

Abstract
Fur (ferric uptake regulator) is a key bacterial protein that regulates iron acquisition and its storage, and modulates the expression of genes involved in the response to different environmental stresses. Although the protein is involved in several regulation mechanisms, and members of the Fur family have been identified in pathogen organisms, the stability and thermodynamic characterization of a Fur protein have not been described. In this work, the stability, thermodynamics and structure of the functional dimeric Fur A from Anabaena sp. PCC 7119 were studied by using computational methods and different biophysical techniques, namely, circular dichroism, fluorescence, Fourier-transform infrared, and nuclear magnetic resonance spectroscopies. The structure, as monitored by circular dichroism and Fourier-transform infrared, was composed of a 40% of α-helix. Chemical-denaturation experiments indicated that Fur A folded via a two-state mechanism, but its conformational stability was small with a value of ΔG = 5.3 ± 0.3 kcal mol−1 at 298 K. Conversely, Fur A was thermally a highly stable protein. The high melting temperature (Tm = 352 ± 5 K), despite its moderate conformational stability, can be ascribed to its low heat capacity change upon unfolding, ΔCp, which had a value of 0.8 ± 0.1 kcal mol−1 K−1. This small value is probably due to burial of polar residues in the Fur A structure. This feature can be used for the design of mutants of Fur A with impaired DNA-binding properties.
INTRODUCTION
Control of iron homeostasis is essential in most biological systems. Iron is required for many cellular processes, but biologically useful iron is very scarce due to its low solubility at physiological pH values (FeIII) or its low abundance (FeII). However, excess of iron is toxic because it catalyses formation of oxygen and nitrogen species (1), which can damage DNA, proteins, or lipids. Accordingly, organisms have evolved efficient mechanisms to store iron in an inert form and to acquire it (2,3). Studies of the iron control homeostasis in bacteria have focused over the last 25 years on the ferric uptake regulator (Fur). Fur represses genes that are involved in iron uptake, under iron-replete conditions, and that are de-repressed when the metal is scarce. The repression mechanism by the Fur of Escherichia coli occurs via a dimeric protein species through inhibition of transcription by blocking the entry of RNA polymerase to the promoter of target genes (4,5). Recently, the control of gene expression by Fur has been extended toward functions including oxidative stress response (6–8), acid resistance (9), toxin production (10,11), and repression of the expression of a small regulatory RNA (10). Probably, Fur is the most important member of a family of regulators, which are all involved in metal-dependent control of gene expression (12–15), but of which no thermodynamic parameters or conformational stability are known.
In cyanobacteria, fur homolog genes have been identified in several species (16–18), but most of the roles of the master regulator Fur are not fully known yet. It is essential to understand in depth the mechanisms of iron regulation in cyanobacteria, since proliferation of cyanobacterial populations, whose growing is regulated by iron availability (19), can alter the environment and public health (20). Biochemical, thermodynamic, and structural analysis of their Fur proteins could provide new insights into their stability, the general stability of dimeric proteins, and how to control unrestrained proliferation of cyanobacteria. We have previously carried out the biochemical analysis of Fur A from Anabaena sp. PCC 7119 (21). This member of the Fur family is 151-residues-long, and contains the amino acid motif H5X2CX2C (where X represents any amino acid) characteristic of other Fur proteins. Fur A tends to oligomerize in solution with the involvement of disulphide bridges: several oligomerization states are observed in the absence of reducing agents, whose different populations depend on protein concentration and ionic strength. In the presence of reducing agents, noncovalently bonded dimeric species are present. Furthermore, Fur A does not bind Zn or other structural metals (21), in contrast to that observed in other members of the family. These features make Fur A a model to carry out structural and thermodynamical studies.
In this work, an extensive structural and thermodynamic characterization of functional dimeric Fur A from Anabaena sp. PCC 7119 were carried out in a wide pH range by using several biophysical techniques, namely, fluorescence, circular dichroism (CD), Fourier transform infrared spectroscopy (FTIR), and nuclear magnetic resonance (NMR). To the best of our knowledge, this is the first study on the conformational stability and thermodynamic characterization of a member of the Fur family. Furthermore, we have modeled the structure of the monomeric Fur A, by using as a template the x-ray structure of a Fur protein (22). This model has allowed us to explain some of the conformational and thermodynamic features of the protein. The different experimental biophysical techniques allowed the determination of the thermodynamic parameters governing the unfolding of Fur A, its secondary structure, its conformational stability and its shape. Our experimental and theoretical findings show that Fur A was mainly composed of α-helical structure. The overall picture, obtained from both chemical- and heat-induced denaturation studies, was consistent with a moderately stable protein at 298 K, which folded via a two-state mechanism. However, the protein was highly resistant toward thermal unfolding (temperature at thermal midpoint, Tm, = 352 ± 5 K), due to a small-capacity heat value (0.8 ± 0.1 kcal mol−1 K−1). This small value is probably due to the presence of buried polar residues in the structure of Fur A. The implications of this small value of the heat capacity change (ΔCp) are discussed in relation to the thermal stability of Fur proteins as opposed to their conformational stability at 298 K.
MATERIALS AND METHODS
Materials
Sodium acetate base and acid, 8-anilino-1-naphtalenesulfonate (ANS), and NaCl were from Sigma (St. Louis, MO). Ultra-pure guanidinium hydrochloride (GdmHCl) was from ICN Biochemicals (Costa Mesa, CA). Dithiothreitol (DTT) was from Apollo Scientific (Stockport, UK). Trypsin proteomics grade was from Sigma. Stock solution of the enzyme was prepared according to manufacturer instructions. Deuterated acetic acid and its sodium salt were from Cambridge Isotope Laboratories (Andover, MA). Standard suppliers were used for all the other chemicals. Water was deionized and purified with a Millipore system (Millipore, Billerica, MA).
Protein expression and purification
Fur A was overexpressed in E. coli BL21-Gold DE3 and purified as described (21). Protein was stored in 10 mM sodium acetate buffer, pH 4.0, and 1 mM DTT to avoid disulphide bridge formation. Protein stocks were run in SDS-PAGE gels and found to be >97% pure. Protein concentration was calculated from the absorbance measured at 280 nm, using the extinction coefficients of model compounds (23).
Fluorescence measurements
Fluorescence spectra were collected in a Cary Eclipse spectrofluorometer (Varian, Cary, NC) interfaced with a Peltier cell, or at the very low protein concentrations explored (0.5 μM), in an Aminco-Bowman SLM 8000 spectrofluorometer (Spectronics Instruments, Urbana, IL) interfaced with a Haake water bath. The slit-widths were equal to 5 nm for the excitation and the emission wavelengths in the Varian spectrofluorimeter, and 8 nm in the Aminco one. Sample concentration was in the range 0.5–6 μM, with 20 or 100 μM of DTT. Under these conditions, Fur A is a dimer, which is the active form of the protein (1,3). An 1- or 0.5-cm-pathlength quartz cells (Hellma, Müllheim/Baden Germany) were used in the Varian and Aminco instruments, respectively. Experiments were acquired at 298 K.
Steady-state fluorescence measurements
Protein samples were excited at 280 and 295 nm in the pH range 2–12. The results at both wavelengths were identical (data not shown). Experiments were recorded between 300 and 400 nm. The signal was acquired for 1 s and the increment of wavelength was set to 1 nm. Blank corrections were made in all spectra. The pH was measured after completion of the experiments with an ultra-thin Aldrich electrode (Sigma-Aldrich, Copenhagen, Denmark) in a Radiometer pH-meter (Westlake, OH). Three-point calibration of the pH-meter was performed using standards from Radiometer. In all cases, buffer concentration was 25 mM. The salts and acids used in buffer preparation were: pH 2.0–3.0, phosphoric acid; pH 3.0–4.0, formic acid; pH 4.0–5.5, acetic acid; pH 6.0–7.0, monosodium di-hydrogen phosphate; pH 7.5–9.0, Tris acid; pH 9.5–11.0, sodium carbonate; and pH 11.5–12.0, sodium phosphate. The samples were kept overnight at 298 K to allow for equilibration.
Exact concentrations of GdmHCl were calculated from the refractive index of the solution (23). Chemical denaturations were fully reversible (data not shown). Every chemical-denaturation described in this work was repeated three times with new samples.
Steady-state ANS binding
Fluorescence spectra were collected in the presence of 100 μM dye at a protein concentration of 2 μM with 100 μM of DTT. Excitation wavelength was 370 nm, and emission was measured from 430 to 700 nm. Stock solutions of ANS were freshly prepared in water, using a molar extinction coefficient of 6.8 × 103 M−1 cm−1 at 370 nm (24,25).
Circular dichroism measurements
CD measurements were carried out in a Jasco J810 spectropolarimeter (JASCO, Tokyo, Japan) fitted with a thermostated cell holder and interfaced with a Neslab RTE-111 water bath (Neslab, Portsmouth, NH). The instrument was periodically calibrated with (+) 10-camphorsulphonic acid. Experiments were acquired at 298 K.
Steady-state experiments
Isothermal wavelength spectra at different values of pH were acquired at a scan speed of 50 nm/min with a response time of 2 s and averaged over four (far-ultraviolet CD) or six (near-UV CD) scans at any pH. Spectra were corrected by subtracting the proper baselines.
The pH- and GdmHCl-denaturation measurements in the far-UV were performed using 15 μM of Fur A in the presence of 100 μM of DTT. Larger protein concentrations (20, 30, and 50 μM) were also used to test for the concentration-dependence of the chemical denaturations. The pathlength of the cell was 0.1 cm (Hellma). Every chemical or pH-denaturation experiment was repeated at least three times with new samples.
Near-UV spectra at the different pH values were acquired using 38 μM of protein with 400 μM of DTT in a 0.5-cm-pathlength cell (Hellma).
The mean residue ellipticity, [Θ], was calculated according to
 |
(1) |
where Θ is the measured ellipticity, l is the pathlength cell (in centimeters), c is the protein concentration (in M), and N is the number of amino acids (151 for Fur A).
Thermal denaturation experiments
Experiments at different pH values and GdmHCl concentrations were performed at constant heating rates of 30 K/h and 60 K/h with a response time of 8 s. Both heating rates yielded the same results (data not shown). Thermal scans were collected in the far-UV region at 222 nm from 298 K to 363 K, in 0.1-cm-pathlength cells, with a total protein concentration of 15 μM. The reversibility of thermal transitions was tested by recording a new scan after cooling down to 283 K. The possibility of drifting of the CD spectropolarimeter was tested by running two samples containing buffer, before and after the thermal experiments. No difference was observed between the scans. Every experiment was repeated at least twice with new samples.
To test for the concentration-dependence of the Tm at 2.5 M GdmHCl, the protein concentration was varied from 20 to 60 μM. We chose this concentration of GdmHCl since the sigmoidal behavior could be easily observed.
Determination of helical content from far-UV CD data
We used two different approaches. Firstly, CD spectral data can be deconvolved by using neural networks to yield the percentages of secondary structure (26). Secondly, a simpler analysis only takes into account the ellipticity at 222 nm by using the expression (27)
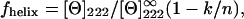 |
(2) |
where the fhelix is the α-helical fraction of the protein, [Θ]222 is the ellipticity at 222 nm, 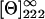 is the ellipticity for an infinite helix at 222 nm (−34,500 deg dmol−1 cm2), k is a wavelength-dependent constant (2.57 at 222 nm), and n is the number of peptide bonds in the protein (150 in Fur A).
is the ellipticity for an infinite helix at 222 nm (−34,500 deg dmol−1 cm2), k is a wavelength-dependent constant (2.57 at 222 nm), and n is the number of peptide bonds in the protein (150 in Fur A).
Analysis of the thermal, pH- and chemical-denaturation curves, and free energy determination
The average emission intensity, 〈λ〉, used to follow the pH- and GdmHCl-denaturation experiments was calculated from (28)
 |
(3) |
where Ii is the fluorescence intensity measured at a wavelength λi.
The pH-denaturation experiments were analyzed assuming that both species, protonated and deprotonated, contributed equally to the fluorescence spectrum,
 |
(4) |
where X is the physical property being observed ([Θ]222, the 〈λ〉, or the fluorescence maximum wavelength), Xa is the physical property being observed at low pH values, Xb is the physical property observed at high pH values, pKa is the apparent ionization constant of the titrating group, and n is the Hill coefficient, which gives a measurement of the cooperativity of the transition. The Hill coefficient was close to 1 in all the curves, except for those of ANS (see Results). The apparent pKa reported was obtained from three measurements in each biophysical technique.
Chemical-denaturation data were obtained by following the [Θ]222, the 〈λ〉 or, at the very low protein concentrations explored, the maximum wavelength. Data were fitted to
 |
(5) |
where XD = αD + βD[D] and XN = αN + βN[D] are the corresponding fractions of the folded and unfolded states, respectively, for which a linear relationship with denaturant is assumed; ΔG is the free energy of denaturation; R is the gas constant; and T is the temperature in K. The curves at different temperatures were analyzed using the linear extrapolation model (LEM): ΔG = m([D]1/2 − [D]) − RT ln(2Ct) (29), where Ct is the molar concentration of the protein expressed in dimer equivalents, m is the slope, [D] is the denaturant concentration, and [D]1/2 is the concentration at the midpoint of the transition.
In Eq. 5, the change in free energy, when temperature is used as a denaturant, is given by the Gibbs-Helmholtz expression (29),
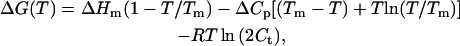 |
(6) |
where ΔHm, the thermal enthalpy change at the thermal midpoint, is the van't Hoff enthalpy change; ΔCp is the heat capacity change; and Tm is the thermal midpoint.
Fitting was carried out by using the general curve fit option of Kaleidagraph (Abelbeck Software, Reading, PA) working on a PC computer.
Nuclear magnetic resonance spectroscopy: diffusion-ordered spectroscopy measurements (DOSY experiments)
NMR experiments were carried out in a Bruker Avance 500 spectrometer (Madison, WI), where the probe temperature was regularly calibrated by using methanol and ethylene glycol (30), and equipped with a 5-mm triple-resonance inverse probe with z-gradients.
Translational self-diffusion measurements were performed using the pulsed-gradient spin-echo NMR method (31,32). The following relationship exists between the translational self-diffusion parameter, D, and the NMR parameters (31–33),
 |
(7) |
where I is the measured peak intensity (or volume) of a particular (or a group of) resonance(s); I0 is the maximum peak intensity of the same (group of) resonance(s) at the smaller gradient strength; D is the translational self-diffusion constant (in cm2 s−1); γ is the gyromagnetic ratio of a proton (2.675 × 104 rad G−1 s−1); δ is the duration (in seconds) of the gradient; G is the strength of the gradient (in G cm−1); and Δ is the time (in seconds) between the two gradients (i.e., the time when the molecule evolves). Data can be plotted as the −ln(I/I0) versus G2 and the slope of the line is 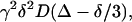 and D can be easily obtained.
and D can be easily obtained.
The Stokes-Einstein equation relates D to the molecular shape via the so-called friction coefficient, f,
 |
(8) |
where T is the temperature and k the Boltzmann constant. The f of a protein is determined by its overall dimensions, hydration, and the rugosity of the surface exposed to water. If it is assumed that the protein adopts a spherical shape, the f is given by
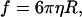 |
(9) |
where η is the viscosity of the solvent and R is the hydrodynamic radius of the sphere. Then, combining Eqs. 8 and 9:
 |
(10) |
The viscosity of a solution is very weakly influenced by the macromolecule component at the low macromolecular concentrations used, and therefore, the viscosity of the solution should be that of the solvent. Solvent viscosity is temperature-dependent according to (33): log η = a + [b/c − T]. The terms a, b, and c are given for a particular 2H2O:H2O ratio. In our conditions, a 100% 2H2O solution, the values are: a = −4.2911, b = −164.97, and c = 174.24. This yields a value of η = 1.253 kg/(cm−1s−1) at 293 K, used in our calculations.
The gradient strength was calibrated using the diffusion rate for the residual proton water line in a sample containing 100% 2H2O in a 5-mm tube, and back-calculating G. This procedure assumes that the diffusion rate for HDO in a 100% 2H2O sample is 1.94 × 10−5 cm2 s−1 at 298 K (34). Experiments were acquired by using the longitudinal eddy-current delay, pulsed gradient-field pulse sequence, with a postgradient eddy-current relaxation delay of 5 ms. Each experiment was averaged over 128 scans and the number of points was 16 K. The strength of the gradients was varied from 2% of the total power of the gradient coil to 95%, and their shape was a sine function. Experiments were acquired at different protein concentrations at pH 4.0 (25 mM deuterated sodium acetate buffer) in 1 mM nondeuterated DTT at 293 K. Protein was concentrated using the Amicon centriprep centrifugal filter devices (Amicon, Ann Arbor, MI; cutoff molecular weight 3500), and the largest protein concentration used was 950 μM. The other concentrations were obtained from dilution of the 950 μM stock in the acetate buffer supplemented with 1 mM nondeuterated DTT. Larger amounts of DTT could not be used since its resonances partly overlap with those of the protein. The duration of the gradient was varied between 3 ms and 2.2 ms, and the time between both gradients was changed between 100 and 150 ms. The most upfield-shifted methyl groups (between 0.8 and 0.0 ppm) were used to measure the changes in intensity.
FTIR experiments
Spectra were acquired on a Bruker FTIR-66S instrument equipped with a deuterated triglycine sulfate detector and fitted with a water bath. The cell container was continuously filled with dry air. Buffer was 50 mM Tris (pH 7), 5 mM DTT, and 200 mM KCl. The contributions of buffer spectra were subtracted, and the resulting spectra were used for analysis. Samples were dried in a Speed Vac concentrator (Savant, Farmingdale, NY) and dissolved in the corresponding buffer. Protein concentration was, in all cases, 720 μM. Protein samples were placed between a pair of CaF2 windows separated by a 50-μm-thick spacer in a Harrick (Ossining, NY) demountable cell.
Three-hundred scans per sample were taken, averaged, apodized with a Happ-Genzel function, and Fourier-transformed to give a final resolution of 2 cm−1. The signal/noise ratio of the spectra was >1000:1. To quantify the different secondary structure components, the amide I band was decomposed into its constituents by curve-fitting (based on a combination of Gaussian and Lorentzian functions). This procedure uses the number and position of bands obtained from the deconvolved (by using a Lorentzian bandwidth of 18 cm−1 and a resolution enhancement factor of 2) and the Fourier derivative spectra (by using a power of 3 and a breakpoint of 0.3) (35,36).
Trypsin digestion experiments
Fur (at a final concentration of 0.59 μg/μl) was mixed with trypsin (at a final concentration of 0.059 μg/μl) in a total volume of 15 μl in 100 mM of buffer (37), at pH values 7, 8, and 9. Samples were incubated at 25°C for 10 min. Digestion was stopped by the addition of 15 μl of SDS-PAGE loading buffer and the resulting samples were heated during 15 min at 110°C. Samples were immediately run on a gel. The intensity of the bands at different times was measured by densitometry. Experiments at any pH were repeated four times.
Homology modeling of the Fur A monomer
The model was based on the crystal structure at 1.80 Å resolution of Fur from Pseudomonas aeruginosa (PDB code 1mzb) (22). Based on the pairwise alignment between Fur A and Fur from P. aeruginosa (PA-Fur), extracted from a multiple alignment of 34 Fur proteins belonging to different species, the target protein (Fur A) was aligned to the three-dimensional template (PA-Fur). Energy minimization was achieved in a two-step process of simulated annealing followed by global energy optimization of buried side chains.
As implemented in ICM (38), the molecular system was described in terms of internal coordinate variables, using a modified ECEPP/3 (39) force field and a distance-dependent dielectric constant, with a value of ɛ = 2 × r. The global energy optimization of buried side chains was performed using the biased-probability Monte Carlo minimization procedure (40). In the biased-probability Monte Carlo global energy optimization method, random conformational changes of the free variables are performed according to a predefined continuous probability distribution (40), followed by a double-energy minimization scheme: local energy of analytical differentiable terms is minimized followed by a calculation of the complete energy including nondifferentiable terms such as entropy and solvation energy. Acceptance or rejection of the total energy is based on the Metropolis criterion (41).
RESULTS
pH-induced structural changes
Steady-state intrinsic fluorescence measurements
Fluorescence was used to monitor the changes in the tertiary structure of the protein. The emission fluorescence spectrum of Fur A between pH 4 and 7, obtained by excitation at 280 nm, showed a maximum at 345 nm, and therefore, the spectrum was dominated by the emission of the sole tryptophan residue (Fig. 1 A). The maximum was blue-shifted toward 339 nm above pH 7. The apparent pKa of this titration was 7.6 ± 0.2. When the changes were followed by the variation of the 〈λ〉, a similar behavior was observed, and the apparent pKa was 7.7 ± 0.2 (Fig. 1 A).
FIGURE 1.

The pH-induced unfolding of Fur A followed by intrinsic and ANS fluorescence. (A) Intrinsic fluorescence: The 〈λ〉 (right axis, solid squares) and the maxima wavelength (left axis, open squares) are represented versus the pH. Protein concentration was 2 μM, in 100 μM of DTT. (B) ANS-binding experiments: The maxima wavelength (left axis, open squares) and the 〈λ〉 (right axis, solid squares) are represented versus the pH. Protein concentration was 2 μM and ANS concentration was 100 μM, in 100 μM of DTT. All the experiments were acquired at 298 K.
As the pH was further increased from 10 to 12, the spectra were red-shifted toward 346 nm, due to basic unfolding (Fig. 1 A). The apparent pKa of this transition could not be determined due to the absence of baseline at high pH values. On the other hand, a blue-shift occurred at acidic values of pH, but the pKa of this transition could not be determined either, since its acidic baseline was not observed. The behavior of 〈λ〉 at those pH values was similar to that of the maximum wavelength (Fig. 1 A).
ANS-binding experiments
ANS-binding was used to monitor the extent of exposure of protein hydrophobic regions. When ANS is bound to solvent-exposed hydrophobic patches of proteins, its quantum yield is enhanced and the maximum of emission is shifted from 520 nm to 480 nm (42,43). At low pH values, the intensity of ANS in the presence of Fur A was enhanced, and the maximum wavelength was 485 nm (Fig. 1 B). As the pH was increased, the maximum wavelength shifted toward 515 nm. Consistent effects were observed when the 〈λ〉 was examined: it was high at low pH values, but it decreased as the pH was raised (Fig. 1 B). The apparent pKa values determined were 7.4 ± 0.2 (from the maximum wavelength) and 7.5 ± 0.2 (from the 〈λ〉), which were similar to those determined by intrinsic fluorescence (see above). However, the Hill indexes were 1.6 ± 0.4 (from the maximum wavelength) and 1.7 ± 0.3 (from the 〈λ〉), suggesting that ANS was reporting the titration of more than one group. This could explain the broadness of the transition observed (Fig. 1 B), when compared to those of the intrinsic fluorescence (Fig. 1 A).
Far-UV CD experiments
The far-UV CD spectrum of Fur A showed a broad minimum at physiological and basic pH values (Fig. 2 A, inset). The behavior of [Θ]222 was similar to those of the 〈λ〉 and the maximum wavelength: as the pH was increased from 2 to 4, the [Θ]222 increased (in absolute value), until a plateau at pH 4 was attained. From pH 7, the ellipticity followed a sigmoidal behavior with a pKa of 8.1 ± 0.5, to finally decrease in absolute value above pH 10 (Fig. 2 A). The percentage of helical structure, as shown by the change in [Θ]222, decreased at high pH values.
FIGURE 2.

Far- and near-UV CD. (A) Far-UV CD, as measured by following the mean residue ellipticity at 222 nm. (Inset) Far-UV CD spectrum of Fur at pH 2.0, 6.0, and 10. Protein concentration was 15 μM in 100 μM of DTT. All the experiments were acquired at 298 K. (B) Near-UV CD of Fur A at pH 7 (continuous line) and pH 10 (dashed line). Protein concentration was 38 μM with 400 μM of DTT. All the experiments were acquired at 298 K.
Near-UV CD experiments
The near-UV spectrum of a protein provides insights on the asymmetric environment of any aromatic residue (44,45). Due to the large amounts of protein used, experiments at selected pH values were carried out. The near-UV spectrum at pH 7 showed an intense band at 278 nm, which changed as the pH was modified: the band was shifted from 280 nm (pH 7) to 297 nm (pH 10) (Fig. 2 B). This band, as shown by protein engineering studies (46,47), can correspond either to the tyrosine or tryptophan residues, although the contribution of the latter is more important.
FTIR experiments
Compared to CD, the main advantage of FTIR is its higher sensitivity to the presence of β-structure, random coil, or some side chains (35,36). However, due to the large amounts of proteins used, only experiments at pH 7 were carried out to determine the percentage of secondary structure. The percentage of α-helical structure was 40%, and that of β-sheet was 33%.
Trypsin digestion of Fur A to map conformational changes at high pH values
Digestion of Fur A was carried out at pH values 7, 8, and 9, where the protein is at the beginning, middle, and end of the transition, respectively (Figs. 1 and 2). The digestion was faster as the pH was increased (Fig. 3). These findings suggest structural changes in this pH range, thus confirming the fluorescence and CD results (see above).
FIGURE 3.

Trypsin digestion experiments. Changes in the Fur A intensity band in a SDS-PAGE gel at different times since the beginning of the reaction digestion. The measurements were repeated four times at the different pH values.
Taken together, these results indicate that at physiological pH, before the basic denaturation, Fur A showed a conformational transition.
Thermal-denaturation measurements
Thermal denaturations were followed by far-UV at pH 3, 4, 7, 10, and 13. No sigmoidal behavior was observed at any pH; instead, the ellipticity decreased in absolute value when the temperature was increased, and at very high temperatures, precipitation occurred (data not shown). However, reversible and sigmoidal curves were observed by the addition of small amounts of GdmHCl (1.00–2.25 M), that do not unfold the protein (see below), at pH 4 (Fig. 4 A); as the concentration of GdmHCl was increased, the Tm decreased. Extrapolation of the data at 0 M GdmHCl yielded a value of Tm = 352 ± 5 K (Fig. 4 B). However, the large errors associated with the calculated ΔHm precluded a reliable estimation of ΔCp.
FIGURE 4.

Thermal denaturation of Fur A. (A) Far-UV CD at pH 4, in the presence of different amounts of GdmHCl by following the change in ellipticity at 222 nm: [GdmHCl] = 1.25 M (open squares), [GdmHCl] = 1.75 M (open circles), and [GdmHCl] = 2.25 M (solid squares). The lines through the data are the fittings to Eqs. 5 and 6. Protein concentration was 15 μM in 100 μM of DTT in all cases. The scale on the y axis is arbitrary. (B) Extrapolation of Tm at zero denaturant concentration from the data in plot A. Error bars are fitting errors to Eqs. 5 and 6. (C) Far-UV CD at pH 4, 2.25 M GdmHCl at 20, and 60 μM. The Tm were 342 ± 1 K for 20 μM and 345.6 ± 0.7 K for 60 μM. The solid lines through the data are the nonlinear least-squares fits to Eq. 5, with the free energy given by Eq. 6. The scale on the y axis is arbitrary.
In experiments at different protein concentrations in 2.25 M GdmHCl, there was a small variation in Tm, suggesting that the unfolding process was a second-order one.
Hydrodynamic properties of Fur A
We have shown that the broad signals of Fur A in one-dimensional NMR experiments hampered any structural determination (21). However, and since Fur A binds at any pH to the gel filtration matrices (probably due to nonspecific interactions with the column) (21), the hydrodynamic properties of Fur A can still be addressed by NMR, by using translational self-diffusion NMR measurements. The diffusion coefficient of Fur A increased linearly as protein concentration was decreased (Fig. 5). Dilution of the protein led to an increase of the translational mobility of the particles in solution, since at lower protein concentrations, the molecular impairment to the translational diffusion was smaller. The extrapolated translational diffusion coefficient, D, at infinite dilution of the protein (i.e., the y axis intercept) was (8.1 ± 0.1) × 10−7 cm2 s−1 at 293 K. The use of Eqs. 8–10 yielded a hydrodynamic radius for a spherical Fur A of 21.6 Å.
FIGURE 5.

DOSY-NMR experiments. (A) The logarithm of the normalized intensity of the most upfield-shifted peaks is shown as a function of the squares of the gradient strength at two selected concentrations: 950 μM (continuous line and open squares) and 475 μM (dotted line and solid squares). The slopes of the plots give the apparent diffusion constant of the molecule in solution at the particular concentration used. (B) NMR diffusion coefficients of Fur A as a function of protein concentration. The bars are fitting errors to the linear equations shown in A. The solid line is the fitting to a linear equation whose y-axis intercept yields the diffusion coefficient in an ideal solution (i.e., at 0 M of protein concentration). The concentration of DTT was 1 mM in all cases at pH 4.
The hydrodynamic radius for an ideal unsolvated spherical molecule can be theoretically calculated considering that the anhydrous molecular volume, 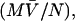 equals the volume of a sphere (48,49),
equals the volume of a sphere (48,49),
 |
where M is the molecular weight of the protein,  is the partial specific volume of the protein, and N is the Avogadro's number. The molecular weight of a monomeric Fur A is 17,259, and
is the partial specific volume of the protein, and N is the Avogadro's number. The molecular weight of a monomeric Fur A is 17,259, and 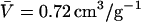 as calculated from amino acid composition (49). Since the functional form of Fur A studied here was a dimer, the above expression led to a hydrodynamic radius of 21.4 Å, which agrees very well with that determined by diffusion measurements. Then, we can conclude that the shape of the dimeric FurA in solution was spherical.
as calculated from amino acid composition (49). Since the functional form of Fur A studied here was a dimer, the above expression led to a hydrodynamic radius of 21.4 Å, which agrees very well with that determined by diffusion measurements. Then, we can conclude that the shape of the dimeric FurA in solution was spherical.
Structure of Fur A by homology modeling
The structural similarity between PA-Fur and Fur A was assumed based on: 1), sequence similarity (∼40%); 2), the near-absence of insertion or deletions in the alignment; and 3), the conserved biological function of both proteins. Furthermore, the helical content of Fur protein in both species is very similar, as concluded from the FTIR and far-UV CD experiments (see Discussion), and the x-ray structure of PA-Fur (22). Due to the lack of strong sequence identity on the dimerization interface of PA-Fur and Fur A, and the lack of sufficient biological and biochemical evidence, any attempt to model the Fur A dimer would not have been realistic enough. Then, the model of the monomer of Fur A will be used in our discussion.
The monomer is composed by two domains (Fig. 6): the N-terminal region is the DNA-binding domain, and is formed by the packing of two helix-turn-helix motifs (α-helix 1 (residues Thr7-Arg16), α-helix 2 (Thr21-Glu33), α-helix 3 (Ser41-Asp52), and α-helix 4 (Ser57-Met71); and the C-terminal region is the dimerization zone, which is formed by a long α-helix (residues Asn112-Lys125) and five β-strands: residues Leu74 to Leu77 (β-strand 1), His85 to Ile88 (β-strand 2), His97 to Cys101 (β-strand 3), Thr107 to Phe110 (β-strand 4), and Thr136 to Ala139 (β-strand 5).
FIGURE 6.

Three-dimensional model of the structure of Fur A. Ribbon representation of the homology model of Fur A based on P. aeruginosa crystal structure (PDB code 1mzb) (22). The helical-rich N-terminus is colored in green and the C-terminus in magenta. Trp18 and its neighboring residues are displayed in stick representation. (Color code: light gray, carbon; blue, nitrogen; red, oxygen; green, sulfur; and dark gray, hydrogen.) Only polar hydrogens are displayed.
The Zn binding pocket in PA-Fur defined by His86, Asp88, Glu107, and His124 was not conserved in Fur A. However, the other Zn binding pocket in PA-Fur (formed by residues His32, Glu80, His89, and Glu100) was fully conserved. Since no Zn has been experimentally detected in Fur A (21), a water molecule was placed in the same position as Zn during the homology model optimization to fill the cavity.
Although most of the charged residues of Fur A were solvent-exposed (considering the monomer conformation), one of the two buried cores was formed by polar residues His39, His85, His96, His98, Glu87, and Glu109. Moreover, a water molecule could be trapped within the cavity defined by the polar residues His39, Glu87, His96, and Glu109.
Conformational stability of Fur A
We used a two-part approach to determine the conformational stability of Fur A. Firstly, protein stability was monitored at several values of pH. Secondly, the thermodynamic parameters governing its thermal unfolding at a selected pH were determined.
Changes in protein stability with the pH
Fluorescence isothermal GdmHCl denaturations at 298 K were carried out at pH 4, 5, 6, 7, 8, and 9. At pH 6, 7, 8, and 9 (close to the conformational transition observed at neutral pH, Figs. 1 and 2 A), the large slopes of the native and unfolding baselines precluded the precise determination of the m- and the GdmHCl concentration at the chemical-denaturation midpoint, i.e., the [GdmHCl]1/2 values (data not shown). Conversely, at pH 4 and 5, sigmoidal curves with a sole transition and steep folding and unfolding baselines were obtained, which yielded the thermodynamical parameters of m = 1.4 ± 0.2 kcal mol−1 M−1 and a [GdmHCl]1/2 = 3.8 ± 0.1 M (at pH 4); and, m = 1.3 ± 0.3 kcal mol−1 M−1 and a [GdmHCl]1/2 = 2.9 ± 0.2 M (at pH 5). It can be observed that the m-value was small and, conversely, the [GdmHCl]1/2 was high for a dimeric protein of this size (50,51).
The GdmHCl chemical-denaturation of Fur A at pH 4 (where the [GdmHCl]1/2 was higher) was also followed by using CD; the analysis of fluorescence and CD unfolding curves showed that the [GdmHCl]1/2- and the m-values were the same, within the experimental error (see Discussion): m = 1.4 ± 0.2 kcal mol−1 M−1 and a [GdmHCl]1/2 = 3.8 ± 0.1 M (for fluorescence); and m = 1.0 ± 0.3 kcal mol−1 M−1 and a [GdmHCl]1/2 = 3.8 ± 0.4 M (for CD). The coincidence of equilibrium denaturation transitions monitored by two different biophysical techniques suggested that the protein followed a two-state mechanism (52).
Chemical-denaturations were also carried out at different protein concentrations. Fluorescence was used to explore the protein concentration range from 0.5 to 5 μM, and CD within the range 15 to 50 μM. There was a protein concentration-dependence of the [GdmHCl]1/2, as it could be expected for the unfolding of a dimeric protein (Fig. 7). However, the steepness of the folded and unfolded baselines, especially important in the CD experiments, precluded a precise determination of the [GdmHCl]1/2-values (Fig. 7).
FIGURE 7.

The GdmHCl-denaturation of Fur A at pH 4 at different protein concentrations. CD raw data (left axis, solid circles) at 50 μM protein concentration and fluorescence raw data (right axis, open circles) at 0.5 μM protein concentration in the presence of DTT (150 μM for CD and 20 μM for fluorescence). Fitting to Eq. 5 resulted in a [GdmHCl]1/2 = 3.9 ± 0.1 M (fluorescence), and [GdmHCl]1/2 = 4.0 ± 0.4 M (CD).
Stability and thermodynamic parameters at pH 4.0
Since it was not possible to obtain the ΔCp from far-UV CD measurements in the presence of GdmHCl, it was necessary to use other approaches. We have used the approach developed by Pace and Laurents (53). However, in Fur A, it was not possible to obtain thermal denaturation data in the absence of denaturant (see above), and then the extrapolated Tm value at 0 M GdmHCl was used, in combination with the values of ΔG obtained by chemical-denaturations at other temperatures.
Since the amounts of protein used in the fluorescence experiments were smaller than those in the CD measurements, the chemical-denaturations of Fur A at several temperatures (293–333 K) were followed by fluorescence at pH 4, where the [GdmHCl]1/2 was higher (see above). The m-values obtained were constant, within the error, in the temperature range explored (Fig. 8 A). Further, there was a good agreement between the data obtained from the isothermal chemical-denaturation measurements in fluorescence and those from the extrapolation of the thermal denaturation experiments in CD (Fig. 8 B). This finding validates the use of the LEM in the analysis of the data, and most importantly, indicates that the same unfolded state of Fur A is being probed by thermal and chemical-denaturation measurements using different biophysical techniques. Furthermore, the results with Fur A suggest that the modification of the Pace and Laurents approach can be used in those proteins that have either a tendency to precipitate at high temperatures or have a high Tm. A bell-shaped curve was observed when the [GdmHCl]1/2 and the ΔG values at each temperature were represented versus the temperature (Fig. 8 B). Fitting of the free-energy curve to Eqs. 5 and 6 yielded the ΔHm, ΔCp, and Tm of the thermal unfolding of Fur A. The temperature dependence of ΔG was consistent with a temperature-independent heat capacity change, ΔCp, of 0.8 ± 0.1 kcal mol−1 K−1, a Tm of 352 ± 1 K (which agrees with that determined previously by extrapolation of the thermal far-UV CD data), and a ΔHm of 53 ± 4 kcal mol−1.
FIGURE 8.

The thermodynamical parameters of the chemical denaturation of Fur A at pH 4. (A) Temperature dependence of the m-value from fluorescence measurements. The errors bars are fitting errors to the LEM. (B) The temperature dependence of the [GdmHCl]1/2 (left side, open squares) and ΔG (right side, solid squares) values. The errors bars are fitting errors to the LEM. The errors are larger at the higher temperatures, because the native baselines in the chemical-denaturation experiments were shorter. The line through the ΔG data is the fitting to Eq. 6. The value of the Tm (right side, where ΔG equals zero) was obtained from the extrapolation of thermal-denaturation experiments (Fig. 4 B). The ΔG values were obtained with the mean of the m-value over all the temperatures (1.3 ± 0.1 kcal mol−1 M−1). The temperature dependence of ΔG was consistent with a temperature-independent heat capacity change, ΔCp, of 0.8 ± 0.1 kcal mol−1 K−1.
DISCUSSION
Structure and pH-induced structural changes of Fur A
Fur A showed a conformational transition with a pKa of 7.6 ± 0.3 (the average of the values measured), as detected by intrinsic fluorescence (Fig. 1 A), CD (Fig. 2), and ANS-fluorescence (Fig. 1 B). The fact that the wavelength of the main band of the near-UV was also affected, together with the observation that the same titration fluorescence curve (Fig. 1 A) was observed when the protein was excited at 295 nm and 280 nm, suggest that Trp18 was monitoring the conformational changes. The titration should involve deprotonation, in principle, of a histidine or cysteine, as suggested by the value of the measured pKa (48,49). However, the experimentally determined pKa (7.6) is closer to the random-coil value of histidine (6.5) than that of a cysteine residue (9.0) (48,49); interestingly enough, similar pKa values for titration of histidine residues have been described in the Fur from E. coli (54). Furthermore, since the closest cysteine residue (Cys133) is 25 Å from the indole moiety, we favor the presence of a histidine as the most plausible explanation for the titration observed. However, there were no histidine residues in the neighborhood of Trp18 (see Fig. 6 for details), and then, we hypothesize that the titration curve could be due to one of the following reasons: 1), there is a shift in the expected pKa of one (or more) of the surrounding lysine, arginine, and/or glutamic residues to Trp18; and 2), the titration is associated to a histidine(s) which cause conformational changes in a region far away from Trp18, but those changes are propagated along the polypeptide chain and they are finally felt by the indole moiety. Since all the titrating residues within a sphere of 12 Å from the Trp18 (Lys10, Glu12, Glu15, Arg16, Arg19, Arg24, Glu25, Tyr62, Arg63, Lys66, and Arg70) are solvent-exposed in the modeled Fur A, and then, the titration midpoints of their side chains should be close to those of random-coil values (4.5 for glutamic, 10.4 for lysine, and ∼12 for arginine residues (48,49)), we did not favor the first proposed hypothesis. Therefore, the more reasonable explanation to understand the entire experimental and computational set of data is the second hypothesis. There are several pieces of evidence that seem to support this argument. Firstly, the closest histidine amino acids to Trp18 are Tyr46-His47 and His85-Tyr86, which are located at 21 Å and 16 Å, respectively; further, the presence of the tyrosine residues could also explain the changes in near-UV (Fig. 2 B). Moreover, the distance between the pair His85-Tyr86 and Trp18 is similar to the largest distance reported in the literature for long-range electrostatic interactions (see (52) and references therein). Secondly, the proteolysis experiments suggest that the rearrangements occur along the whole polypeptide chain, since all the digestion sites of trypsin (Arg and Lys residues) were more solvent-accessible after transition occurred. We can speculate that the rearrangements might disrupt the four-helix bundle at the N-terminal domain, thus solvent-exposing the whole DNA-binding site of Fur A. Whatever the exact nature of the rearrangements, these were so dramatic that they precluded the determination of the chemical-denaturation parameters at pH values close to the titration midpoint.
The tertiary and secondary structure of Fur A also changed at acidic and basic pH values, due to acidic and basic denaturation. None of these changes were detected by ANS-binding experiments, which suggests that upon acidic or basic unfolding the solvent-exposed hydrophobic patches were not close enough to bind the fluorescent probe. Then, it seems, from the CD (Fig. 2) and fluorescence results (Fig. 1), that the structure of Fur A remained unaltered between pH 4 and 7, but outside this interval, the structure was altered by the basic and acid denaturation, and further by the conformational transition around physiological pH.
The CD spectral data were deconvolved by using neural networks (26) to yield a 43% of α-helix at pH 7. A simpler analysis, which takes into account the ellipticity at 222 nm (27), led to a 37% of helical structure. Both percentages agree quite well with the value obtained by FTIR deconvolution: 40%. It is interesting to compare these values with the percentages of secondary structure observed in other members of the Fur family. At the best of our knowledge, only the crystal structures of PA-Fur (22) and that of Rhizobium leguminosarum (55) have been resolved at pH 7. Both structures are similar and show dimeric species, where each monomer is composed of two domains (see Results). In the monomer of PA-Fur, 60 amino acids (i.e., 44%), out of 135, are involved in α-helical structure (22), and a similar percentage (41%) is observed in Fur from R. leguminosarum. These findings suggest that in other members of the Fur family the percentage of helical structure will remain the same, probably because that structure is necessary for the proper function of the protein: binding of DNA through the helical regions (3). The experimental resolution of the three-dimensional structure of other members of the Fur family, in the DNA- bound and free forms, will validate these hypotheses.
The folding of Fur A
Two equilibrium unfolding mechanisms have been described in oligomeric proteins, exposed to high temperatures or high concentrations of chemical denaturants (50,51): 1), dissociation followed by unfolding of the native or partially unfolded species; and 2), dissociation occurring concomitantly with unfolding of the monomers. In this work, we investigated the energetics of dimeric Fur A by thermal and chemical-denaturation. Fur A showed a single sigmoidal transition in the explored concentration range from 0.5 (fluorescence) to 60 μM (far-UV CD) at most pH values. This indicates that dissociation occurred concomitantly to monomer denaturation, and then, Fur followed the second unfolding mechanism.
We observed a protein-concentration dependence in the measured thermodynamic parameters either in the chemical (Fig. 7) and thermal denaturations (Fig. 4). A protein concentration-dependence should be always observed in a second-order process according to the rules of Thermodynamics. However, in a dissociation reaction, the lower the dissociation constant is, the smaller the protein concentration-dependence observed at the standard concentrations used in the biophysical techniques (i.e., in the range of μM). This tendency has been experimentally described and discussed for: 1), the thermal unfolding transition of the tetrameric SecB (56) (whose dissociation constant is in the order of nM), where at protein concentrations in the range of 50–60 μM, the variations in Tm are of 1°C; and, 2), the GdmHCl chemical-denaturation of the factor-for-inversion-stimulation protein (FIS, whose dissociation constant is in the order of pM; see (57)), where the differences among [GdmHCl]1/2-values, in the protein concentration range 1–10 μM, are <0.1 M. In the Fur family, there are not measurements of the dissociation constant of the dimeric species, but since the affinity of the active dimeric species for DNA is in the nM range (58,59), it is reasonable to assume that the dissociation constant for the formation of the dimeric protein is in the nanomolar-to-picomolar range. Then, this would imply that the variation in the thermal or chemical denaturation midpoints of Fur A, in the μM range of protein concentration, should be very small. Our chemical-denaturation experimental data, due to the steepness of the unfolding and folding baselines, had an experimental uncertainty of 0.3 M in the [GdmHCl]1/2-values (Fig. 7), thus precluding any reliable conclusion; conversely, the thermal denaturation data clearly showed a protein-concentration dependence in Tm (Fig. 4 C). However, it could be argued that, as it happens in the dimerization domain of the HIV-1 (60), some of the biophysical probes were spectroscopically silent to dimer chemical-dissociation (because the monomer has essentially the same structure in the monomeric or dimeric species), and then a non-concentration-dependence behavior in the [GdmHCl]1/2-value would be observed. Although we cannot rule that either fluorescence or CD were spectroscopically silent to Fur A chemical dissociation, the steepness of the baselines at any of the concentrations explored (0.5–50 μM) make us favor the experimental uncertainty as the most plausible reason of the impossibility of determining an unambiguous protein concentration-dependence in the [GdmHCl]1/2-values.
Conformational stability versus high thermal- and chemical-denaturation midpoints
Although the conformational stability of Fur A at 298 K and pH 4 was not very high (5.3 ± 0.3 kcal mol−1), the protein showed a remarkable stability upon thermal- (i.e., a high Tm) and chemical-denaturation (i.e., a large [GdmHCl]1/2). Then, a question can be raised, where did this high stability come from? This feature can be explained by the small values of m- and ΔCp.
The thermal stability of a protein can be attained either by a large maximum in the ΔG value, or by a low ΔCp (or by both reasons together); either factor makes the free energy curve intercept the x axis (the T axis in Fig. 8 B) at high values. In Fur A, the intercept with the x axis was high (Fig. 8 B) not because of the large stability of the protein (that is, not because there was a large ΔG maximum), but because of its very low ΔCp (0.8 ± 0.1 kcal mol−1 K−1). A similar explanation has been suggested to account for the high thermal midpoint values of hyperthermophilic proteins (61,62).
The m-value and ΔCp are both measurements of the difference in solvent-accessible surface area between the unfolded and native state. However, whereas ΔCp comes from the release of water when nonpolar groups are buried, the m-value indicates the difference in the number of unspecific denaturant binding sites between both states (63–66). In Fur A, the m-value was smaller than that of most globular proteins with a similar number of residues and following a two-state folding mechanism (65); furthermore, the ΔCp was also rather low for a protein of similar size (29,65). These deviations suggest that factors other than simple burial of the hydrophobic surface must contribute to the values of m and ΔCp. For instance, it has been suggested that: 1), proteins with an elongated shape have lower values of m, because of higher solvent exposure in the native state (52); 2), electrostatic interactions can reduce the value of ΔCp (61,62,67); and 3), the presence of residual structure in the unfolded state can also contribute to decrease the value of ΔCp (68). We can rule out the first explanation, since the dimeric Fur A had a spherical shape, as suggested by the DOSY-NMR measurements (Fig. 5). The exact value of the contribution of the desolvation of polar groups to ΔCp can be estimated based on the modeled structure (Fig. 6). For instance, one of the two buried cores is formed by polar residues, namely His39, His85, Glu87, His96, His98, and Glu109; further, a water molecule can be trapped within the cavity defined by residues His39, Glu87, His96, and Glu109. Then, it is tempting to suggest that desolvation of those polar residues, as it happens in the ribosomal protein L30e (61), might make a large positive contribution to the value of ΔCp . These findings suggest that the burial of charged side chains is an alternative to reduce the value of ΔCp not only in thermophilic proteins (61) but also in mesophilic ones. However, with the techniques described in this work, we cannot rule out that the presence of residual structure in the unfolded state of Fur A could also contribute to reduce the value of ΔCp. Thus, it is possible that the last two explanations contribute jointly to the decrease of ΔCp.
Those features can be used in the design of Fur A mutants with increased thermal stability and impaired dimerization capabilities, and then, according to the proposed model of the Fur function (1–3) (where dimer formation is necessary for DNA binding), impaired DNA-binding ability. Assuming that the main contribution to ΔCp comes from the amount of nonpolar surface exposed upon unfolding, the thermal stability of the protein could be enhanced by conservative mutations designed to increase the ratio of polar to nonpolar area buried in the folded state of the protein, while keeping mostly unaffected their intrinsic stability. If those mutants were also involved in the DNA-binding interface, they could be used as regulators of the wild-type Fur activity. These hypotheses are being further investigated in our laboratories by using protein-engineering techniques.
Acknowledgments
The authors declare that they do not have any competing interest.
We thank Miguel R. Moreno for his help in acquisition of FTIR experiments. We deeply thank May García, María del Carmen Fuster, Javier Casanova, Helena López, Eva Hernández, and María T. Garzón for excellent technical assistance. We thank the two anonymous reviewers and the editor handling the manuscript for their insights and suggestions.
This work was supported by grants from Ministerio de Sanidad y Consumo (No. FIS 01/0004-02); Ministerio de Educación y Ciencia (No. CTQ2004-04474); Generalitat Valenciana (No. GV04B-402); and by an institutional grant by URBASA to J.L.N.; and from Ministerio de Educación y Ciencia (No. BM2000-1001) and the Training and Mobility of Researchers program (No. ERB-4001GT963549) to M.F.F. and M.L.P. J.A.H. and F.N.B. were supported by two predoctoral “Formación de Profesorado Universitario” fellowships (from Ministerio de Educación y Ciencia). E.H.G. was the recipient of a predoctoral fellowship from Generalitat Valenciana.
This article is dedicated to the memory of Jörg Meier, who passed away in August 2005.
José A. Hernández and José L. Neira contributed equally to this work.
José A. Hernández's present address is Plant and Microbiological Biology Department, 211 Koshland Hall, University of California at Berkeley; Berkeley, CA 94720.
References
- 1.Braun, V., and H. Killmann. 1999. Bacterial solutions to the iron supply problem. Trends Biochem. Sci. 24:104–109. [DOI] [PubMed] [Google Scholar]
- 2.Litwin, C. M., and S. B. Calderwood. 1993. Role of iron in regulation of virulence genes. Clin. Microbiol. Rev. 6:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ratledge, C., and L. G. Dover. 2000. Iron metabolism in pathogenic bacteria. Annu. Rev. Microbiol. 54:881–941. [DOI] [PubMed] [Google Scholar]
- 4.Escolar, L., V. de Lorenzo, and J. Pérez-Martín. 1997. Metalloregulation in vitro of the aerobactin promoter of Escherichia coli by the Fur (ferric uptake regulator) protein. Mol. Microbiol. 26:799–808. [DOI] [PubMed] [Google Scholar]
- 5.Dubrac, S., and D. Touati. 2000. Fur positive regulation of iron superoxide dismutase in Escherichia coli: functional analysis of the sodB promoter. J. Bacteriol. 182:3802–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zahrt, T. C., J. Song, J. Siple, and V. Deretic. 2001. Mycobacterial FurA is a negative regulator of catalase-peroxidase gene katG. Mol. Microbiol. 39:1174–1185. [DOI] [PubMed] [Google Scholar]
- 7.Hall, H. K., and J. W. Foster. 1996. The role of Fur in the acid tolerance response of Salmonella typhimurium is physiologically and genetically separable from its role in iron acquisition. J. Bacteriol. 178:5683–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Escolar, L., J. Pérez-Martín, and V. de Lorenzo. 1999. Opening the iron box: transcriptional metalloregulation by the Fur protein. J. Bacteriol. 181:6223–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stojiljkovic, I., A. J. Baumler, and K. Hantke. 1994. Fur regulation in Gram-negative bacteria. Identification and characterization of new iron-regulated Escherichia coli genes by a fur titration assay. J. Mol. Biol. 236:531–545. Erratum in 1994. J. Mol. Biol. 240:271. [DOI] [PubMed] [Google Scholar]
- 10.Horsburgh, M. J., E. Ingham, and S. J. Foster. 2001. In Staphylococcus aureus, Fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J. Bacteriol. 183:468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massé, E., and S. Gottesman. 2002. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl. Acad. Sci. USA. 99:4620–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaballa, A., and J. D. Hellmann. 1998. Identification of a zinc-specific metalloregulatory protein Zur, controlling zinc transport operon in Bacillus subtilis. J. Bacteriol. 180:5815–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bsat, N., A. Herbig, L. Casillas-Martínez, P. Setlow, and J. D. Hellman. 1998. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulation. Mol. Microbiol. 29:189–198. [DOI] [PubMed] [Google Scholar]
- 14.Qi, Z., I. Hamza, and M. R. O'Brian. 1999. Heme is an effector molecule for iron-dependent degradation of the bacterial iron response regulator (Irr) protein. Proc. Natl. Acad. Sci. USA. 96:13056–13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stojiljkovic, I., and K. Hantke. 1995. Functional domains of the Escherichia coli ferric uptake regulator protein. Mol. Gen. Genet. 247:199–205. [DOI] [PubMed] [Google Scholar]
- 16.Ghassemian, M., and N. A. Strauss. 1996. Fur regulates the expression of iron-stress genes in cyanobacterium Synechococcus sp. strain PCC 7942. Microbiology. 142:1469–1476. [DOI] [PubMed] [Google Scholar]
- 17.Kaneko, T., S. Sato, H. Kotani, A. Tanaka, E. Asamizu, Y. Nakamura, N. Miyajima, M. Hirosawa, M. Sugiura, S. Sasamoto, T. Kimura, T. Hosouchi, A. Matsuno, A. Muraki, N. Nakazaki, K. Naruo, S. Okumura, S. Shimpo, C. Takeuchi, T. Wada, A. Watanabe, M. Yamada, M. Yasuda, and S. Tabata. 1996. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 3:109–136. [DOI] [PubMed] [Google Scholar]
- 18.Bes, M. T., J. A. Hernández, M. L. Peleato, and M. F. Fillat. 2001. Cloning, overexpression and interaction of recombinant Fur from the cyanobacterium Anabaena PCC 7119 with isiB and its own promoter. FEMS Microbiol. Lett. 194:187–192. [DOI] [PubMed] [Google Scholar]
- 19.Lukac, M., and R. Aegerter. 1993. Influence of trace metals on growth and toxin production of Microcystis aeruginosa. Toxicon. 31:293–305. [DOI] [PubMed] [Google Scholar]
- 20.Boyd, P. W., A. J. Watson, C. S. Law, E. R. Abraham, T. Trull, R. Murdoch, D. C. Bakker, A. R. Bowie, K. O. Buesseler, H. Chang, M. Charette, P. Croot, K. Downing, R. Frew, M. Gall, M. Hadfield, J. Hall, M. Harvey, G. Jameson, J. LaRoche, M. Liddicoat, R. Ling, M. T. Maldonado, R. M. McKay, S. Nodder, S. Pickmere, R. Pridmore, S. Rintoul, K. Safi, P. Sutton, R. Strzepek, K. Tanneberger, S. Turner, A. Waite, and J. Zeldis. 2000. A mesoscale phytoplankton bloom in the polar southern ocean stimulated by iron fertilization. Nature. 407:695–702. [DOI] [PubMed] [Google Scholar]
- 21.Hernández, J. A., M. T. Bes, M. F. Fillat, J. L. Neira, and M. L. Peleato. 2002. Biochemical analysis of the recombinant Fur (ferric uptake regulator) protein from Anabaena PCC 7119: factors affecting its oligomerization state. Biochem. J. 366:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pohl, E., J. C. Haller, A. Mijovilovich, W. Meyer-Klaucke, E. Garman, and M. L. Vasil. 2003. Architecture of a protein central to iron homeostasis: crystal structure and spectroscopic analysis of the ferric uptake regulator. Mol. Microbiol. 47:903–915. [DOI] [PubMed] [Google Scholar]
- 23.Pace, C. N., and J. M. Scholtz. 1997. Measuring the conformational stability of a protein. In Protein Structure, 2nd Ed. T.E. Creighton, editor. Oxford University Press, Oxford, UK. 253–259.
- 24.Mann, C. J., and C. R. Matthews. 1993. Structure and stability of an early folding intermediate of Escherichia coli Trp aporepressor measured by far-UV stopped-flow circular dichroism and 8-anilino-1-naphthalene sulfonate binding. Biochemistry. 32:5282–5290. [DOI] [PubMed] [Google Scholar]
- 25.Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy, 2nd Ed. Plenum Press, New York.
- 26.Andrade, M. A., P. Chacón, J. J. Merelo, and F. Morán. 1993. Evaluation of secondary structure of proteins from UV circular dichroism using an unsupervised learning neural network. Protein Eng. 6:383–390. [DOI] [PubMed] [Google Scholar]
- 27.Zurdo, J., J. M. Sanz, C. González, M. Rico, and J. P. G. Ballesta. 1997. The exchangeable yeast ribosomal acidic protein YP2β shows characteristics of a partly folded state under physiological conditions. Biochemistry. 36:9625–9635. [DOI] [PubMed] [Google Scholar]
- 28.Royer, C. A. 1995. Fluorescence spectroscopy. In Protein Stability and Folding. B.A. Shirley, editor. Humana Press, Totowa, NJ. 65–89.
- 29.Backmann, J., G. Schäfer, L. Wyns, and H. Bönisch. 1998. Thermodynamics and kinetics of unfolding of the thermostable trimeric adenylate kinase from the archeon Sulfolobus acidocaldarius. J. Mol. Biol. 284:817–833. [DOI] [PubMed] [Google Scholar]
- 30.Martin, M. L., J.-J. Delpuech, and G. J. Martin. 1980. Practical NMR Spectroscopy. Heyden, London, UK. 330–341.
- 31.Price, W. S. 1997. Pulse-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: part I. Basic theory. Concepts Magn. Reson. 9:299–336. [Google Scholar]
- 32.Price, W. S. 1998. Pulse-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: part II. Experimental aspects. Concepts Magn. Reson. 10:197–237. [Google Scholar]
- 33.Lapham, J., J. P. Rife, P. B. Moore, and D. M. Crothers. 1997. Measurement of diffusion constants for nucleic acids by NMR. J. Biomol. NMR. 10:255–262. [DOI] [PubMed] [Google Scholar]
- 34.Stejskal, E. O., and J. E. Tanner. 1965. Spin diffusion measurements: spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 42:288–292. [Google Scholar]
- 35.Barth, A., and C. Zscherp. 2002. What vibrations tell us about proteins. Q. Rev. Biophys. 35:369–430. [DOI] [PubMed] [Google Scholar]
- 36.Jackson, M., and H. H. Mantsch. 1995. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 30:95–120. [DOI] [PubMed] [Google Scholar]
- 37.Sipos, T., and J. R. Merkel. 1970. An effect of calcium ions on the activity, heat, stability and structure of trypsin. Biochemistry. 9:2766–2775. [DOI] [PubMed] [Google Scholar]
- 38.Molsoft LLC. ICM Manual, 3rd Ed. Molsoft LLC, La Jolla, CA.
- 39.Nemethy, G., K. D. Gibson, K. A. Palmer, C. N. Yoon, M. G. Paterlini, A. Zagari, S. Rumsey, and H. A. Scheraga. 1992. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J. Phys. Chem. 96:6472–6484. [Google Scholar]
- 40.Abagyan, R., and M. Totrov. 1994. Biased probability Monte-Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235:983–1002. [DOI] [PubMed] [Google Scholar]
- 41.Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21:1087–1092. [Google Scholar]
- 42.Stryer, L. 1965. The interaction of a naphthalene sulfonate dye with apomyoglobin and apohemoglobin: a fluorescent probe of non-polar binding site. J. Mol. Biol. 13:482–495. [DOI] [PubMed] [Google Scholar]
- 43.Semisotnov, G. V., N. A. Rodionova, O. I. Razgulyaev, V. N. Uversky, A. F. Gripas, and R. I. Gimanshin. 1991. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 31:119–128. [DOI] [PubMed] [Google Scholar]
- 44.Woody, R. W. 1995. Circular dichroism. Methods Enzymol. 246:34–71. [DOI] [PubMed] [Google Scholar]
- 45.Kelly, S. M., and N. C. Price. 2000. The use of circular dichroism in the investigation of protein structure and function. Curr. Prot. Peptide Sci. 1:349–384. [DOI] [PubMed] [Google Scholar]
- 46.Vuillumier, S., J. Sancho, R. Loewenthal, and A. R. Fersht. 1993. Circular dichroism studies of barnase and its mutants: characterization of the contribution of aromatic side chains. Biochemistry. 32:10303–10313. [DOI] [PubMed] [Google Scholar]
- 47.Freskgärd, P. O., L. G. Märtensson, P. Jonasson, B. H. Jonsson, and U. Carlsson. 1994. Assignment of the contribution of the tryptophan residues to the circular dichroism spectrum of human carbonic anhydrase II. Biochemistry. 33:14281–14288. [DOI] [PubMed] [Google Scholar]
- 48.Cantor, C. R., and P. R. Schimmel. 1980. Biophysical Chemistry. W. H. Freeman and Company, New York.
- 49.Creighton, T. E. 1993. Proteins. Structures and Macromolecular Properties, 2nd Ed. W. H. Freeman, New York.
- 50.Neet, K. E., and D. E. Timm. 1994. Conformational stability of dimeric proteins: quantitative studies by equilibrium denaturation. Protein Sci. 3:2167–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaenicke, R., and H. Lillie. 2000. Folding and association of oligomeric and multimeric proteins. Adv. Protein Chem. 53:329–401. [DOI] [PubMed] [Google Scholar]
- 52.Fersht, A. R. 1999. Structure and Mechanism in Protein Science. A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman, New York.
- 53.Pace, C. N., and D. V. Laurents. 1989. A new method for determining the heat capacity change for protein folding. Biochemistry. 28:2520–2525. [DOI] [PubMed] [Google Scholar]
- 54.Saito, T., D. Duly, and R. J. P. Williams. 1991. The histidines of the iron-uptake regulator protein. Eur. J. Biochem. 197:39–42. [DOI] [PubMed] [Google Scholar]
- 55.Kolade, O. O., P. Bellini, M. Wexler, A. W. B. Johnston, J. G. Grossmann, and A. M. Hemmings. 2002. Structural studies of the fur protein from Rhizobium leguminosarum. Biochem. Soc. Trans. 30:771–774. [DOI] [PubMed] [Google Scholar]
- 56.Panse, V. G., C. P. Swaminatham, J. J. Aloor, A. Surolia, and R. Varadarajan. 2000. Unfolding thermodynamics of the tetrameric chaperone SecB. Biochemistry. 39:2362–2369. [DOI] [PubMed] [Google Scholar]
- 57.Topping, T. B., D. A. Hoch, and L. M. Gloss. 2004. Folding mechanism of FIS, the intertwined, dimeric factor for inversion stimulation. J. Mol. Biol. 335:1065–1081. [DOI] [PubMed] [Google Scholar]
- 58.Althaus, E. W., C. E. Outten, K. E. Olson, H. Cao, and T. V. O'Halloran. 1999. The ferric uptake regulation (Fur) repressor is a Zn-regulated protein. Biochemistry. 38:6559–6569. [DOI] [PubMed] [Google Scholar]
- 59.Zelezhnova, E. E., J. H. Crosa, and R. G. Brennan. 2000. Characterization of the DNA- and metal-binding properties of Vibrio angillarum Fur reveals conservation of a Zn2+ ion. J. Bacteriol. 182:6262–6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mateu, M. G. 2002. Conformational stability of dimeric and monomeric forms of the C-terminal domain of human immunodeficiency virus-1 capsid protein. J. Mol. Biol. 318:519–531. [DOI] [PubMed] [Google Scholar]
- 61.Lee, C.-F., M. D. Allen, M. Bycroft, and K.-B. Wong. 2005. Electrostatic interactions contribute to reduce heat capacity change of unfolding in a thermophilic ribosomal protein L30e. J. Mol. Biol. 348:419–431. [DOI] [PubMed] [Google Scholar]
- 62.Rees, D. C., and A. D. Robertson. 2001. Some thermodynamic implications for the thermostability of proteins. Protein Sci. 10:1187–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spolar, R. S., J. R. Livingstone, and M. T. Record. 1992. Use of liquid hydrocarbon and amide transfer data to estimate contributions to thermodynamic functions of protein folding from the removal of nonpolar and polar surface from water. Biochemistry. 31:3947–3955. [DOI] [PubMed] [Google Scholar]
- 64.Makhatadze, G. I., and P. L. Privalov. 1990. Heat capacity of proteins. I. Partial molar heat capacity of individual amino acids in aqueous solution: hydration effect. J. Mol. Biol. 213:375–384. [DOI] [PubMed] [Google Scholar]
- 65.Myers, J. K., C. N. Pace, and J. M. Scholtz. 1995. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4:2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Otzen, D. E., and M. Oliveberg. 2004. Correspondence between anomalous m- and ΔCp-values in protein folding. Protein Sci. 13:3253–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mogensen, J. E., H. Ipsen, J. Holm, and D. E. Otzen. 2004. Elimination of a misfolded folding intermediate by a single point mutation. Biochemistry. 43:3357–3367. [DOI] [PubMed] [Google Scholar]
- 68.Robic, S., M. Guzmán-Casado, J. M. Sánchez-Ruiz, and S. Marqusee. 2003. Role of residual structure in the unfolded state of a thermophilic protein. Proc. Natl. Acad. Sci. USA. 100:11345–11349. [DOI] [PMC free article] [PubMed] [Google Scholar]