. The relationship rises up the buffering curve closer to saturation and [Ca]SR increases 66%. Under these conditions, a large amount of the Ca is free and unbuffered. The large rise in [Ca]SR causes the RyR to be more likely to spontaneously release Ca increases propensity for DADs (Fig. 4 C).
If the RyR [Ca]i affinity is included with the other adrenergic effects, [Ca]SRT increases less (35% higher than control), closer to the middle of the buffering range and farther from the DAD threshold. The enhanced Ca transient occurs with a greater fractional release with a more modest increase of [Ca]SRT. The overall effect is to stabilize the cell at a lower [Ca]SR without compromising its ability to increase the size of the Ca transient.
Effects of increased RyR Ca affinity upon the diseased myocyte
In HF, altered Ca transport results in decrease in [Ca]SRT, which is a major factor in systolic dysfunction (36–39). Fig. 5, A and B, shows reduced Ca transients, decreased [Ca]SRT, and reduced fractional release obtained by simulating some of the key changes in Ca transport in HF. Here we doubled INCX activity and decreased JSRCaP by 50%. This decreased Ca transients, [Ca]SRT, and fractional release (Fig. 5, A and B), consistent with data in rabbit myocytes (2). However, this decrease in fractional release is not seen in published data in rabbit HF myocytes where fractional release was equivalent to control (40,39); however, see Diaz et al. (41). To further investigate this effect, we also increase Jleak during HF. This might occur via either cAMP- or Ca-calmodulin-dependent protein kinase (PKA or CaMKII) induced increase in RyR [Ca]i sensitivity (21,22) in a manner like that in Fig. 3 B where the digital cell is exposed to low [caffeine]. The [Ca]SRT drops further, but the fractional release is near the control level at steady state (similar to experimental data (36,38,39)). Note that the same fractional release at the lower [Ca]SRT observed in HF (and in the simulation here) results in smaller [Ca]i transients. Fig. 5 C shows that these simple Ca transport changes are also sufficient to reproduce the [Ca]SRT dependence of SR Ca leak measured experimentally (Fig. 5 C, inset (20)).
FIGURE 5.

Effects of luminal regulation in the heart failure myocyte. (A) Steady-state Ca transients for a control cell, one where HF is simulated with and without altered RyR Ca affinity. (B) Steady-state [Ca]SRT and fractional SR Ca release for these same conditions. (C) Diastolic SR Ca leak-load relationship in the model compares favorably with our measurements in isolated rabbit myocytes (inset from Shannon et al. (20)).
Altered RyR gating and arrhythmogenesis
Enhanced SR Ca leak in HF coupled with other cellular changes, such as decreased inward rectifier current (IK1) by 49% and transient outward current (Ito) by 39%, can also result in triggered arrhythmias (38). The INCX, Ito, and IK1 changes will tend to depolarize the membrane and destabilize the diastolic membrane potential (42,38). DADs are a consequence of increased [Ca]SR. As in Fig. 5 C, Jleak increases nonlinearly as [Ca]SRT rises (especially in HF), getting extremely steep at high [Ca]SRT. Thus, a small increase in [Ca]SRT can cause a large increase in Jleak into the junctional space that activates inward INCX (and to a lesser extent ICl(Ca)), which depolarizes the membrane toward threshold.
The model results in Fig. 6 A resemble experimental data (38). Digital HF myocytes were stimulated to steady state at different frequencies, then stimulation was stopped and DADs and spontaneous action potentials (APs) were monitored. DADs were not seen at baseline at 2 Hz in control (not shown) or HF (Fig. 6 A, left), but in “isoproterenol” (i.e., double ICa, SR Ca pump affinity, and RyR affinity) DADs and triggered APs were seen upon cessation of 1-Hz stimulation (Fig. 6 A, middle). If we exclude RyR Ca-sensitization in HF, DAD-triggered APs with isoproterenol are not seen until 2 Hz (Fig. 6 A, right). Thus increased RyR Ca affinity increased the likelihood of reaching threshold and causing a triggered AP.
FIGURE 6.

Altered RyR gating and arrythmogenesis. (A) Stimulation of steady-state AP and Ca transients followed by a period of rest in digital HF cell at 2 Hz (left), with isoproterenol at 1 Hz (middle), and with isoproterenol at 2 Hz with control RyR Ca affinity (right). (B) [Ca]cleft does not rise until the AP rises in HF simulations. (C) [Ca]cleft (left) and to a lesser extent [Ca]SL (right) rise before the AP in digital HF cells with isoproterenol at 1 Hz.
Fig. 6, B and C, shows data on an expanded scale. In normal cells and the HF case in Fig. 6 B, SR Ca release does not occur until the AP rises and ICa is activated. However, when DADs occur the rise in local [Ca]i in the cleft ([Ca]cleft) precedes and drives the spontaneous depolarization (subthreshold and AP; Fig. 6 C, left). Notably, even before the DAD initiation, spontaneous local SR Ca release is occurring during diastole before net depolarization, yet an AP is not triggered until the stimulus (injected current) occurs at the 1-s cycle length. This cyclical SR Ca release observable at 1 Hz may be an emergent SR Ca-dependent pacemaker that is being overdriven by 1-Hz stimulation. However, it causes a series of DADs upon termination of stimulation (which decline in frequency and amplitude as [Ca]SRT declines). Fig. 6 C (right) shows that the SR Ca release that is sufficient to cause a subthreshold DAD (after the two DAD-induced APs, is undetectable on the global Ca transient ([Ca]i), barely apparent in the sarcolemmal region ([Ca]SL), but is mechanistically clear on the cleft Ca transient ([Ca]cleft).
DISCUSSION
We modeled the regulation of SR Ca release by [Ca]SR and by altered RyR function, and have addressed physiological and pathophysiological consequences. This has provided novel insight into SR Ca regulation in cardiac myocytes.
Extensive experimental work has demonstrated that increased [Ca]SRT activates the RyR at rest and during ECC (7,1,2,18,14) and the decline in [Ca]SR may also be critical in regulating the shutoff of SR Ca release (17,43). There is also strong experimental evidence for a sort of autoregulation of ECC, where alterations of RyR regulation have only transient effects on Ca transients (27,13,34). Diastolic SR Ca leak due to altered RyR gating can also decrease [Ca]SRT and this can influence the force-frequency relationship and contractility in HF (21,20). Here we use computer simulations to test our understanding of the function of these processes in the myocyte.
Model characteristics
A mathematical model was used to circumvent the experimental complexity of ECC that makes it difficult to isolate individual processes (e.g., RyR regulation by [Ca]SR) without affecting numerous other processes simultaneously. Though the model is not a substitute for actual data, we have used it here as a supplement to direct further inquiry into questions regarding the role of regulation of RyR activity under physiological and pathophysiological conditions. This was done in some cases by performing experiments in silico that cannot currently be accomplished otherwise at the bench.
The Shannon-Bers model (28) has key features relevant to our aims: 1), the model has luminal Ca regulation of the RyR. 2), It has a subsarcolemmal space that accounts for a higher [Ca] just under the SL membrane, but outside the cleft (44,29). 3), It has several improvements in Ca flux balance and INCX simulation (45). These characteristics allow evaluation of SR Ca release regulation and Jleak in the complicated process of ECC.
We chose to represent the RyR gating scheme as a four-state Markovian model similar to that described by Stern et al. (30). Unlike schemes that are designed to represent channel activity in bilayers, this design has a strong inactivation component that allows termination of locally controlled regenerative release. Though common pool models have been shown to be unstable in modeling cardiac ECC (46), it is important to note that this inactivation component along with the control by the Ca within the SR makes this scheme workable without resorting to more spatially accurate local control models that require more computational power. Note that we were very careful to document both the ICa dependence and the [Ca]SRT dependence of release (Figs. 5 A and 6 C (28) and to assure that we could replicate relevant experimental data from the literature (Figs. 2 B and 3, A and B) in an effort to confirm that the model was appropriate for our investigation.
Also of import is the maintenance of a balance of fluxes so that the proper amount of SR Ca pump and Na-Ca exchange activity is simulated, resulting in a reasonable level of depolarization of the SL membrane in response to a defined level of SR Ca leak (Fig. 6). It is this regard, as well as in the design of the various aspects of diastolic Ca transport, including the addition of a physiological subsarcolemmal compartment, which can be fairly said to be the strength of the model (28).
Physiological role of diastolic SR Ca release
Although some results suggested that diastolic JSRCaRel was rather small (47,48,35), other work has shown that JSRCaRel is relatively high, especially as [Ca]SRT increases (49,27,14). Intuitively this large diastolic SR Ca leak seems harmful to the cell, as it must be balanced by futile SR Ca-ATPase activity to maintain [Ca]SRT. Such a scheme would seem to waste energy. So we are left with the question: Does this seemingly wasteful system have a purpose?
One possible explanation relates to the “force-frequency” data in Fig. 1. A normal Jleak gives a reasonably positive force frequency (even without additional physiological effects, e.g., phosphorylation of effector proteins). However, reducing Jleak to near zero makes this relationship negative. This is because during longer diastole the cell has more time to approach the thermodynamically limiting Ca gradient (ΔG = RT ln([Ca]SR/[Ca]i)) that SERCA can generate without leak (32). As diastole becomes shorter, [Ca]SR cannot reach this limit (see Fig. 1 B). Net SR Ca loss occurs during diastole, and the more frequent beats bring more Ca in driving SR Ca-ATPase perforce to increase [Ca]SRT. The data would suggest that the SR Ca leak is, and indeed must be, relatively large and that a benefit of high SR Ca leak may be to maintain a reserve upon which the myocyte may draw during physiological demands to increase Δ[Ca]i, complementing the effects of frequency-dependent acceleration of relaxation (50) and sympathetic activation on ICa, SR Ca-ATPase, and RyR (see below). In fact, when Jleak is doubled, the force-frequency relationship becomes more even more steeply positive. However, if leak becomes too large the Ca transient may be adversely affected.
Autoregulation with altered RyR regulation
Autoregulation of Ca transient amplitude (34) is readily reproduced in the model, for both shifts in RyR Ca sensitivity and increased SR Ca buffering (Figs. 2 and 3). Increased RyR Ca sensitivity (e.g., caffeine or phosphorylation) increases SR Ca release and Δ[Ca]i at the first beat, but this increases Ca extrusion via INCX and decreases Ca entry via Ca-dependent ICa inactivation. Thus, the amount of Ca in the cell and [Ca]SRT is reduced, which causes the next SR Ca release to be smaller. If it is still higher than control, there will be further cell and SR Ca loss, until the Ca influx and efflux are again in balance with little change in steady-state Ca transient amplitude (but higher fractional SR Ca release). The converse applies for things that depress RyR Ca sensitivity (e.g., tetracaine). Notably, luminal Ca regulation of the RyR is not required for this autoregulation, but it does speed the responsiveness of this process and tightens its control.
Increased SR Ca buffering slows the decline in [Ca]SR during release and the time-to-peak of the [Ca]i transient is prolonged. The results are consistent with those of Terentyev et al. (16), and with [Ca]SR playing a role in terminating SR Ca release during ECC. However, in our model increased intra-SR Ca buffering only transiently increased Ca transient amplitude, whereas sustained enhancement was seen experimentally (16). The reason for this discrepancy is unclear. One possibility is that termination of release in our model depends on both increased [Ca]cleft and reduced [Ca]SR (28) and we may have too much reliance on one or the other. For instance, a greater dependence on [Ca]i-dependent inactivation (as opposed to [Ca]SR) would cause the autoregulatory [Ca]i transient response in Fig. 2 B to be more like that in Fig. 3 B (right), where luminal SR Ca dependence is eliminated and the response to low [caffeine] (which increases RyR [Ca]i affinity) is slower and less complete. Reality is likely somewhere between the two extremes. Clearly we do not understand these issues sufficiently, yet. Identifying this is a benefit of this modeling approach.
In any case, luminal Ca regulation of RyR may serve to stabilize [Ca]SR and limit changes in both systolic and diastolic release. Eventually, very high leak or complete block of RyR-dependent Ca release will prevent the SR from participating in ECC. However, without SR Ca release the amount of Ca influx via ICa and INCX increases, and in most species (except perhaps rat and mouse) this limits the reduction in Ca transients.
The role of alteration of RyR-dependent flux in normal cells
RyR phosphorylation by PKA or CaMKII can enhance channel opening and [Ca]i-sensitivity (CaMKII (51)), although details are controversial (52,53) and we will not distinguish these here. Nevertheless, this effect alone is unlikely to alter Ca transients, as seen for low [caffeine] in Fig. 3 B (26). This raises the question, what is the physiological relevance of enhanced RyR Ca-sensitivity upon phosphorylation?
One explanation is that RyR phosphorylation may increase the initial rate of SR Ca release, even if it does not change the total amount released (54). Indeed, the model reproduces this effect in the presence of caffeine (not shown). However, Fig. 4 A raises another possibility. When RyR Ca affinity is increased, ECC is enhanced in that the same gain is attained at lower [Ca]SRT (and [Ca]SR). Fig. 4 B shows that this may allow inotropy, while keeping SR [Ca] on the stable part of the SR Ca buffering curve. Increased ICa and JSRCaP increase [Ca]SRT, which would otherwise move up the SR Ca buffering curve closer to saturation, and where spontaneous Ca release and arrhythmias occur. Thus, RyR regulation may be an integral part of the myocyte response to PKA or CaMKII (along with altered ICa, SR Ca uptake, frequency, etc.).
Pathophysiological effects of diastolic Ca release
Under conditions like HF where [Ca]SRT may be low for other reasons as well (e.g., reduced SR Ca-pump and enhanced INCX function), enhanced RyR Ca affinity may further decrease [Ca]SRT, accentuating the resultant systolic dysfunction (19,20). In this case the RyR effect may be divorced from the synergistic effects of enhanced ICa and SR Ca uptake that occur with physiological increases in heart rate and sympathetic activation. Increased Jleak in HF does not explain the reduced positive force-frequency relationship seen in HF, as suggested in Fig. 1, but it may contribute to both reduced [Ca]SRT and arrhythmogenesis in HF.
The reduction in [Ca]SRT in HF depends on at least three factors (reduced SR Ca-pump function, enhanced NCX, and increased SR Ca leak) and our simulations here suggest a more prominent contribution of SR Ca leak than our previous analysis because, instead of simply increasing a passive leak, we have explicitly increased the Ca affinity of the RyR in the model. Thus, the resultant [Ca]SRT decrease is a function of both the decreased net SR Ca uptake due to the diastolic leak and the increased systolic release and subsequent autoregulatory effects (e.g., Fig. 3 B).
Increased RyR Ca sensitivity also results in a lower leak-limited [Ca]SRT, as shown most graphically in Fig. 5 C. This could limit contractile reserve. Digital HF myocytes also exhibited an increased propensity for DADs, as observed in real rabbit HF myocytes (38). These simulations also show subthreshold Ca-dependent automaticity in HF cells, apparent first in [Ca]cleft (analogous to that in normal sinoatrial node cells (55)) and these may be precursors to triggered ventricular arrhythmias (Fig. 6). It is interesting to note that this increased propensity appears despite the fact that the increased Ca-sensitivity of the RyR actually drops the [Ca]SRT even more than in the digital cell without this change. In this respect, it may be the combination of higher diastolic SR Ca release with the already higher Na-Ca exchange activity and reduced IK1 channel density that finally destabilizes the membrane to the point where DADs trigger APs. Absence of any of these changes likely reduces the chances of extrasystolic release substantially, as the reduction of diastolic release to normal levels does here (Fig. 6).
In conclusion, we have investigated the physiological and pathophysiological consequences of SR Ca leak within cardiac ventricular myocytes. The mathematical model that reproduces many key characteristics of cellular experimental data has allowed clearer understanding (but also new questions) about the roles of luminal SR Ca regulation and SR Ca leak in normal and pathophysiological conditions. Though a high leak would appear on the surface to be wasteful of cellular energy, closer examination introduces the possibility that it may serve to maintain a reserve from which the cell can draw by increasing beat frequency. An increased RyR affinity may also serve to keep SR Ca within a stable buffering range but during HF this mechanism may turn into a disadvantage, resulting in electrical destabilization of the SL membrane and generation of arrhythmias. RyR regulation may therefore benefit the cell physiologically but may turn into a detriment under pathophysiological conditions.
Acknowledgments
The authors have no conflicts of interest to disclose.
This work was supported by grants from the National Institutes of Health (HL-71893 to T.R.S. and HL-30077 and HL-64724 to D.M.B.).
APPENDIX: CONDITIONAL CHANGES IN PARAMETERS
Symbols and abbreviations refer to equations in Shannon et al. (28)
Isoproterenol
Wherever isoproterenol was simulated in the article the following parameters were changed from the standard values in Shannon et al. (28):
| Parameter | Normal value | Isoproterenol value |
| Kd(Ca) of troponin C | 0.6 μM | 0.9 μM |
| PCa of LTCC | 5.4 × 10−4 | 10.8 × 10−4 |
| Kmf of SR Ca pump | 0.246 μM | 0.123 μM |
| KoCa RyR (28) | 10 mM−2ms−1 | 20 mM−2ms−1 |
In addition, the following were changed from their normal formulation (as in Shannon et al. (28)) to shift the peak of the IKs IV curve 40 mV negative:
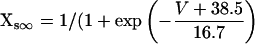 |
 |
The peak of ICa IV curve was also shifted 5 mV negative in the presence of isoproterenol:
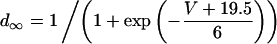 |
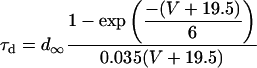 |
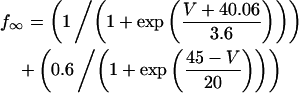 |
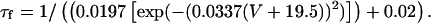 |
Cystic fibrosis transmembrane conductance regulator (CFTR) current, which becomes prominent under high β-adrenergic tone, was also added when the digital cell was simulated with isoproterenol:
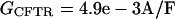 |
 |
where ECl is the reversal potential for chloride.
Heart failure
When simulating the condition of chronic heart failure within the model, the following were changed from their normal values:
| Parameter | Normal value | Heart failure value |
| GTO,slow | 0.020 mS/μF | 0.013 mS/μF |
| GTO,fast | 0.06 mS/μF | 0.04 mS/μF |
| Vmax, Na-Ca exchange | 9 A/F | 18 A/F |
| Vmax, SR Ca pump | 286 μmol/l cyto/s | 172 μmol/l cyto/s |
| kleak | 5.35 × 10−6/ms | 16.58 × 10−6/ms |
| GKs for IKs | – | Reduced 40% |
| GK1 for IK1 | – | Reduced 50% |
| KoCa RyR (28) | 10 mM−2ms−1 | 30 mM−2ms−1 |
Parameters used for specific figures
Fig. 1
The Δ[Ca]i-frequency relationship was simulated. When Jleak was normal, kleak was 5.348 × 10−6/ms. The breakdown of this total diastolic SR Ca leak as the sum of the passive leak (JSRCaLeak) and the diastolic Ca release through the RyR (JSRCaRel) at the different frequencies was as follows:
| Hz | Jleak (μmol/l cyto/ms) | Diastolic JSRCaRel (μmol/l cyto/ms) | JSRCaLeak (μmol/l cyto/ms) |
| 0.1 | 0.0032 | 0.0004 | 0.0028 |
| 0.25 | 0.0034 | 0.0005 | 0.0029 |
| 0.5 | 0.0035 | 0.0006 | 0.0030 |
| 1 | 0.0036 | 0.0006 | 0.0030 |
| 2 | 0.0038 | 0.0008 | 0.0031 |
| 3 | 0.0041 | 0.0010 | 0.0031 |
When Jleak was double, kleak was set to 16.58e − 6/ms and the leak fluxes were as described in the following table:
| Hz | Jleak (μmol/l cyto/ms) | Diastolic JSRCaRel (μmol/l cyto/ms) | JSRCaLeak (μmol/l cyto/ms) |
| 0.1 | 0.0060 | 0.0003 | 0.0058 |
| 0.25 | 0.0077 | 0.0005 | 0.0071 |
| 0.5 | 0.0090 | 0.0009 | 0.0081 |
| 1 | 0.0102 | 0.0013 | 0.0088 |
| 2 | 0.0111 | 0.0018 | 0.0093 |
| 3 | 0.0115 | 0.0021 | 0.0095 |
Finally, when Jleak was <1 μmol/l cyto/ms, kleak was set to 0/ms, and the breakdown was as follows:
| Hz | Jleak (μmol/l cyto/ms) | Diastolic JSRCaRel (μmol/l cyto/ms) | JSRCaLeak (μmol/l cyto/ms) |
| 0.1 | 0.0003 | 0.0003 | 0 |
| 0.25 | 0.0002 | 0.0002 | 0 |
| 0.5 | 0.0002 | 0.0002 | 0 |
| 1 | 0.0002 | 0.0002 | 0 |
| 2 | 0.0004 | 0.0004 | 0 |
| 3 | 0.0006 | 0.0006 | 0 |
Fig. 2 A
At 10 s into the simulation, RyR parameters were held constant and luminal SR Ca was no longer able to affect them. Steady-state parameters with SR lumenal regulation at this transition were Ko,SRCa = 2.41, Ki,SRCa = 0.245, peak Po = 0.0016.
Fig. 2 B
The steady-state parameters before [CSQN] was increased in this figure at 10 s were: Bmax for CSQN = 2.7 mM, Ko,SRCa = 2.41, Ki,SRCa = 0.245, and peak Po = 0.0016. After this point, [CSQN] was increased and a new steady state was reached. The parameters at the new steady state were: Bmax = 8.1 mM, Ko,SRCa = 2.22, Ki,SRCa = 0.235, and peak Po = 0.0013.
Fig. 3 A
Tetracaine was added 10 s into the simulation (i.e., RyR conductance, ks, was reduced). The steady-state parameters before this addition were: ks = 25.0, Ko,SRCa = 2.41, Ki,SRCa = 0.245, and peak Po = 0.0016. Steady-state parameters after tetracaine were: ks = 8.0, Ko,SRCa = 3.68, Ki,SRCa = 0.30, and peak Po = 0.0010.
Fig. 3 B
Similar to Fig. 4 A, caffeine was added 10 s into the simulation (i.e., RyR Ca affinity was increased). External [Ca] was reduced to 1.1 mM as in the actual data from the literature (26). Steady-state parameters before caffeine were Ko,Ca = 10.0, Ko,SRCa = 1.79, Ki,SRCa = 0.0.212, peak Po = 0.0003. Values after caffeine were: Ko,Ca = 15.0, Ko,SRCa = 2.25, Ki,SRCa = 0.19, peak Po = 0.0006.
Fig. 4 A
The gain of ECC with different RyR affinities. For control: Ko,Ca = 10.0, Ki,Ca = 0.5; for double the control Ca affinity: Ko,Ca = 20.0, Ki,Ca = 0.5; and for triple the RyR affinity: Ko,Ca = 30.0, Ki,Ca = 0.5.
Fig. 4 B
The SR Ca buffering curve and the position on that curve during diastole at steady state for each condition. When the simulation was done with isoproterenol, all of the changes mentioned in the table above were made. When isoproterenol was simulated along with the increased RyR Ca affinity that would be expected with PKA phosphorylation, Ko,Ca = 20.0 and Ki,Ca = 0.5.
Fig. 5
For control and HF simulations with control RyR Ca affinity, kleak = 5.348e − 6/ms. When HF was simulated with an increased RyR Ca affinity kleak = 16.58e − 6/ms.
Fig. 6
HF 2Hz
RyR Ca-dependent parameters were Ko,Ca = 30.0, Ki,Ca = 0.5, Ko,SRCa = 4.42, and Ki,SRCa = 0.192. The resulting SR Ca leak could be broken down as (μmol/l cyto/ms) Jleak = 0.0076, diastolic JSRCaRel = 0.0012, and JSRCaLeak = 0.0064.
HF+iso 1Hz
Similar to above, RyR parameters under this condition were Ko,Ca = 30.0, Ki,Ca = 0.5, Ko,SRCa = 12.23, and Ki,SRCa = 0.319. The resulting SR Ca leak was (μmol/l cyto/ms): Jleak = 0.046, diastolic JSRCaRel = 0.0356, and JSRCaLeak = 0.011.
HF+iso (no change on RyR) 2Hz
Finally, RyR parameters in the panels where this condition was simulated were Ko,Ca = 10.0, Ki,Ca = 0.5, Ko,SRCa = 4.1172, and Ki,SRCa = 0.32083 and the resulting SR Ca leak (μmol/l cyto/ms): Jleak = 0.0148, diastolic JSRCaRel = 0.0041 and JSRCaLeak = 0.011.
Abbreviations used: ECC, excitation-contraction coupling; [Ca]cleft, [Ca] in the junctional cleft; [Ca]i, bulk cytoplasmic [Ca]; [Ca]SL, [Ca] in the SL submembrane compartment; [Ca]SR, free SR [Ca]; [Ca]SRT, total SR [Ca]; AP, action potential; CICR, Ca-induced Ca release; DAD, delayed afterdepolarization; HF, heart failure; RyR, ryanodine receptor; SR, sarcoplasmic reticulum.
References
- 1.Gyorke, I., and S. Gyorke. 1998. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys. J. 75:2801–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shannon, T. R., K. S. Ginsburg, and D. M. Bers. 2000. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys. J. 78:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terentyev, D., S. Viatchenko-Karpinski, H. H. Valdivia, A. L. Escobar, and S. Gyorke. 2002. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ. Res. 91:414–420. [DOI] [PubMed] [Google Scholar]
- 4.Sitsapesan, R., and A. J. Williams. 1994. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+. J. Membr. Biol. 137:215–226. [DOI] [PubMed] [Google Scholar]
- 5.Gyorke, I., N. Hester, L. R. Jones, and S. Gyorke. 2004. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 86:2121–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isenberg, G., and S. Han. 1994. Gradation of Ca(2+)-induced Ca2+ release by voltage-clamp pulse duration in potentiated guinea-pig ventricular myocytes. J. Physiol. 480:423–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bassani, J. W., W. Yuan, and D. M. Bers. 1995. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 268:C1313–C1319. [DOI] [PubMed] [Google Scholar]
- 8.Spencer, C. I., and J. R. Berlin. 1995. Control of sarcoplasmic reticulum calcium release during calcium loading in isolated rat ventricular myocytes. J. Physiol. 488:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dettbarn, C., and P. Palade. 1997. Ca2+ feedback on “quantal” Ca2+ release involving ryanodine receptors. Mol. Pharmacol. 52:1124–1130. [DOI] [PubMed] [Google Scholar]
- 10.Santana, L. F., E. G. Kranias, and W. J. Lederer. 1997. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J. Physiol. 503:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spencer, C. I., and J. R. Berlin. 1997. Calcium-induced release of strontium ions from the sarcoplasmic reticulum of rat cardiac ventricular myocytes. J. Physiol. 504:565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huser, J., D. M. Bers, and L. A. Blatter. 1998. Subcellular properties of [Ca2+]i transients in phospholamban-deficient mouse ventricular cells. Am. J. Physiol. 274:H1800–H1811. [DOI] [PubMed] [Google Scholar]
- 13.Trafford, A. W., M. E. Diaz, G. C. Sibbring, and D. A. Eisner. 2000. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J. Physiol. 522:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shannon, T. R., K. S. Ginsburg, and D. M. Bers. 2002. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ. Res. 91:594–600. [DOI] [PubMed] [Google Scholar]
- 15.Diaz, M. E., S. C. O'Neill, and D. A. Eisner. 2004. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ. Res. 94:650–656. [DOI] [PubMed] [Google Scholar]
- 16.Terentyev, D., S. Viatchenko-Karpinski, H. H. Valdivia, A. L. Escobar, and S. Gyorke. 2002. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ. Res. 91:414–420. [DOI] [PubMed] [Google Scholar]
- 17.Terentyev, D., S. Viatchenko-Karpinski, I. Gyorke, P. Volpe, S. C. Williams, and S. Gyorke. 2003. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc. Natl. Acad. Sci. USA. 100:11759–11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukyanenko, V., S. Viatchenko-Karpinski, A. Smirnov, T. F. Wiesner, and S. Gyorke. 2001. Dynamic regulation of sarcoplasmic reticulum Ca(2+) content and release by luminal Ca(2+)-sensitive leak in rat ventricular myocytes. Biophys. J. 81:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neary, P., A. M. Duncan, S. M. Cobbe, and G. L. Smith. 2002. Assessment of sarcoplasmic reticulum Ca(2+) flux pathways in cardiomyocytes from rabbits with infarct-induced left-ventricular dysfunction. Pflugers Arch. 444:360–371. [DOI] [PubMed] [Google Scholar]
- 20.Shannon, T. R., S. M. Pogwizd, and D. M. Bers. 2003. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res. 93:592–594. [DOI] [PubMed] [Google Scholar]
- 21.Marx, S. O., S. Reiken, Y. Hisamatsu, T. Jayaraman, D. Burkhoff, N. Rosemblit, and A. R. Marks. 2000. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 101:365–376. [DOI] [PubMed] [Google Scholar]
- 22.Maier, L. S., T. Zhang, L. Chen, J. DeSantiago, J. H. Brown, and D. M. Bers. 2003. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92:904–911. [DOI] [PubMed] [Google Scholar]
- 23.Wehrens, X. H., S. E. Lehnart, S. R. Reiken, and A. R. Marks. 2004. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94:e61–e70. [DOI] [PubMed] [Google Scholar]
- 24.Currie, S., C. M. Loughrey, M. A. Craig, and G. L. Smith. 2004. Calcium/calmodulin-dependent protein kinase II delta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem. J. 377:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bers, D. M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 26.Trafford, A. W., G. C. Sibbring, M. E. Diaz, and D. A. Eisner. 2000. The effects of low concentrations of caffeine on spontaneous Ca release in isolated rat ventricular myocytes. Cell Calcium. 28:269–276. [DOI] [PubMed] [Google Scholar]
- 27.Overend, C. L., S. C. O'Neill, and D. A. Eisner. 1998. The effect of tetracaine on stimulated contractions, sarcoplasmic reticulum Ca2+ content and membrane current in isolated rat ventricular myocytes. J. Physiol. 507:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shannon, T. R., F. Wang, J. Puglisi, C. Weber, and D. M. Bers. 2004. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys. J. 87:3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber, C. R., V. Piacentino III, K. S. Ginsburg, S. R. Houser, and D. M. Bers. 2002. Na(+)-Ca(2+) exchange current and submembrane [Ca(2+)] during the cardiac action potential. Circ. Res. 90:182–189. [DOI] [PubMed] [Google Scholar]
- 30.Stern, M. D., L. S. Song, H. Cheng, J. S. Sham, H. T. Yang, K. R. Boheler, and E. Rios. 1999. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 113:469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maier, L. S., D. M. Bers, and B. Pieske. 2000. Differences in Ca(2+)-handling and sarcoplasmic reticulum Ca(2+)-content in isolated rat and rabbit myocardium. J. Mol. Cell. Cardiol. 32:2249–2258. [DOI] [PubMed] [Google Scholar]
- 32.Shannon, T. R., and D. M. Bers. 1997. Assessment of intra-SR free [Ca] and buffering in rat heart. Biophys. J. 73:1524–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pieske, B., L. S. Maier, D. M. Bers, and G. Hasenfuss. 1999. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ. Res. 85:38–46. [DOI] [PubMed] [Google Scholar]
- 34.Eisner, D. A., and A. W. Trafford. 2000. No role for the ryanodine receptor in regulating cardiac contraction? News Physiol. Sci. 15:275–279. [DOI] [PubMed] [Google Scholar]
- 35.Shannon, T. R., K. S. Ginsburg, and D. M. Bers. 2000. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys. J. 78:322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pogwizd, S. M., M. Qi, W. Yuan, A. M. Samarel, and D. M. Bers. 1999. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 85:1009–1019. [DOI] [PubMed] [Google Scholar]
- 37.Hobai, I. A., and B. O'Rourke. 2001. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 103:1577–1584. [DOI] [PubMed] [Google Scholar]
- 38.Pogwizd, S. M., K. Schlotthauer, L. Li, W. Yuan, and D. M. Bers. 2001. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ. Res. 88:1159–1167. [DOI] [PubMed] [Google Scholar]
- 39.Piacentino, V., C. R. Weber, X. Chen, J. Weisser-Thomas, K. B. Margulies, D. M. Bers, and S. R. Houser. 2003. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 92:651–658. [DOI] [PubMed] [Google Scholar]
- 40.Bers, D. M., S. M. Pogwizd, and K. Schlotthauer. 2002. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res. Cardiol. 97(Suppl 1):I36–I42. [DOI] [PubMed] [Google Scholar]
- 41.Diaz, M. E., H. K. Graham, and A. W. Trafford. 2004. Enhanced sarcolemmal Ca2+ efflux reduces sarcoplasmic reticulum Ca2+ content and systolic Ca2+ in cardiac hypertrophy. Cardiovasc. Res. 62:538–547. [DOI] [PubMed] [Google Scholar]
- 42.Schlotthauer, K., and D. M. Bers. 2000. Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ. Res. 87:774–780. [DOI] [PubMed] [Google Scholar]
- 43.Shannon, T. R., T. Guo, and D. M. Bers. 2003. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 93:40–45. [DOI] [PubMed] [Google Scholar]
- 44.Trafford, A. W., M. E. Diaz, and D. A. Eisner. 1998. Ca-activated chloride current and Na-Ca exchange have different timecourses during sarcoplasmic reticulum Ca release in ferret ventricular myocytes. Pflugers Arch. 435:743–745. [DOI] [PubMed] [Google Scholar]
- 45.Weber, C. R., K. S. Ginsburg, K. D. Philipson, T. R. Shannon, and D. M. Bers. 2001. Allosteric regulation of Na/Ca exchange current by cytosolic Ca in intact cardiac myocytes. J. Gen. Physiol. 117:119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stern, M. D. 1992. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 63:497–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bassani, R. A., and D. M. Bers. 1995. Rate of diastolic Ca release from the sarcoplasmic reticulum of intact rabbit and rat ventricular myocytes. Biophys. J. 68:2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ginsburg, K. S., C. R. Weber, and D. M. Bers. 1998. Control of maximum sarcoplasmic reticulum Ca load in intact ferret ventricular myocytes. Effects of thapsigargin and isoproterenol. J. Gen. Physiol. 111:491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gyorke, S., V. Lukyanenko, and I. Gyorke. 1997. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J. Physiol. 500:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeSantiago, J., L. S. Maier, and D. M. Bers. 2002. Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J. Mol. Cell. Cardiol. 34:975–984. [DOI] [PubMed] [Google Scholar]
- 51.Wehrens, X. H., S. E. Lehnart, S. R. Reiken, and A. R. Marks. 2004. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94:e61–e70. [DOI] [PubMed] [Google Scholar]
- 52.Bers, D. M., D. A. Eisner, and H. H. Valdivia. 2003. Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Circ. Res. 93:487–490. [DOI] [PubMed] [Google Scholar]
- 53.Marks, A. R. 2003. A guide for the perplexed: towards an understanding of the molecular basis of heart failure. Circulation. 107:1456–1459. [DOI] [PubMed] [Google Scholar]
- 54.Ginsburg, K. S., and D. M. Bers. 2004. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J. Physiol. 556:463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vinogradova, T. M., Y. Y. Zhou, V. Maltsev, A. Lyashkov, M. Stern, and E. G. Lakatta. 2004. Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ. Res. 94:802–809. [DOI] [PubMed] [Google Scholar]