

Abstract
Partially folded intermediates have been frequently observed in equilibrium and kinetic protein folding studies. However, folding intermediates that exist at the native side of the rate-limiting step are rather difficult to study because they often evade detection by conventional folding kinetic methods. Here, we demonstrated that a laser-induced temperature-jump method can potentially be used to identify the existence of such post-transition or hidden intermediates. Specifically, we studied two cross-linked variants of GCN4-p1 coiled-coil. The GCN4 leucine zipper has been studied extensively and most of these studies have regarded it as a two-state folder. Our static circular dichroism and infrared data also indicate that the thermal unfolding of these two monomeric coiled-coils can be adequately described by an apparent two-state model. However, their temperature-jump-induced relaxation kinetics exhibit non-monoexponential behavior, dependent upon sequence and temperature. Taken together, our results support a folding mechanism wherein at least one folding intermediate populates behind the main rate-limiting step.
INTRODUCTION
The thermal unfolding transition of many small, single domain proteins, when monitored by a spectroscopic signal, is often cooperative and can be modeled by two-state thermodynamics. This, however, does not imply that a protein that exhibits two-state equilibrium folding-unfolding behaviors folds without any detectable kinetic intermediates (1–3). Recently, Bai and co-workers have shown that even proteins that exhibit two-state folding kinetics when studied by stopped-flow techniques may actually fold via post-transition or hidden intermediates (4). For example, using a native hydrogen exchange method (5) in conjunction with a protein engineering approach, they have shown that Rd-apocyt b562, a redesigned four-helix bundle protein that exhibits two-state folding thermodynamics as well as first-order stopped-flow folding kinetics, actually folds via a series of partially folded structural ensembles or hidden intermediates that exist at the native side of the rate-limiting step (6). Although such hidden intermediates are certainly important to protein folding, they often evade detection by conventional kinetic methods (4). Using GCN4 coiled-coil as an example, here we show that the laser-induced temperature-jump (T-jump) infrared (IR) method can be used to investigate the existence of such hidden intermediates.
The T-jump technique uses a burst of energy to heat up a sample solution within a very short period of time, e.g., a few nanoseconds. As a result, the sudden increase in temperature induces a population-redistribution among conformational ensembles that are initially at equilibrium. Hence, the time course in which the nonequilibrium state evolves toward a new equilibrium position, which is determined by the final temperature, contains information regarding the kinetics of folding and unfolding. For example, for a simple two-state scenario, the relaxation in response to a T-jump follows first-order kinetics and the relaxation rate constant is simply the sum of the folding and unfolding rate constants (7,8). However, for a folding process that involves intermediates, the relaxation kinetics become non-monoexponential. Therefore, rapid T-jump techniques can potentially be used to distinguish folding processes involving one or multiple free energy barriers, even though the observation of first-order relaxation kinetics does not necessitate that folding proceeds through only one free energy barrier (1,9).
To demonstrate the utility of using T-jump IR spectroscopy to probe the so-called hidden intermediates in apparent two-state folders, we studied the folding kinetics of two variants of the GCN4 leucine zipper derived from the yeast transcription activator GCN4 (10). The folding stability and kinetics of the GCN4 leucine zipper and related coiled-coils have been studied extensively in the past (11–21). These studies provided invaluable insights into our understanding of many important factors that govern the folding mechanism and stability of the coiled-coil structural motif as well as protein folding in general. In particular, the first-order folding kinetics observed in stopped-flow circular dichroism (CD) and fluorescence studies generally support the idea that the folding of coiled-coils exhibits two-state-like behavior, wherein only one dominating free-energy barrier separates the unfolded monomers from the folded dimer, and the bimolecular reaction is essentially coupled with the overall folding of the coiled-coil structure. On the other hand, a number of equilibrium thermodynamic studies (14,21) suggested that the folding of coiled-coils may involve one or two additional steps at the native side of the major folding barrier. Therefore, the coiled-coil motif appears to be an appropriate model system for exploring the existence of post-transition intermediates.
Specifically, we studied two cross-linked variants of the GCN4-p1 leucine zipper, similar to those used by Sosnick and co-workers (15,22). These covalently linked homodimers, with a cross-linker at either the N-terminus (GCN4-p1n″) or C-terminus (GCN4-p1c″), were prepared by linking two monomers together via a disulfide bond formed between two cysteine residues (15,23). An advantage of cross-linking two helices together is that it eliminates the bimolecular collision process encountered by the dimeric forms of GCN4-p1, thereby simplifying the interpretation of the experimental results. In addition, Sosnick and co-workers (15,22) have recently shown that an unstructured cross-linker can lead to a structurally more homogeneous folding transition state for the folding of coiled-coils, compared to that of unlinked monomers.
The T-jump-induced relaxation kinetics of these monomeric coiled-coils, obtained by monitoring the amide I′ band of the polypeptide backbone, indeed show non-monoexponential behavior. At relatively low temperatures, the relaxation is fast and can be modeled by single-exponential kinetics, whereas around the thermal melting temperature the relaxation kinetics become slower and can best be described by a double-exponential function. Taken together, these results support the idea that the folding of these coiled-coils involves at least one post-transition intermediate, demonstrating the applicability of the T-jump IR method in revealing the microscopic detail of a folding process.
MATERIALS AND METHODS
Peptide synthesis and purification
The GCN4 peptides used in the current study have the following sequences with the cross-linkers underlined:
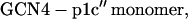 |
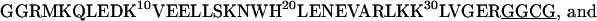 |
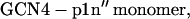 |
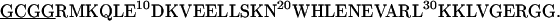 |
Both peptides were synthesized using the standard solid-phase 9-fluorenylmethoxycarbonyl method. Before purification, the Cys was reduced to the thiol by reaction with Tris(2-carboxyethyl)-phosphine hydrochloride (TCEP·HCl), and the peptides were purified by reverse-phase HPLC with a C4 column. The corresponding homodimers (i.e., GCN4-p1c″ and GCN4-p1n″) were prepared by cross-linking the individual polypeptide chains by stirring the peptide solution at pH 7.8 overnight. The cross-linked peptides were purified with reverse-phase HPLC again and characterized by MALDI-TOF mass spectrometry. The identity of these peptides was further verified by size-exclusion chromatography. Specifically, peptide solutions at ∼1 mM concentration and pH 7.0 were loaded onto a Pharmacia Sephadex Peptide column running a pH 7.0 buffer at 1 mL/min (Pharmacia, Peapack, NJ). Both peptides elute from the column as a single species corresponding to the desired molecular weight (data not shown).
Sample preparation
As described in detail elsewhere (7), multiple rounds of lyophilization were performed to remove the trifluoroacetic acid from peptide synthesis and also the exchangeable protons of the peptides. For CD and IR experiments, the solution was prepared by directly dissolving the lyophilized samples in either 20 mM (CD) or 50 mM (IR) potassium phosphate D2O buffers (pH* 7.0), respectively. The concentration of the sample was determined by absorbance at 280 nm, using an extinction coefficient of 11,200 cm−1 M−1. The final concentration was ∼10 μM for CD and ∼0.5 mM for IR, respectively.
Circular dichroism (CD) spectroscopy
Both wavelength scan and thermal melting CD curves were collected on an Aviv 62A DS circular dichroism spectrometer (Aviv Associates, Lakewood, NJ) with a 1-mm sample cuvette.
The temperature-dependent mean residue ellipticities at 222 nm, i.e., [θ](T), were further analyzed according to an apparent two-state model, namely,
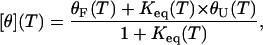 |
(1) |
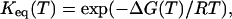 |
(2) |
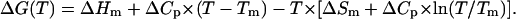 |
(3) |
Here, θF(T) is the pre-transition baseline, θU(T) is the post-transition baseline, Keq(T) is the equilibrium constant, Tm = ΔHm/ΔSm is the thermal melting temperature, ΔHm is the enthalpy change at Tm, ΔSm is the entropy change at Tm, and ΔCp is the heat capacity change, which has been assumed here to be temperature-independent. In the fit, θF(T) was treated as a linear function of temperature (i.e., θF (T) = a + bT, where a and b are constants), whereas θU(T) was determined from the CD signals of GCN4-p1c″ and GCN4-p1n″ in 5 M guanidinium hydrochloride solution.
Fluorescence spectroscopy
Fluorescence spectra were obtained on a Fluorolog 3.10 spectrofluorometer (Jobin Yvon Horiba, Edison, NJ) with 1-nm spectral resolution (excitation and emission) and a 1-cm quartz sample cuvette. Temperature was regulated using a TLC 50 Peltier temperature controller (Quantum Northwest, Spokane, WA). The sample was prepared by directly dissolving lyophilized peptides into 40 mM phosphate buffer (pH 7) and the final concentration was ∼9 μM. The temperature-dependent fluorescence spectra (300–400 nm) of the tryptophan residue in both GCN4-p1c″ and GCN4-p1n″ were collected from 5 to 90°C, in a step of 5°C, with an excitation wavelength of 270 nm.
Fourier transform infrared (FTIR) spectroscopy
Static FTIR spectra were collected on a Magna-IR 860 spectrometer (Nicolet Instrument, Madison, WI) equipped with a HgCdTe detector using 2-cm−1 resolution. A homemade CaF2 sample cell that was divided into two compartments with a 52-μm Teflon spacer and mounted on a programmable translation stage was used to allow separate measurements of the sample and the reference under identical conditions. Temperature control with ±0.2°C precision was obtained by a thermostated copper block. Typically, 256 scans were averaged to generate one spectrum.
Time-resolved T-jump IR spectroscopy
The time-resolved T-jump IR apparatus has been described in detail elsewhere (7). Briefly, the 1.9-μm T-jump pulse was generated by Raman-shifting the fundamental output of a Q-switched Nd:YAG laser in a mixture of H2 and Ar pressurized at 750 psi. The T-jump-induced transient absorbance changes were measured by a continuous-wave IR diode laser (Laser Components, Hudson, NH) in conjunction with a 50-MHz HgCdTe detector and a digital oscilloscope (Tektronix, Wilsonville, OR). As in the static FTIR experiments, a thermostated, two-compartment sample cell with 130-μm path length was used to allow the separate measurements of the sample and buffer under identical conditions. The measurements on buffer provide information for both the background subtraction and the T-jump amplitude determination. The latter was achieved by using the T-jump-induced absorbance change of the D2O buffer solution at the probing frequency ν, ΔA(ΔT, ν), and the equation ΔA(ΔT, ν) = a(ν) × ΔT + b(ν) × ΔT2, where ΔT corresponds to the difference between the final (Tf) and initial (Ti) temperatures, and a(ν) and b(ν) are constants that were determined by analyzing the temperature dependence of the FTIR spectra of the buffer. In the current study, the T-jump amplitudes were characterized to be between 8 and 10°C.
RESULTS
CD spectroscopy
At ambient temperatures, both GCN4-p1c″ and GCN4-p1n″ give rise to a far-UV CD spectrum that is characteristic of α-helical proteins, with double minima at 208 and 222 nm, respectively (data not shown). In addition, their thermal folding-unfolding transitions, obtained by monitoring the ellipticity at 222 nm as a function of temperature, can be described by a two-state model (Fig. 1), and the resulting thermodynamic parameters are similar to those obtained on GCN4-p1 (20,21). A closer inspection of these data, however, indicates that the introduction of a disulfide tether at the N-terminal region leads to a more stable monomeric coiled-coil, as indicated by the higher Tm observed for GCN4-p1n″. This result is consistent with previous suggestions that the C-terminal region of the GCN4-p1 peptide plays a more important role in stabilizing the coiled-coil dimer (24,25).
FIGURE 1.

Mean residue ellipticities of GCN4-p1c″ (open triangle) and GCN4-p1n″ (open circle) in water at 222 nm as a function of temperature. The data for GCN4-p1n″ have been offset by −4000 deg cm2 dmol−1 for clarity. Fitting these data to an apparent two-state model (solid lines), i.e., Eqs. 1–3, yields the following thermodynamic parameters: for GCN4-p1c″, ΔHm = 45.8 kcal mol−1, ΔSm = 127 cal K−1 mol−1, ΔCp = 839 cal K−1 mol−1, and Tm = 87.9°C; and for GCN4-p1n″, ΔHm = 52.6 kcal mol−1, ΔSm = 145 cal K−1 mol−1, ΔCp = 909 cal K−1 mol−1, and Tm = 89.7°C.
FTIR spectroscopy
The thermal unfolding transition of these coiled-coils was further studied by FTIR spectroscopy. Specifically, the amide I′ band of these peptides, which arises mainly from the stretching vibration of backbone carbonyls and is an established indicator of protein secondary structures because of its sensitivity to structural parameters (26), was collected between 5 and 95°C. As shown (Fig. 2 a), the resolution-enhanced FTIR spectrum (27) of GCN4-p1c″ at 5.1°C exhibits four resolvable spectral features, centered at ∼1606, 1636, 1650, and 1672 cm−1, respectively. The 1606 cm−1 band is frequently assigned to amino acid side chains, whereas the bands centered at ∼1636 and 1650 cm−1 are due to fully and/or partially hydrated and dehydrated helical amides (28–30), respectively. The band at 1672 cm−1 arises from the carboxylate of the residual trifluoroacetic acid. As indicated (Fig. 2 b), the hydrated helical band at 1632 cm−1 shows a pretransition baseline, followed by a thermal unfolding transition similar to that reported by CD spectroscopy. Although several factors, such as changes in the solvation status of hydrated helical amides (28,29) as well as helix fraying, could contribute to this pretransition baseline, it is expected that these changes will take place on a timescale that is too fast to be resolved by the current setup (see below). Similarly, the intensity at 1666 cm−1, which is associated with the formation of thermally unfolded conformations, exhibits comparable temperature-dependence (Fig. 2 b). Finally, it is worth pointing out that these FTIR spectra indicate that no detectable (by the current method) aggregation takes place even at the highest temperature (i.e., 94.5°C), as judged by the lack of the distinct spectral features of aggregates at 1618 and 1683 cm−1.
FIGURE 2.

(a) FTIR spectra of GCN4-p1c″ coiled-coil (in the amide I′ region) at 5.1°C (solid line) and 94.5°C (dashed line), respectively. Also shown are the corresponding FSD spectra of these data. The band narrowing was achieved by the Fourier self-deconvolution (FSD) method (27) with k = 2 and full-width at half-maximum = 18 cm−1. (b) Optical density change (ΔOD) of the amide I′ band of GCN4-p1c″ versus temperature at frequencies used in T-jump experiments, i.e., 1632 cm−1 (open square), and 1666 cm−1 (open triangle), respectively.
Fluorescence study
Following Dragan and Privalov (21), we have also studied the thermal unfolding transition of GCN4-p1c″ and GCN4-p1n″ by monitoring the Trp fluorescence. For both peptides, the fluorescence emission spectrum of Trp is peaked at ∼358 nm (data not shown), indicating that the Trp residue in these peptides is located at a position that is largely exposed to solvent. As expected, increasing temperature results in a decrease in the Trp fluorescence intensity (Fig. 3). Although part of this decrease is certainly due to the intrinsic temperature-dependence of the fluorescence quantum yield of the fluorophore, a conformational process was also believed to contribute to this decrease. Indeed, further analysis of these data using the method of Dragan and Privalov (21) shows that part of this decrease can be ascribed to a thermal transition and, similar to that observed for dimeric GCN4-p1 (21), this thermal event has a Tm of ∼50°C and is not detected in the CD and IR measurements. Although Trp fluorescence has been frequently used in protein conformational studies, a rigorous interpretation of its change is not always straightforward. As for the present case, the thermal event reported by the Trp residues may correspond to a global conformational event, such as that proposed by Dragan and Privalov (21), or local side-chain dynamics leading to higher energy rotamer states.
FIGURE 3.

Trp fluorescence intensity (open circle) of GCN4-p1c″ at 350 nm as a function of temperature. The value λex = 270 nm. The solid line is a two-state fit using the method of Privalov and co-workers (21). The resulting thermodynamic parameters, i.e., ΔHm = 19.3 kcal mol−1, ΔSm = 60 cal K−1 mol−1, ΔCp = 40 cal K−1 mol−1, and Tm = 49.9°C, are similar to those obtained by Dragan and Privalov on GCN4-p1 (21).
T-jump IR study
The T-jump-induced relaxation kinetics of the GCN4-p1c″ coiled-coil were monitored by time-resolved IR spectroscopy (7). As shown (Fig. 4), the relaxation exhibits two distinct phases and also depends on the final temperature. The fast phase, which is unresolved in time due to the 10–15 ns response time of the IR detection system, is likely due to the temperature-induced spectral changes discussed above. Other factors, such as helix-fraying (21) and imperfect background subtraction may also contribute to this phase. On the other hand, the slow phase is on the microsecond timescale and thereby well resolved. However, the kinetics of the slow phase depend on the final temperature in an unusual manner. For example, at a relatively low final temperature, the slow relaxation phase can be modeled by first-order kinetics with a time constant of ∼10 μs, whereas at a relatively high final temperature, such as those at or above Tm, this slow relaxation phase becomes non-monoexponential, suggestive of a deviation from a simple, two-state folding pathway. Specifically, these non-monoexponential relaxations can be modeled by a double-exponential function with one time constant similar to that obtained at low final temperatures and another one of ∼100 μs (Fig. 5). Although we cannot completely rule out the possibility that even longer relaxation events exist, the fact that the kinetic amplitude at the longest decay time matches quite well with the equilibrium amplitude (data not shown) suggests that there is no significant change of the IR absorbance occurring on the millisecond or longer timescales. In the following discussion, we only discuss the slow relaxation phase and assign the kinetic component that is observed at all temperatures as the fast component (∼10 μs) and the kinetic component that is observed only at high final temperatures as the slow component (∼100 μs).
FIGURE 4.

Two T-jump-induced relaxation traces (open circles) of GCN4-p1c″ measured at 1632 cm−1. The T-jumps were 64.7–73.7°C and 79.4–87.8°C, respectively. The data corresponding to the T-jump of 79.4–87.8°C have been scaled, as indicated. The smooth lines are fits of these data to the following function: ΔOD(t) = A × [1–B1 × exp(−t/τ1) – B2 × exp(−t/τ2)], with A = −0.018, B1 = 0.21, B2 = 0.083, τ1 = 8.3 μs, and τ2 = 35.2 μs for the T-jump relaxation trace of 64.7–73.7°C; and A = −0.035, B1 = 0.17, B2 = 0.44, τ1 = 5.9 μs, and τ2 = 104 μs for the T-jump relaxation trace of 79.4–87.8°C. The corresponding residuals for the T-jump relaxation trace of 79.4–87.8°C were given in a. Also shown (inset) is the fit of the T-jump relaxation trace of 79.4–87.8°C to the following function: ΔOD(t) = A×[1–B1×exp(−t/τ)], with A = −0.035, B = 0.6, and τ1 = 76 μs. Similarly, the corresponding residuals obtained from this fit were given in b.
FIGURE 5.

T-jump-induced relaxation rates of GCN4-p1c″ (solid symbols) and GCN4-p1n″ (open symbols) versus final temperature. The T-jump amplitude of each measurement was between 8 and 10°C.
The T-jump-induced relaxation of GCN4-p1n″ is similar to that of GCN4-p1c″, although its overall relaxation kinetics are somewhat slower than those of GCN4-p1c″ (Fig. 5).
DISCUSSION
Our current understanding of the folding kinetics of GCN4 coiled-coils is based on extensive time-resolved studies employing stopped-flow fluorescence (11,13,15,22) and CD (19,20,31) techniques. The majority of these studies reported first-order folding kinetics and thereby supported the idea that the folding of coiled-coils exhibits two-state-like folding behavior, wherein only one dominating free energy barrier separates the unfolded monomers from the folded dimer. Additionally, a number of equilibrium unfolding studies (20,32) also suggest that an apparent two-state model can adequately describe the thermal as well as chaotropic folding-unfolding transitions of GCN4 coiled-coils.
In agreement with previous CD studies on dimeric GCN4 coiled-coils, our CD results also show that the thermal unfolding transition of both GCN4-p1c″ and GCN4-p1n″ can be modeled by two-state thermodynamics. On the contrary, our time-resolved IR studies suggest that the folding pathway of these monomeric coiled-coils is nevertheless more complicated than a process involving only two conformational ensembles, as indicated by the non-monoexponential relaxation kinetics of these molecules in response to a T-jump. Since a two-state protein can only give rise to single-exponential folding/unfolding kinetics, the observation of non-monoexponential kinetics in a T-jump unfolding experiment certainly suggests that the relaxation involves two or more equilibria. Although other factors, such as downhill folding or diffusion (33,34), can also lead to non-monoexponential relaxations, a recent study by Meisner and Sosnick (35) has shown that the folding of a cross-linked GCN4 coiled-coil similar to GCN4-p1c″ is barrier-limited. Therefore, we tentatively attributed the observed non-monoexponentiality in the present case to folding intermediates (36,37).
The idea that the folding of the GCN4 coiled-coil involves intermediates has been put forward before. For example, several equilibrium studies, employing either NMR spectroscopy (14,38,39) or calorimetric (21) method, have suggested that the thermal unfolding transition of the GCN4 leucine zipper and related coiled-coils may actually proceed via several distinct steps. For example, Dragan and Privalov (21) have studied in detail the thermal unfolding process of the leucine zipper coiled-coil by employing several techniques, such as CD spectroscopy, spectrofluorimetry, and heat capacity scanning calorimetry. They concluded that the thermal unfolding transition of the leucine zipper coiled-coil consists of several stages. The first transition starts at the very beginning of heating from 0.8°C and is associated with unfolding or fraying of the N-terminus of the leucine zipper. The second transition, which influences the emission properties of a tryptophan residue placed in the central part of the zipper, takes place at a considerably higher temperature (∼50°C) and was hypothesized to be associated with some repacking of the coiled-coil. Finally, this second transition is followed at higher temperatures by the concentration-dependent cooperative unfolding/dissociation of the two strands. Additionally, simulations carried out by Mohanty et al. (18) also suggested that the thermal unfolding transition in the GCN4 leucine zipper (GCN4-lz) cannot be rigorously described by a two-state model. Rather, their results indicated that there is an initial loss of helix content within the dimer, which is followed by the chain dissociation.
Moreover, Holtzer et al. (14) have shown that the local thermal unfolding temperatures of the GCN4-lzK coiled-coil, probed by NMR spectroscopy, can be quite different. The computational study of Klimov and Thirumalai (40) also indicated that a local unfolding event can take place at a temperature that is different from the two-state thermal melting temperature. Consistent with this picture and also the results of Dragan and Privalov (21), the Trp fluorescence of GCN4-p1c″ reports a Tm of ∼50°C, which is very different from those measured by CD and IR (Fig. 6). Although it is tempting to relate the thermal event reported by the Trp fluorescence to the fast relaxation component observed in the T-jump IR experiments, such an assignment is probably not valid simply because this kinetic component persists even to the highest final temperature reached in the experiment, i.e., ∼94°C, whereas the thermal unfolding transition associated with the Trp fluorescence is practically over by 80°C (Fig. 6). Furthermore, the lowest final temperature at which we could observe a relaxation signal is ∼65°C, which is also inconsistent with such an assignment. Otherwise, the onset final temperature at which a T-jump signal can be observed would be much lower than 65°C. Finally, if the transition revealed by the Trp fluorescence is indeed due to the repacking of the coiled-coil strands as suggested by Dragan and Privalov (21), it is quite likely that this process involves only minimal, if any, helicity change. Thus, this process would evade detection by the infrared method employed here because the amide I′ band of polypeptides is mostly sensitive to secondary structural elements, e.g., helices. In other words, it is difficult to use the current IR method to distinguish transient and/or accumulative intermediates containing the same or similar secondary structural contents, even if their tertiary packings are different.
FIGURE 6.

The thermal unfolding events of GCN4-p1c″ reported by Trp fluorescence and CD spectroscopy, as indicated. The open triangles correspond to the percentages of the amplitude of the fast kinetic component (∼10 μs) obtained from the T-jump experiments at the corresponding final temperatures.
Although our findings seemingly contradict the results of several stopped-flow experiments, the discrepancy can be easily resolved if we attribute the observed non-monoexponential T-jump relaxation kinetics to a folding intermediate that exists at the native side of the rate-limiting step. It is easy to show that such a post-transition or hidden intermediate cannot be detected by conventional stopped-flow folding experiments (4). If we ignore the step reported by the Trp fluorescence, which is insensitive to the IR probe employed here, a phenomenological kinetic scheme that can describe the IR relaxation kinetics is
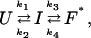 |
where U is the unfolded state, I is a partially folded intermediate, and F* corresponds to the folded state ensemble that may contain different conformations, and ki is the microscopic rate constant of individual steps. Apparently, in response to a perturbation, e.g., a T-jump, the above reaction scheme will produce a relaxation that is characterized by a double-exponential function with two rate constants that are nonlinear combinations of ki (41). Although one cannot use the measured rate constants alone to determine the relative order of the conformational events involved, a correct assessment may be made by knowing how their amplitudes vary with temperature. For example, in the current case, the T-jump IR signal associated with the fast kinetic component (∼10 μs) dominates at low final temperatures (Fig. 6), where the population of F* also dominates, suggesting that this relaxation corresponds to a population redistribution process taking place between I and F* and, supposedly, I is more stable than U. Because of the lack of some critical information, such as the temperature-dependent free energy change for each step as well as the helicity of the intermediate, a rigorous yet meaningful analysis of those T-jump data is not attainable. However, to show qualitatively that the three-state model presented in the text can explain the trend in the amplitude of the fast/slow kinetic component over the temperature range of the data obtained, we assumed that:
The helicity of the intermediate is 30% of that of the folded state;
At 360.2 K (reference temperature), ΔHU–I = −28.4 kcal mol−1, ΔSU–I = −79.3 cal K−1 mol−1, ΔCpU–I = −566.9 cal K−1 mol−1, ΔHI–F = −19.3 kcal mol−1, ΔSI–F = −53.1 cal K−1 mol−1, and ΔCpI–F = −332.9 cal K−1 mol−1; and
k1 = 7500 s−1 and k3 = 90,000 s−1, which are temperature-independent.
Under these conditions, the percentage of the fast component of the double-exponential relaxation kinetics in response to a T-jump of 10°C, solved based on the method of Parker et al. (41), shows the following trend: 81% (60°C), 61% (70°C), 44% (80°C), and 25% (90°C); the values given in parentheses are final temperatures. This trend is consistent with, albeit qualitatively, that observed in the experiment. In addition, the much faster relaxation rate of this process also suggests that the free energy barrier between I and F* is smaller than that between U and I. In other words, I is an intermediate that is behind the rate-limiting folding step, i.e., from U to I.
Although we feel confident attributing the slow kinetic component (∼100 μs) to the cooperative association-dissociation of the two coiled-coil strands, i.e., shift in the equilibrium between U and I, the nature of the fast kinetic component is rather difficult to assess. Since this process is visible to the current IR method, it should involve changes at the secondary structural level. One possibility is that the partially folded intermediate, I, is only structured in the region that is close to the cross-linker and, as a result, the fast kinetic component reflects the growth-dissolution of a loosely packed, helical coiled-coil structure ensemble. If we were to assume that the Trp fluorescence indeed reports a global folding-unfolding event, then a final repacking process, which involves mostly side chains and therefore affects the Trp fluorescence, is needed to take this loosely packed conformation to the fully folded state. This hypothesis seems to be consistent with a recent study of Sosnick and co-workers (15,22), who have shown that an unstructured cross-linker can lead to a structurally more homogeneous folding transition state by enhancing the nucleation probability near the linker. Therefore, it would be interesting to study the T-jump-induced relaxation behavior of the dimeric version of the GCN4-p1 coiled-coil because it has been shown that the 10-residue sequence at the C-terminus plays a critical role in triggering helix initiation and tertiary structure formation (19).
Although results from this study certainly suggest that folding intermediates exist on the folding pathway of coiled-coils, their conformations are difficult to assess. In addition, it is also quite difficult to calculate the corresponding microscopic folding and unfolding rates from the measured relaxation kinetics because the temperature-dependent free energy change associated with each folding step is not known. In principle, isotope-editing technique (7,42,43) can help to elucidate the nature of these hidden intermediates. Such studies are currently underway. In addition, T-jump studies employing Trp fluorescence as a probe can potentially help us to understand the nature of the thermal transition at ∼50°C.
Finally, it is worthwhile pointing out that the probe-dependent thermal unfolding behavior of GCN4-p1c″ and GCN4-p1n″ has also been observed for other proteins. For example, Yang et al. (44) have recently shown that the thermal unfolding temperature of a small β-hairpin, Trpzip2, ranges from 15 to 150°C, depending on the probe. They attributed such behavior to the roughness of the folding energy landscape. On the other hand, a downhill folding scenario (45) can also produce probe-dependent thermal unfolding transitions (46), although it is quite likely that this is not the case for the coiled-coils studied here (35).
CONCLUSIONS
In summary, we have studied the thermal stability and folding kinetics of two cross-linked variants of GCN4-p1 leucine zipper using static CD, fluorescence, and infrared spectroscopies as well as time-resolved T-jump infrared technique. Our results suggest that the folding of these coiled-coils is not a simple two-state process. Instead, they are consistent with a minimal, three-step folding pathway whereby the collision of two strands first leads to the formation of a partially helical intermediate, followed by a fast propagation process during which the entire group of native helices are formed. Finally, a reorganization process, probably corresponding to the repacking of side chains, leads to the formation of the fully folded conformation. Although more studies are required to provide the molecular details regarding the folding pathways of these coiled-coils, our results nevertheless suggest that the folding mechanism of a protein can be more complex than that suggested from equilibrium measurements, and also demonstrated the utility of using the T-jump method to reveal microscopic features of apparent two-state folders.
Acknowledgments
We gratefully acknowledge financial support from the National Science Foundation (grant No. CHE-0094077 and No. MR00-79909).
Ting Wang and Wai Leung Lau contributed equally to this work.
References
- 1.Sanchez, I. E., and T. Kiefhaber. 2003. Evidence for sequential barriers and obligatory intermediates in apparent two-state protein folding. J. Mol. Biol. 325:367–376. [DOI] [PubMed] [Google Scholar]
- 2.Daggett, V., and A. Fersht. 2003. The present view of the mechanism of protein folding. Nat. Rev. Mol. Cell Biol. 4:497–502. [DOI] [PubMed] [Google Scholar]
- 3.Kamagata, K., M. Arai, and K. Kuwajima. 2004. Unification of the folding mechanisms of non-two-state and two-state proteins. J. Mol. Biol. 339:951–965. [DOI] [PubMed] [Google Scholar]
- 4.Bai, Y. 2003. Hidden intermediates and Levinthal paradox in the folding of small proteins. Biochem. Biophys. Res. Commun. 305:785–788. [DOI] [PubMed] [Google Scholar]
- 5.Bai, Y., T. R. Sosnick, L. Mayne, and S. W. Englander. 1995. Protein folding intermediates: native-state hydrogen exchange. Science. 269:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takei, J., W. Pei, D. Vu, and Y. Bai. 2002. Populating partially unfolded forms by hydrogen exchange-directed protein engineering. Biochemistry. 41:12308–12312. [DOI] [PubMed] [Google Scholar]
- 7.Huang, C.-Y., Z. Getahun, Y. Zhu, J. W. Klemke, W. F. DeGrado, and F. Gai. 2002. Helix formation via conformation diffusion search. Proc. Natl. Acad. Sci. USA. 99:2788–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang, T., Y. Zhu, Z. Getahun, D. Du, C.-Y. Huang, W. F. DeGrado, and F. Gai. 2004. Length-dependent helix-coil transition kinetics of nine alanine-based peptides. J. Phys. Chem. B. 108:15301–15310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagen, S. J. 2003. Exponential decay kinetics in “downhill” protein folding. Proteins. 50:1–4. [DOI] [PubMed] [Google Scholar]
- 10.O'Shea, E. K., J. D. Klemm, P. S. Kim, and T. Alber. 1991. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science. 254:539–544. [DOI] [PubMed] [Google Scholar]
- 11.Wendt, H., A. Baici, and H. R. Bosshard. 1994. Mechanism of assembly of a leucine zipper domain. J. Am. Chem. Soc. 116:6973–6974. [Google Scholar]
- 12.Wendt, H., C. Berger, A. Baici, R. M. Thomas, and H. R. Bosshard. 1995. Kinetics of folding of leucine zipper domains. Biochemistry. 34:4097–4107. [DOI] [PubMed] [Google Scholar]
- 13.Sosnick, T. R., S. Jackson, R. R. Wilk, S. W. Englander, and W. F. DeGrado. 1996. The role of helix formation in the folding of a fully α-helical coiled coil. Proteins. 24:427–432. [DOI] [PubMed] [Google Scholar]
- 14.Holtzer, M. E., E. G. Lovett, D. A. d'Avignon, and A. Holtzer. 1997. Thermal unfolding in a GCN4-like leucine zipper: 13Cα NMR chemical shifts and local unfolding curves. Biophys. J. 73:1031–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moran, L. B., J. P. Schneider, A. Kentsis, G. A. Reddy, and T. R. Sosnick. 1999. Transition-state heterogeneity in GCN4 coiled coil folding studied by using multisite mutations and crosslinking. Proc. Natl. Acad. Sci. USA. 96:10699–10704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia, Y., D. S. Talaga, W. L. Lau, H. S. M. Lu, W. F. DeGrado, and R. M. Hochstrasser. 1999. Folding dynamics of single GCN4 peptides by fluorescence resonant energy transfer confocal microscopy. Chem. Phys. 247:69–83. [Google Scholar]
- 17.Myers, J. K., and T. G. Oas. 1999. Reinterpretation of GCN4-p1 folding kinetics: partial helix formation precedes dimerization in coiled coil folding. J. Mol. Biol. 289:205–209. [DOI] [PubMed] [Google Scholar]
- 18.Mohanty, D., A. Kolinski, and J. Skolnick. 1999. De novo simulations of the folding thermodynamics of the GCN4 leucine zipper. Biophys. J. 77:54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zitzewitz, J. A., B. Ibarra-Molero, D. R. Fishel, K. L. Terry, and C. R. Matthews. 2000. Preformed secondary structure drives the association reaction of GCN4-p1, a model coiled-coil system. J. Mol. Biol. 296:1105–1116. [DOI] [PubMed] [Google Scholar]
- 20.Ibarra-Molero, B., G. I. Makhatadze, and C. R. Matthews. 2001. Mapping the energy surface for the folding reaction of the coiled-coil peptide GCN4-p1. Biochemistry. 40:719–731. [DOI] [PubMed] [Google Scholar]
- 21.Dragan, A. I., and P. L. Privalov. 2002. Unfolding of a leucine zipper is not a simple two-state transition. J. Mol. Biol. 321:891–908. [DOI] [PubMed] [Google Scholar]
- 22.Krantz, B. A., and T. R. Sosnick. 2001. Engineered metal binding sites map the heterogeneous folding landscape of a coiled coil. Nat. Struct. Biol. 8:1042–1047. [DOI] [PubMed] [Google Scholar]
- 23.O'Shea, E. K., R. Rutkowski, and P. S. Kim. 1989. Evidence that the leucine zipper is a coiled coil. Science. 243:538–542. [DOI] [PubMed] [Google Scholar]
- 24.Lumb, K. J., C. M. Carr, and P. S. Kim. 1994. Subdomain folding of the coiled coil leucine zipper from the bZIP transcriptional activator GCN4. Biochemistry. 33:7361–7367. [DOI] [PubMed] [Google Scholar]
- 25.Kammerer, R. A., T. Schulthess, R. Landwehr, A. Lustig, J. Engel, U. Aebi, and M. O. Steinmetz. 1998. An autonomous folding unit mediates the assembly of two-stranded coiled coils. Proc. Natl. Acad. Sci. USA. 95:13419–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38:181–364. [DOI] [PubMed] [Google Scholar]
- 27.Kauppinen, J. K., D. J. Moffatt, H. H. Mantsch, and D. G. Cameron. 1981. Fourier self-deconvolution: a method for resolving intrinsically overlapped bands. Appl. Spectrosc. 35:271–276. [Google Scholar]
- 28.Zhu, Y., D. O. V. Alonso, K. Maki, C.-Y. Huang, S. J. Lahr, V. Daggett, H. Roder, W. F. DeGrado, and F. Gai. 2003. Ultrafast folding of α3D: a de novo designed three-helix bundle protein. Proc. Natl. Acad. Sci. USA. 100:15486–15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu, Y., X. Fu, T. Wang, A. Tamura, S. Takada, J. G. Saven, and F. Gai. 2004. Guiding the search for a protein's maximum rate of folding. Chem. Phys. 307:99–109. [Google Scholar]
- 30.Walsh, S. T. R., R. P. Cheng, W. W. Wright, D. O. V. Alonso, V. Daggett, J. M. Vanderkooi, and W. F. DeGrado. 2003. The hydration of amides in helices; a comprehensive picture from molecular dynamics, IR, and NMR. Protein Sci. 12:520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zitzewitz, J. A., O. Bilsel, J. B. Luo, B. E. Jones, and C. R. Matthews. 1995. Probing the folding mechanism of a leucine-zipper peptide by stopped-flow circular-dichroism spectroscopy. Biochemistry. 34:12812–12819. [DOI] [PubMed] [Google Scholar]
- 32.Thompson, K. S., C. R. Vinson, and E. Freire. 1993. Thermodynamic characterization of the structural stability of the coiled-coil region of the bZIP transcription factor GCN4. Biochemistry. 32:5491–5496. [DOI] [PubMed] [Google Scholar]
- 33.Sabelko, J., J. Ervin, and M. Gruebele. 1999. Observation of strange kinetics in protein folding. Proc. Natl. Acad. Sci. USA. 96:6031–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang, W. Y., and M. Gruebele. 2004. Folding λ-repressor at its speed limit. Biophys. J. 87:596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meisner, W. K., and T. R. Sosnick. 2004. Barrier-limited, microsecond folding of a stable protein measured with hydrogen exchange: implications for downhill folding. Proc. Natl. Acad. Sci. USA. 101:15639–15644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jemth, P., S. Gianni, R. Day, B. Li, C. M. Johnson, V. Daggett, and A. R. Fersht. 2004. Demonstration of a low-energy on-pathway intermediate in a fast-folding protein by kinetics, protein engineering, and simulation. Proc. Natl. Acad. Sci. USA. 101:6450–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roder, H., K. Maki, H. Cheng, and M. C. R. Shastry. 2004. Rapid mixing methods for exploring the kinetics of protein folding. Methods. 34:15–27. [DOI] [PubMed] [Google Scholar]
- 38.d'Avignon, D. A., G. L. Bretthorst, M. E. Holtzer, and A. Holtzer. 1998. Site-specific thermodynamics and kinetics of a coiled-coil transition by spin inversion transfer NMR. Biophys. J. 74:3190–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.d'Avignon, D. A., G. L. Bretthorst, M. E. Holtzer, and A. Holtzer. 1999. Thermodynamics and kinetics of a folded-folded′ transition at valine-9 of a GCN4-like leucine zipper. Biophys. J. 76:2752–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klimov, D. K., and D. Thirumalai. 2002. Is there a unique melting temperature for two-state proteins? J. Comput. Chem. 23:161–165. [DOI] [PubMed] [Google Scholar]
- 41.Parker, M. J., J. Spencer, and A. R. Clarke. 1995. An integrated kinetic analysis of intermediates and transition states in protein folding reactions. J. Mol. Biol. 253:771–786. [DOI] [PubMed] [Google Scholar]
- 42.Huang, C.-Y., Z. Getahun, T. Wang, W. F. DeGrado, and F. Gai. 2001. Time-resolved infrared study of the helix-coil transition using 13C-labeled helical peptides. J. Am. Chem. Soc. 123:12111–12112. [DOI] [PubMed] [Google Scholar]
- 43.Decatur, S. M., and J. Antonic. 1999. Isotope-edited infrared spectroscopy of helical peptides. J. Am. Chem. Soc. 121:11914–11915. [Google Scholar]
- 44.Yang, W. Y., J. W. Pitera, W. C. Swope, and M. Gruebele. 2004. Heterogeneous folding of the Trpzip hairpin: full atom simulation and experiment. J. Mol. Biol. 336:241–251. [DOI] [PubMed] [Google Scholar]
- 45.Onuchic, J. N., Z. Luthey-Schulten, and P. G. Wolynes. 1997. Theory of protein folding: the energy landscape perspective. Annu. Rev. Phys. Chem. 48:545–600. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Mira, M. M., M. Sadqi, N. Fischer, J. M. Sanchez-Ruiz, and V. Munoz. 2002. Experimental identification of downhill protein folding. Science. 298:2191–2195. [DOI] [PubMed] [Google Scholar]