

Abstract
In 5% (v/v) trifluoroethanol, pH 5.5, 25°C one of the acylphosphatases from Drosophila melanogaster (AcPDro2) forms fibrillar aggregates that bind thioflavin T and Congo red and have an extensive β-sheet structure, as revealed by circular dichroism. Atomic force microscopy indicates that the fibrils and their constituent protofilaments have diameters compatible with those of natural amyloid fibrils. Spectroscopic and biochemical investigation, carried out using near- and far-UV circular dichroism, intrinsic and 1-anilino-8-naphthalenesulfonic acid-derived fluorescence, dynamic light scattering, and enzymatic activity assays, shows that AcPDro2 has, before aggregation, a secondary structure content packing around aromatic and hydrophobic residues, hydrodynamic diameter, and catalytic activity indistinguishable from those of the native protein. The native protein was found to have the same conformational stability under native and aggregating conditions, as determined from urea-induced unfolding. The kinetic analysis supports models in which AcPDro2 aggregates initially without need to unfold and subsequently undergoes a conformational change into amyloid-like structures. Although fully or partially unfolded states have a higher propensity to aggregate, the residual aggregation potential that proteins maintain upon complete folding can be physiologically relevant and be directly involved in the pathogenesis of some protein deposition diseases.
INTRODUCTION
A number of pathological conditions are associated with the conversion of a specific protein or peptide from its soluble state into well organized fibrillar aggregates generally referred to as amyloid or amyloid-like fibrils (1–4). These fibrils generally consist of 2–6 protofilaments, each ∼2–5 nm in diameter, that generally twist together to form fibrils that are typically 7–13 nm wide (5,6). In each individual protofilament the protein molecules are arranged to form β-strands that run perpendicular to the long axis of the fibril or protofilament (5,7). The fibrils have the ability to bind specific dyes such as thioflavin T (ThT) and Congo red (CR) (8–10), although the specificity of binding of CR to amyloid and the resulting yellow-green birefringence has recently been questioned (11,12).
In some of the pathological conditions associated with formation of amyloid-like structures the precursor protein is normally folded, in its soluble state, into a globular unit with persistent secondary structure and long-range interactions. Examples include β2-microglobulin, transthyretin, lysozyme, immunoglobulin light chain, superoxide dismutase, ataxin-3, cystatin c, and gelsolin (3,13,14). Such proteins spend most of their lifetime in a folded state, but can transiently adopt a partially or totally unfolded state, for example during biosynthesis, translocation or stress conditions. Partially or totally unfolded states can also be in equilibrium with the native state even under nonstress conditions; this may provide a pool of protein molecules that can initiate and propagate the aggregation process, especially when the native structure is destabilized by mutations that shift the equilibrium toward the unfolded state (14,15).
A major question that needs to be answered is whether amyloid formation is initiated by such partially or totally unfolded states that are only occasionally populated, or whether it directly involves the native state. The observations that unfolded states have a greater propensity than folded states to aggregate, and that amyloid formation is often associated with genetic mutations that can effectively destabilize the native state, have led to the proposal that a conformational change is the first essential step in amyloidogenesis, at least for those diseases associated with such globular proteins (3,16,17). However, recent observations have questioned the generality of this mechanism. The pathogenic variant of ataxin-3, the protein associated with spinocerebellar ataxia type-3, does not appear to have a destabilized native structure, which leads to the proposal that the pathway for fibril formation is distinct from that of unfolding (18). Amyloid formation by insulin is preceded by an oligomerization of the protein in which a native-like content of α-helical structure is retained almost completely (19). Within a group of variants of the protein S6 from Thermus thermophilus, no significant correlation was found between the rate of fibril formation, under conditions in which a quasinative state was populated before aggregation, and the unfolding rate or conformational stability (20). Aggregation of the acylphosphatase from Sulfolobus solfataricus into amyloid protofibrils is more rapid than unfolding under conditions in which the native state is thermodynamically more stable than the major partially unfolded state (21). All of these observations suggest that an association of protein molecules in their native-like states can be the first event in the aggregation of some globular proteins, with the structural conversion into an amyloid conformation occurring subsequently.
The study of amyloid formation under solution media in which the protein is initially in a native-like conformation is therefore of vital importance to understand aggregation pathways that may occur in vivo. Our ability to distinguish between mechanisms in which partial or full unfolding is a first step and others in which oligomerization precedes a structural reorganization is important not just to clarify the pathogenesis of specific protein deposition diseases, but also to recognize the dangerous pathways from which proteins generally need to escape to remain soluble in vivo. In this work, we investigate the aggregation mechanism of a novel acylphosphatase from Drosophila melanogaster (AcPDro2), a 102-residue protein domain having a ferredoxin-like topology in its native state (22,23). We will show that AcPDro2 self-assembles under conditions in which the native structure is highly populated and has a conformation no less stable than that measured under native conditions in which aggregation is not observed. The kinetic data rule out models in which a partial unfolding of the protein is required to start aggregation.
MATERIALS AND METHODS
Materials
Thioflavin T (ThT), trifluoroethanol (TFE), dithiothreitol (DTT), urea, Congo red (CR) and 8-anilino-1-naphthalenesulfonic acid (ANS) were purchased from Sigma-Aldrich (St. Louis, MO). Trifluoroacetic acid was purchased from Biosolve (Valkenswaard, The Netherlands). Formic acid was purchased from Merck (Darmstadt, Germany). Benzoylphosphate was synthesized and purified as described (24). AcPDro2 was expressed and purified as described previously (22) and stored at 4°C in 5 or 50 mM acetate buffer, 2 mM DTT, pH 5.5. Protein concentration was determined spectrophotometrically using an ɛ280 value of 1.09 ml mg−1 cm−1. Before starting each experiment, the protein solution was centrifuged at 18,000 rpm for 3–5 min and filtered using 0.02 μm Anotop 10 filters (Whatman International, Maidstone, UK).
ThT assay
AcPDro2 was incubated at a concentration of 0.4 mg ml−1 in 50 mM acetate buffer, 2 mM DTT, pH 5.5, at 25°C in the presence of concentrations of TFE ranging from 0 to 20% (v/v). At regular times 60 μl aliquots of each sample were added to 440 μl of a solution containing 25 μM ThT, 25 mM phosphate buffer, pH 6.0. The steady-state fluorescence values of the resulting samples were measured at 25°C using a 2 × 10-mm-pathlength quartz cuvette and a Perkin-Elmer (Wellesley, MA) LS 55 spectrofluorimeter equipped with a thermostated cell holder attached to a Haake (Karlsruhe, Germany) F8 water bath. The excitation and emission wavelengths were 440 and 485 nm, respectively (9). ThT fluorescence was plotted versus time and fitted to single exponential functions of the form
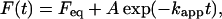 |
(1) |
where F(t) is the ThT fluorescence at time t, Feq is the maximum ThT fluorescence obtained at the end of the observed exponential phase, A is the amplitude of the fluorescence change, and kapp is the apparent rate constant. Another set of data was collected after incubating the protein in 50 mM formiate buffer, 5% (v/v) TFE, 25 mM NaCl, 2 mM DTT, pH 3.5, at 25°C.
CR assay
AcPDro2 was incubated at a concentration of 0.4 mg ml−1 for ∼60 h in 50 mM acetate buffer, 2 mM DTT, pH 5.5, at 25°C, in the presence of 0 or 5% (v/v) TFE. In both cases, a 60 μl aliquot was mixed with a 440-μl solution containing 20 μM CR, 5 mM phosphate buffer, 150 mM NaCl, pH 7.4, in a reduced volume, 5-mm-pathlength quartz cuvette. Corresponding solutions of CR without protein and protein with no CR were used as controls. Optical absorption spectra were acquired from 400 to 700 nm by an Ultrospec 2000 spectrophotometer (Pharmacia Biotech, Cambridge, UK). The spectrum obtained with buffer only was subtracted from all acquired spectra.
Tapping mode atomic force microscopy
AcPDro2 was incubated at a concentration of 0.4 mg ml−1 in 50 mM acetate buffer, 5% (v/v) TFE, 2 mM DTT, pH 5.5, 25°C. Aliquots of 20 μl were withdrawn at various times, deposited on freshly cleaved mica substrates and dried under mild vacuum for 30 min. Tapping mode atomic force microscopy (TM-AFM) images were acquired in air using a Dimension 3000 microscope (Digital Instruments, Veeco, Santa Barbara, CA) equipped with a “G” scanning head (maximum scan size 100 μm). Single-beam uncoated silicon cantilevers were used (type RTESP, Veeco, Santa Barbara, CA). The drive frequency was ∼300 kHz; the scan rate was between 0.3 and 0.7 Hz.
Dynamic light scattering
Samples were prepared at final AcPDro2 concentrations ranging from 0.3 to 1.0 mg ml−1 in 50 mM acetate buffer, 2 mM DTT, pH 5.5, at 25°C, in the presence of 0% and 5% TFE. Each protein sample was prepared by diluting a stocked protein sample and was immediately centrifuged (18, 000 rpm for 5 min) and filtered (0.02 μm) shortly before acquiring its size distribution to remove any preexisting aggregates and dust particles. A low-volume (45 μl) black quartz cuvette with 10 mm light path was used. Data were obtained with a Zetasizer Nano S dynamic light scattering (DLS) instrument (Malvern Instruments, Worcestershire, UK) setting the appropriate viscosity and refractive index parameters for each solution and keeping the temperature at 25°C during the measurements by means of a Peltier thermostating system.
Far-ultraviolet circular dichroism
Samples of AcPDro2 were prepared at final concentrations ranging from 0.01 to 0.8 mg ml−1 in 5 mM acetate buffer, 2 mM DTT, pH 5.5, at 25°C, in the presence of 0–5% and 80% TFE, or 5 M urea with no TFE. Far- ultraviolet (UV) circular dichroism (CD) spectra were acquired at 25°C immediately after diluting the stocked protein sample into these solutions. Quartz cuvettes with pathlengths ranging from 1 to 10 mm were used, depending on protein concentrations. A Jasco (Tokyo, Japan) J-810 spectropolarimeter equipped with a thermostated cell holder attached to a Thermo Haake C25P water bath was used. Each spectrum of the protein was recorded from 190 to 260 nm as the average of four scans, blank-subtracted, processed using the adaptive smoothing method (25) and normalized to mean residue ellipticity.
Near-UV CD
Samples of AcPDro2 were prepared at final protein concentrations of 0.2 mg ml−1 in 5 mM acetate buffer, 2 mM DTT, pH 5.5, at 25°C, in the presence of 0% and 5% (v/v) TFE, or 4.5 M urea. Near-UV CD spectra were acquired at 25°C from 250 to 350 nm, immediately after diluting the stocked protein sample into these solutions. A 10-mm-pathlength quartz cuvette and the CD instrument described in the Far-UV CD section were used. Due to the low signal/noise ratio, spectra for the protein samples in 0% TFE and in 4.5 M urea were recorded as the average of 40 separate scans, whereas 10 scans were averaged for the protein sample in 5% TFE. Each spectrum was blank-subtracted, processed using the adaptive smoothing method (25), and normalized to mean residue ellipticity.
Intrinsic fluorescence
AcPDro2 was diluted to a final concentration of 0.02 mg ml−1 in 5 mM acetate, 2 mM DTT, pH 5.5, at 25°C and in the presence of various denaturants. Fluorescence spectra were acquired immediately after dilution using the cuvette and instrumental apparatus described in the ThT assay section. Excitation wavelength was set at 280 nm and emission range was from 300 to 450 nm. Each spectrum was recorded as the average of two scans and smoothed using a moving average function with the statistical mean of seven points.
ANS fluorescence
AcPDro2 was diluted to a final concentration of 0.02 mg ml−1 in 55 μM ANS, 50 mM acetate, 2 mM DTT, pH 5.5, at 25°C. We prepared two samples containing 0 and 5% TFE, respectively. Fluorescence spectra were acquired immediately after dilution using the same cell and equipment described in the ThT assay section. Excitation wavelength was set at 380 nm and emission range was from 410 to 630 nm. Spectra were also acquired for the corresponding solutions in the absence of protein. Each spectrum was the average of two scans and was smoothed using a moving average function with the statistical mean of seven points. ANS concentration was determined spectrophotometrically using an ɛ375 value of 8000 ml mmol−1 cm−1.
Enzymatic activity
Enzymatic activity of AcPDro2 was tested in the presence of TFE concentrations ranging from 0 to 5%, using a final protein concentration of 0.5 μg ml−1 and 4 mM benzoylphosphate as a substrate, as described by Ramponi et al. (26). Other experimental conditions were 50 mM acetate buffer, 2 mM DTT, pH 5.5, at 25°C. Activity was measured with a Perkin-Elmer λ 4 B UV-Vis spectrophotometer. The noncatalyzed spontaneous hydrolysis of benzoylphosphate was subtracted from all measurements. Measurements were performed immediately after addition of TFE, without any incubation time.
Equilibrium unfolding
Unfolding of AcPDro2 was studied at equilibrium in the presence of different concentrations of TFE, ranging from 0 to 5% (v/v). For each TFE concentration 30 samples were prepared containing 0.02 mg ml−1 protein in 50 mM acetate buffer, 2 mM DTT, pH 5.5, and various urea concentrations ranging from 0.1 to 8 M. The samples were left to equilibrate for a few minutes at 25°C. Fluorescence spectra were acquired at 25°C using the cuvette and instrumental apparatus described in the ThT assay section, with a wavelength range between 300 and 450 nm and an excitation wavelength at 280 nm. Each spectrum was recorded as the average of two scans and smoothed using a moving average function with the statistical mean of seven points. The fluorescence at 348 nm was plotted versus urea concentration and the resulting plot was fitted to the equation described by Santoro and Bolen (27) to determine the free energy difference between the unfolded and folded states of the protein in the absence of urea (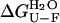 ), the dependence of ΔGU−F on urea concentration (m value) and the midpoint of unfolding (Cm).
), the dependence of ΔGU−F on urea concentration (m value) and the midpoint of unfolding (Cm).
Stopped-flow kinetics
Folding and unfolding of AcPDro2 were studied with a Bio-logic (Claix, France) SFM-3 stopped-flow device coupled to a fluorescence detection system and thermostated at 25°C with a Neslab (Newington, NH) RTE-200 water-circulating bath. The excitation wavelength was 280 nm, and the fluorescence emitted above 320 nm was monitored using a bandpass filter. All the experiments were performed in 50 mM acetate, 2 mM DTT, pH 5.5, at a final protein concentration of 0.02 mg ml−1. In a first set of unfolding and folding experiments the protein was initially in 0 and 6 M urea, respectively. Both reactions were initiated by 10-fold dilutions. Unfolding and folding kinetics were studied at final urea concentrations of 5.4 and 0.6 M, respectively, in the presence of TFE concentrations ranging from 0 to 8.25% (v/v). The dead time was 10.44 ms. The unfolding and refolding traces were fitted to single and double exponential functions, respectively, of the form
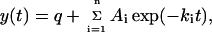 |
(2) |
where y(t) is the fluorescence at time t, q is the fluorescence value at equilibrium, Ai and ki are the amplitude and rate constant of the ith phase, respectively, and n is the number of observed phases. In a second set of experiments, the unfolding and folding kinetics were monitored at final urea concentrations ranging from 1.80 M to 7.20 M for unfolding and from 0.50 M to 3.20 M for folding. Finally, in a third set of experiments the protein was unfolded in 2 M urea, 10 mM trifluoroacetic acid, pH 2.0. Folding was initiated by a 20-fold dilution in 50 mm acetate buffer, pH 5.5, 2 mM DTT, 25°C, at final urea concentrations ranging from 0.1 to 1.8 M. These experiments were performed both in 0% and 5% (v/v) TFE. In the second and third sets of experiments the analysis was carried out at final TFE concentrations of 0 and 5% (v/v). The unfolding and refolding traces were fitted to Eq. 2 and the natural logarithm of ki was plotted versus urea concentration for both the analyses carried out in 0 and 5% TFE. The resulting plots were fitted using the following equation, edited by Jackson and Fersht (28):
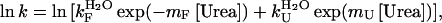 |
(3) |
where 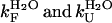 are the rate constants for folding and unfolding in the absence of urea, respectively and mF and mU are the dependencies of ln kF and ln kU on urea concentration, respectively.
are the rate constants for folding and unfolding in the absence of urea, respectively and mF and mU are the dependencies of ln kF and ln kU on urea concentration, respectively.
RESULTS
Formation of amyloid-like structures at low concentrations of TFE
AcPDro2 was incubated at a concentration of 0.4 mg ml−1 in 50 mM acetate buffer, 2 mM DTT, pH 5.5, at 25°C, at TFE concentrations ranging from 0 to 20% (v/v). After 2 and 24 h, the protein samples were subjected to the ThT assay (9). All protein samples incubated at TFE concentrations >4–5% (v/v) were able to increase significantly the fluorescence of ThT, whereas no significant aggregation was found to occur at lower TFE concentrations after 24 h (Fig. 1 a). Aggregation appears to be slightly lower in 20% than in 50% (v/v) TFE, with no significant differences in ThT binding after 2 h and 24 h (data not shown).
FIGURE 1.

(a) AcPDro2 aggregation measured by ThT fluorescence, after 2 h (○) and 24 h (•) of incubation in 0–20% (v/v) TFE. (b and c) CR assay on AcPDro2 aggregates. The protein was preincubated for ∼60 h in two separate samples containing 0% TFE (b) and 5% TFE (c). Spectra were acquired for protein in phosphate buffer (▴, “spectrum 1”), CR (•, “spectrum 2”), and protein in CR-phosphate buffer (▪, “spectrum 3”). The insets show the respective difference spectra, calculated by subtracting spectra 1 and 2 from spectrum 3. (d) Far-UV CD spectra of AcPDro2 in 5% TFE at protein concentrations of 0.4 (▴), 0.2 (•), and 0.1 mg ml−1 (▪). Spectra were acquired immediately after (solid lines) and 48 h after (dashed lines) incubation under these conditions.
The protein samples incubated for 60 h in 0 and 5% (v/v) TFE were also subjected to the CR assay (8). The spectra acquired for CR in the presence and absence of protein preincubated in buffer without TFE are superimposable (Fig. 1 b). The difference spectrum, obtained by subtracting the spectra of protein only and CR only from the spectrum of CR with protein, does not show a peak at 540 nm, indicating that the protein is not able to bind to the dye and cause the characteristic red shift from 490 to 540 nm (Fig. 1 b, inset). By contrast, the spectrum acquired for CR in the presence of protein preincubated in 5% (v/v) TFE exhibited a significant red shift relative to the spectrum of CR alone (Fig. 1 c). The higher absorbance values found in the former at all wavelengths arises from the ability of the protein aggregates to scatter the incident light, as shown by the spectrum acquired for the protein only in the absence of CR (Fig. 1 c). The difference spectrum, obtained as described above for the analysis in the absence of TFE, shows a peak at 540 nm, as expected for CR bound to ordered aggregates (Fig. 1 c, inset).
Secondary structure of the aggregates was monitored using far-UV CD (Fig. 1 d). Spectra were acquired using two sets of protein samples, each at 0.1, 0.2, and 0.4 mg ml−1 in 5% (v/v) TFE. Although the first set was preincubated for 48 h, the other was tested immediately after the addition of TFE. The CD spectra of the aggregated protein are significantly different from those obtained immediately after the addition of TFE when the aggregates are not yet present (Fig. 1 d). This indicates that a conformational change is associated with AcPDro2 aggregation. The CD spectra of the aggregated protein display a single minimum at ∼220–224 nm, typical of aggregates having β-sheet structure (29). The peak is generally at 215–218 nm, but undergoes a red shift due to light-scattering phenomena arising from the aggregates.
Protein samples incubated in 5% TFE were analyzed using TM-AFM. Since the sample was dried to facilitate its adhesion to the mica substrate, the measured aggregate sizes reported below are reduced with respect to fully hydrated conditions, the shrinking factor being 2.0–2.5, as previously evaluated (30). After 24 h of incubation, aggregates with a twisted, fibrillar appearance are observed (Fig. 2 a). The fibrils have a height of 2.8 ± 0.2 nm, with the height reduced at their ends to 1.8 ± 0.1 nm. The typical fibril length is 50–200 nm. After 48 h of incubation, longer fibrils (typically from 300 nm to 1 μm) are found (Fig. 2 b). Twisted fibrils with a height of 3.6 ± 0.1 nm result from the intertwining of thinner filaments whose diameter exhibits a bimodal distribution with peaks around 1.2 and 2.3 nm and mean value 1.9 ± 0.1 nm. Given the shrinking factor of 2.0–2.5, the 3.6-nm-high fibrils and their constituent 1.9-nm-high filaments have widths consistent with amyloid fibrils and protofilaments, respectively. Overall, the aggregates formed from AcPDro2 analyzed here appear to have morphological, structural, and tinctorial properties typical of amyloid-like structures.
FIGURE 2.

TM-AFM images (amplitude data; insets, height data) of AcPDro2 aggregates obtained in 5% TFE after 24 h (a) and 48 h (b). Image sizes are (a) 1.0 × 0.8 μm, inset 380 × 320 nm; (b) 1.9 × 1.3 μm, inset 300 × 270 nm. The Z ranges for the height data reported in the insets are (a) 15 nm and (b) 10 nm. The aggregate sizes reported in the Results section were obtained by measuring the aggregate height in cross section in the TM-AFM images. The apparent size deduced visually from the image turns out to be overestimated because of the broadening effects caused by the AFM tip in the scan plane (30).
AcPDro2 is initially in its native-like state
Next we investigated the conformational state adopted by AcPDro2 in 5% (v/v) TFE before the aggregates form. This was technically feasible due to the relatively slow kinetics of the process that allowed a biophysical investigation to be carried out before the aggregates accumulate significantly. DLS measurements were performed on protein samples with and without 5% (v/v) TFE added immediately before the measurements. Fig. 3 a shows two representative size distributions by light-scattering intensity obtained at a protein concentration of 0.5 mg ml−1, in 0% (solid line) and 5% (dashed line) TFE. The peaks at 3.77 ± 0.13 and 4.10 ± 0.13 nm observed in 0 and 5% (v/v) TFE, respectively, are consistent with the hydrodynamic diameter of native and monomeric AcPDro2, as determined by the x-ray crystallographic structure (23) (errors reported in the manuscript indicate standard deviations). All pairs of measurements, carried out at different protein concentrations, show that the apparent hydrodynamic diameters measured in 5% (v/v) TFE are within error of those measured in the absence of TFE (Fig. 3 a, inset). This result led us to the conclusion that a monomeric form of AcPDro2, with a compactness similar to that of the native protein, is the most represented in solution before the aggregation process starts.
FIGURE 3.

(a) Size distributions by light-scattering intensity of AcPDro2 in 0% (solid line) and immediately after addition of 5% (dashed line) TFE. The inset shows the apparent diameters measured at different protein concentrations in 0% (•) and 5% (○) TFE. (b) Far-UV CD spectra of AcPDro2 in 0% TFE (solid line), 5% TFE (dashed line), 80% TFE (•), and 5 M urea without TFE (♦). (c) Near-UV CD spectra of AcPDro2 in 0% TFE (solid line), 5% TFE (dashed line) and 4.5 M urea without TFE (♦). In both the far- and near-UV analyses, the spectra were acquired a few seconds after dilution under the conditions described to focus the analysis on the initial states present before aggregation. (d) Intrinsic fluorescence spectra of AcPDro2 in 0% TFE (solid line), 5% TFE (dashed line), 80% TFE (•), and 5 M urea without TFE (♦). The spectra were acquired a few seconds after dilution under the conditions described to focus the analysis on the initial states present before aggregation. The inset shows the intrinsic fluorescence at 352 nm versus the percentage of TFE in the solution. (e) Fluorescence spectra of 55 μM ANS in the presence of 0.02 mg ml−1 AcPDro2 in 0% TFE (solid line with solid circles) and 5% TFE (dashed line with solid triangles). Spectra of free ANS in the absence of protein were also recorded as controls in 0% TFE (solid line) and 5% TFE (dashed line). Difference spectra, obtained by subtracting the ANS spectra recorded in the absence of protein from the corresponding ones acquired in its presence, are also shown in 0% TFE (○) and 5% TFE (▵). (f) Enzymatic activity of AcPDro2, tested by adding the protein to a final concentration of 0.5 μg ml−1 to solutions containing 4 mM benzoylphosphate. “I.U./l” indicates international units of activity per liter.
The secondary structure content of AcPDro2 before aggregation was investigated using far-UV CD. The CD spectra of AcPDro2, recorded immediately after the addition of 1, 2, 3, 4, and 5% (v/v) TFE, are very similar to that obtained for the native protein in the absence of TFE. Fig. 3 b shows that the spectra in 0 and 5% (v/v) are highly superimposable, both having a large negative band at 205–235 nm, typical of an α/β protein. This result shows that the protein does not undergo any detectable change in secondary structure after the addition of small quantities of TFE, even at a concentration able to induce aggregation. By contrast, the CD spectra acquired in 80% (v/v) TFE and in 5 M urea with no TFE indicate major changes in secondary structure composition under these conditions (Fig. 3 b).
The near-UV CD spectra are also highly superimposable when recorded for protein samples in 0 and 5% TFE (Fig. 3 c). The broad peak at 262–280 nm and the two sharp peaks at 286 and 295 nm are present in both spectra, indicating that no modifications in protein structure packing, particularly in anisotropy of aromatic residues, seem to occur after the addition of 5% TFE. By contrast, the near-UV CD spectrum of the protein sample denatured in 4.5 M urea, recorded as a control, shows that AcPDro2 has lost its native packing after denaturation (Fig. 3 c).
Modifications in the chemical environment around tryptophan residues were also monitored with fluorescence spectroscopy (Fig. 3 d). AcPDro2 has two tryptophan residues at positions 42 and 68, respectively. The intrinsic fluorescence spectra of the protein in 0–5% (v/v) TFE are very similar to each other, as shown for the two representative spectra in 0 and 5% TFE (Fig. 3 d). We found no significant differences for the wavelength of maximum emission (λmax = 350 nm) in 0–5% TFE. The weak increase of fluorescence emission from 0 to 5% TFE correlates linearly with TFE concentration (Fig. 3 d, inset); hence, it is most likely attributable to solvent effects rather than to a conformational change. On the other hand, the spectra recorded in 80% (v/v) TFE and 5 M urea with no TFE show a noticeable loss in total fluorescence emission and a red shift of the λmax value from 350 nm to 355 nm (80% TFE) and 358 nm (5 M urea).
Possible variations in the solvent exposure of hydrophobic clusters after the addition of TFE have been further investigated using ANS. This dye is known to bind to clustered hydrophobic residues that are exposed to the solvent in a protein and hence to give rise to a blue shift and an increase in fluorescence emission intensity (31,32). Both the protein samples in 0 and 5% (v/v) TFE were found to cause very weak and very similar increases of fluorescence emission of ANS (Fig. 3 e). The difference spectra, obtained in each case by subtracting the spectrum of free ANS from that recorded in the presence of protein, exhibit λmax values of 480 and 490 nm for the protein in 0% and 5% (v/v) TFE, respectively, a blue shift from the λmax of 515 nm for free ANS (Fig. 3 e). This indicates that small fractions of ANS molecules are bound to the protein, in both 0 and 5% TFE. The weak peaks shown in the difference spectra may perhaps be attributed to the ability of ANS to bind to cationic groups of the protein (33), since at pH 5.5 AcPDro2 has a net charge of +6. However, the fact that a similar relative increase of ANS fluorescence is caused by the protein in the two conditions rules out that additional hydrophobic clusters become superficial after the addition of small amounts of TFE.
The acylphosphatase activity of AcPDro2 was also determined in the presence of low concentrations of TFE, ranging from 0 to 5% (v/v). The catalytic activity of acylphosphatases requires the correct spatial positioning of residues that are relatively distant in the sequence, namely Arg-27, Asn-45, and the residues of the loop 19GRVQGV24 (34). We found that the ability of AcPDro2 to hydrolyze the acylphosphate bond did not change significantly within the range of TFE concentration tested (Fig. 3 f). Overall, the analysis presented here indicates that the protein populated under aggregating conditions, but before aggregation occurs, has structural and catalytic properties indistinguishable from those of the native protein under conditions in which it resists aggregation.
Absence of destabilization under aggregating conditions
Thermodynamic stability of native AcPDro2 has been monitored by equilibrium denaturation experiments carried out at TFE concentrations ranging from 0 to 5% (v/v). We followed unfolding of AcPDro2 at equilibrium using urea as a chemical denaturant and intrinsic fluorescence as a spectroscopic probe. Fig. 4 a shows the unfolding curves normalized to the fraction of folded protein with and without 5% (v/v) TFE. The unfolding transitions appear to occur at similar urea concentrations and with similar cooperativities in the two solution conditions (Fig. 4 a). The main thermodynamic parameters of the unfolding reaction were inferred at all TFE concentrations tested from the equilibrium curves assuming a two-state model and using the equation described by Santoro and Bolen (27). The free energy change upon unfolding in the absence of urea (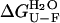 ) and the midpoint of unfolding (Cm) were found to remain substantially unchanged at the different TFE concentrations used (Fig. 4 b). We concluded that addition of up to 5% TFE does not lead to a substantial thermodynamic destabilization of the native structure of AcPDro2.
) and the midpoint of unfolding (Cm) were found to remain substantially unchanged at the different TFE concentrations used (Fig. 4 b). We concluded that addition of up to 5% TFE does not lead to a substantial thermodynamic destabilization of the native structure of AcPDro2.
FIGURE 4.

(a) Equilibrium urea unfolding curves of AcPDro2 in 0% (•) and 5% (♦) TFE. The lines represent the best fits to the two-state equation described by Santoro and Bolen (27), for 0% TFE (solid line) and 5% TFE (dashed line). (b) Variations in 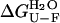 (•) and Cm (○) with TFE concentration. Error bars represent standard deviations. (c) Rate constants for folding in 0.6 M urea (•) and unfolding in 5.4 M urea (○) of AcPDro2 as a function of TFE concentration. (d) Rate constants for folding and unfolding of AcPDro2 as a function of urea concentration, in 0% TFE (lower trace) and 5% TFE (upper trace). Solid and open symbols indicate folding and unfolding, respectively. Solid diamonds and solid circles indicate experiments performed by unfolding the protein in 6 M urea at pH 5.5, whereas solid squares and solid triangles refer to 2 M urea and pH 2.0 as initial unfolding conditions.
(•) and Cm (○) with TFE concentration. Error bars represent standard deviations. (c) Rate constants for folding in 0.6 M urea (•) and unfolding in 5.4 M urea (○) of AcPDro2 as a function of TFE concentration. (d) Rate constants for folding and unfolding of AcPDro2 as a function of urea concentration, in 0% TFE (lower trace) and 5% TFE (upper trace). Solid and open symbols indicate folding and unfolding, respectively. Solid diamonds and solid circles indicate experiments performed by unfolding the protein in 6 M urea at pH 5.5, whereas solid squares and solid triangles refer to 2 M urea and pH 2.0 as initial unfolding conditions.
Folding and unfolding kinetics of AcPDro2 were monitored using a stopped-flow device coupled to a fluorescence detection system. In a first set of experiments the folding and unfolding rate constants were measured at TFE concentrations ranging from 0 to 9% (v/v). In all cases folding was monitored after a 10-fold dilution of the protein denatured in 6 M urea to reach a final urea concentration of 0.6 M. Two exponential phases were observed. The first phase, corresponding to the major folding phase, shows a marked positive dependence on TFE concentration and reaches the maximum rate at ∼6–8% TFE (Fig. 4 c, solid circles). This behavior is typical of all proteins studied so far and has been attributed to the fact that folding rate reaches a maximum when the formation of secondary structure is optimal and close to native-like levels (35). The unfolding reaction was monitored by a 10-fold dilution of the native protein in solutions containing 6 M urea to reach a final urea concentration of 5.4 M urea. Unfolding appears to be a monophasic process in all recorded traces. The unfolding rate constant appears to have a strong positive dependence on TFE concentration, reaching no apparent maximum in the range 0–9% TFE (Fig. 4 c, open circles).
In a second set of experiments the folding and unfolding rate constants were determined in 0 and 5% (v/v) TFE in the presence of various urea concentrations (see Materials and Methods for experimental details). The resulting chevron plots are shown in Fig. 4 d for the analyses carried out in both the presence and absence of 5% TFE. In 5% TFE the chevron plot is characterized by a noticeable downward curvature at 0–0.6 M urea, indicating that the major fast phase for folding has a decreasing dependence on urea concentration as the latter decreases. This phenomenon is often referred to as a rollover and probably reflects the accumulation of a partially folded or misfolded state during folding at these low urea concentrations (36).
Both chevron plots, determined in 0 and 5% TFE, were fitted to Eq. 3 to calculate the values for the two rate constants in the absence of urea (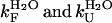 , respectively). The data points between 0 and 0.6 M urea in 5% TFE were obviously excluded from the fitting procedure.
, respectively). The data points between 0 and 0.6 M urea in 5% TFE were obviously excluded from the fitting procedure. 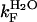 was found to be 7.5 (± 0.3) s−1 and 28.6 (± 2.4) s−1 in 0 and 5% TFE, respectively.
was found to be 7.5 (± 0.3) s−1 and 28.6 (± 2.4) s−1 in 0 and 5% TFE, respectively. 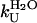 was found to be 4.2 (± 0.8) × 10−3 s−1 and 1.7 (± 0.5)·10−2 s−1 in 0 and 5% TFE, respectively. The
was found to be 4.2 (± 0.8) × 10−3 s−1 and 1.7 (± 0.5)·10−2 s−1 in 0 and 5% TFE, respectively. The 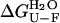 values, obtainable from equilibrium experiments, can also be obtained from kinetic data using
values, obtainable from equilibrium experiments, can also be obtained from kinetic data using
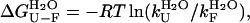 |
(4) |
where R and T are the universal gas constant and temperature, respectively. 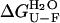 was found to be 18.5 (± 0.6) kJ mol−1 in the absence of TFE and 18.4 (± 1.1) kJ mol−1 in 5% (v/v) TFE. These values appear to be within experimental error to those obtained from the equilibrium chemically induced denaturation curves (compare with data in Fig. 4 b). The observation that folding and unfolding are both accelerated by ∼4 times upon the addition of small quantities of TFE provides an explanation as to why the conformational stability of AcPDro2 (i.e.,
was found to be 18.5 (± 0.6) kJ mol−1 in the absence of TFE and 18.4 (± 1.1) kJ mol−1 in 5% (v/v) TFE. These values appear to be within experimental error to those obtained from the equilibrium chemically induced denaturation curves (compare with data in Fig. 4 b). The observation that folding and unfolding are both accelerated by ∼4 times upon the addition of small quantities of TFE provides an explanation as to why the conformational stability of AcPDro2 (i.e., 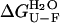 ) is not changed from 0 to 5% TFE.
) is not changed from 0 to 5% TFE.
Aggregation mechanism of AcPDro2
The results shown in the previous paragraphs show that AcPDro2 is able to form amyloid-like fibrils and protofilaments under conditions in which a native-like structure of the protein is initially populated and does not appear thermodynamically destabilized relative to the unfolded or partially folded state. One issue that is important to address is whether the fibrils (Agg) form from the partially unfolded or totally unfolded state (I) that is scarcely populated but in rapid equilibrium with the native state (N) through the folding and unfolding rate constants (Fig. 5, Scheme 1). Alternatively, the protein may fibrillate via a pathway in which various molecules of native AcPDro2 assemble and subsequently undergo a structural reorganization to form the observed amyloid-like fibrils (Fig. 5, Scheme 2). If Scheme 1 is correct to describe the aggregation process of AcPDro2 under the investigated conditions, the apparent rate constant for aggregation (kobs) results from a combination of the unfolding and folding rate constants (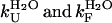 ) and of the rate constant for the conversion of the partially unfolded ensemble into aggregates (kagg). Kinetic inspection of the system and matrix algebra (37) leads to
) and of the rate constant for the conversion of the partially unfolded ensemble into aggregates (kagg). Kinetic inspection of the system and matrix algebra (37) leads to
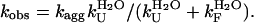 |
(5) |
FIGURE 5.

Two possible mechanisms of AcPDro2 aggregation. The protein may form fibrils from a totally or partially unfolded state (I) that is scarcely populated but in rapid equilibrium with the native state (N) through the folding and unfolding rate constants (Scheme 1). Alternatively various molecules of native AcPDro2 may assemble and subsequently undergo a structural reorganization to form the observed amyloid-like fibrils (Scheme 2). The analysis shows that Scheme 2 is more appropriate for AcPDro2 aggregation.
To determine kobs, AcPDro2 was incubated at a concentration of 0.4 mg ml−1 in 5% TFE. The steady-state fluorescence of ThT was measured at different time points in the presence of aliquots of the protein sample that had undergone aggregation in the absence of ThT. The plot of ThT fluorescence versus time was fitted to Eq. 1 to determine the kobs value (Fig. 6). The experiment and analysis were repeated three times and the kinetic profiles were found to be superimposable and reproducible. The fitting procedure yielded values of 2.9 ± (0.3)·10−5, 2.4 ± (0.4)·10−5, and 2.5 ± (0.2)·10−5 s−1 for the kobs value in the three cases.
FIGURE 6.

Time course of aggregation of 0.4 mg ml−1 AcPDro2 in 5% TFE. The zero time point refers to the addition of TFE to the protein solution. The experiment has been repeated three times, although only one representative set of data is shown. The solid line through the data represents the best fit of the data shown in the figure to a single exponential function (Eq. 1). The two dashed lines represent best fits for the other two sets of data. The figure also shows data for experiments in which AcPDro2 was incubated under the same conditions in the absence of TFE (solid circles) and in 5% TFE at pH 3.5 (crosses).
Although the 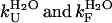 values have been determined under the conditions explored here (1.7 (± 0.5)·10−2 and 28.6 (± 2.4) s−1, respectively), the kagg value is difficult to determine experimentally under such conditions because the nonaggregated protein initially populates a native-like rather than unfolded or partially unfolded state. This problem was circumvented, however, with two approaches. First the kagg value has been calculated using the algorithm edited by DuBay et al. (38), allowing the rate constant for aggregation to be calculated for any unstructured amino acid sequence at a desired protein concentration, pH, and ionic strength. The value of kagg for AcPDro2 under the conditions investigated here is predicted to be 4.5 (± 3.0) × 10−5. Second, the kagg value has been determined under conditions in which the protein is substantially unfolded. A pH-titration experiment showed that AcPDro2 is >95% unfolded in 5% TFE at pH <4 (data not shown). The kinetic profile of the aggregation process has therefore been followed at pH 3.5 (see Materials and Methods for details). Aggregation was found to be very slow with no aggregates formed to any significant extent for up to 24 h (Fig. 6). An experimental bound can be kagg < 2 × 10−6. This bound has been determined in 5% (v/v) TFE at pH 3.5 and is not, therefore, correct for the conditions of interest here (5% TFE, pH 5.5). This bound can be corrected using the previously determined relationship between kagg and net charge (39), leading to an experimental bound of <2 × 10−5 s−1 for kagg in 5% TFE, pH 5.5, with an order of magnitude similar to the predicted value.
values have been determined under the conditions explored here (1.7 (± 0.5)·10−2 and 28.6 (± 2.4) s−1, respectively), the kagg value is difficult to determine experimentally under such conditions because the nonaggregated protein initially populates a native-like rather than unfolded or partially unfolded state. This problem was circumvented, however, with two approaches. First the kagg value has been calculated using the algorithm edited by DuBay et al. (38), allowing the rate constant for aggregation to be calculated for any unstructured amino acid sequence at a desired protein concentration, pH, and ionic strength. The value of kagg for AcPDro2 under the conditions investigated here is predicted to be 4.5 (± 3.0) × 10−5. Second, the kagg value has been determined under conditions in which the protein is substantially unfolded. A pH-titration experiment showed that AcPDro2 is >95% unfolded in 5% TFE at pH <4 (data not shown). The kinetic profile of the aggregation process has therefore been followed at pH 3.5 (see Materials and Methods for details). Aggregation was found to be very slow with no aggregates formed to any significant extent for up to 24 h (Fig. 6). An experimental bound can be kagg < 2 × 10−6. This bound has been determined in 5% (v/v) TFE at pH 3.5 and is not, therefore, correct for the conditions of interest here (5% TFE, pH 5.5). This bound can be corrected using the previously determined relationship between kagg and net charge (39), leading to an experimental bound of <2 × 10−5 s−1 for kagg in 5% TFE, pH 5.5, with an order of magnitude similar to the predicted value.
Substitution of the experimentally determined values of 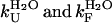 , and of the predicted value of kagg in Eq. 5 leads to a value of 2.65 (± 1.95) × 10−8 s−1 for kobs, three orders of magnitude lower than that determined experimentally (kobs = 2.6 (± 0.3) × 10−5 s−1). Using the experimental bound <2 × 10−5 s−1 for kagg, a bound of <1 × 10−8 s−1 is obtained for kobs, again far from that determined from the kinetic profile. This discrepancy indicates that Scheme 1 in Fig. 5 is not correct and that AcPDro2 can aggregate without unfolding into a fully or partially unfolded state under these conditions.
, and of the predicted value of kagg in Eq. 5 leads to a value of 2.65 (± 1.95) × 10−8 s−1 for kobs, three orders of magnitude lower than that determined experimentally (kobs = 2.6 (± 0.3) × 10−5 s−1). Using the experimental bound <2 × 10−5 s−1 for kagg, a bound of <1 × 10−8 s−1 is obtained for kobs, again far from that determined from the kinetic profile. This discrepancy indicates that Scheme 1 in Fig. 5 is not correct and that AcPDro2 can aggregate without unfolding into a fully or partially unfolded state under these conditions.
DISCUSSION
It is generally believed that globular proteins need to unfold, at least partially, to aggregate into amyloid or amyloid-like fibrils (3,16,17,40). Such a “conformational change hypothesis” is widely supported by a large body of experimental data. Proteins normally adopting a compact and well defined three-dimensional fold have a higher propensity to aggregate under conditions that promote their partial unfolding, such as high temperature, high pressure, low pH, or moderate concentrations of organic solvents (41–44). In addition, for some diseases in which the aggregating proteins normally adopt folded conformations, evidence suggests that a destabilization of the native structure, resulting in an increase in the population of nonnative states, is one of the mechanisms of action through which the pathogenicity of natural mutations is mediated; this was shown for proteins such as human lysozyme, transthyretin, superoxide dismutase, and the immunoglobulin light chain (14,15,45,46). Amyloid formation by proteins that have no link to human disease is also promoted by destabilizing mutations both in vitro and in vivo (44,47,48).
Although the “conformational change hypothesis” is undoubtedly correct to describe the aggregation mechanism of many globular proteins, the results presented here for AcPDro2, and additional evidence reported recently for other proteins, question the general applicability of this theory. Under the conditions of pH, ionic strength, and temperature investigated here, AcPDro2 is stable in its native conformation in the absence of TFE, with a free energy change of unfolding (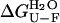 ) of ∼18 kJ mol−1 and no apparent aggregates formed after 24 h. In the presence of a small concentration of TFE, such as 5% (v/v), the protein assembles into fibrillar aggregates able to bind ThT and CR and having extensive β-sheet structure. Under these conditions, AcPDro2 maintains a native-like conformation before aggregation. Indeed, the protein has a secondary structure content, packing around aromatic and hydrophobic residues, hydrodynamic diameter, and catalytic activity indistinguishable from those of the native protein kept under conditions that do not promote aggregation. More importantly, the thermodynamic stability of the native conformation was found to be similar under native and aggregating conditions, with folding and unfolding both appearing accelerated to similar extents.
) of ∼18 kJ mol−1 and no apparent aggregates formed after 24 h. In the presence of a small concentration of TFE, such as 5% (v/v), the protein assembles into fibrillar aggregates able to bind ThT and CR and having extensive β-sheet structure. Under these conditions, AcPDro2 maintains a native-like conformation before aggregation. Indeed, the protein has a secondary structure content, packing around aromatic and hydrophobic residues, hydrodynamic diameter, and catalytic activity indistinguishable from those of the native protein kept under conditions that do not promote aggregation. More importantly, the thermodynamic stability of the native conformation was found to be similar under native and aggregating conditions, with folding and unfolding both appearing accelerated to similar extents.
In a previous study we have shown that the N-terminal domain of the Escherichia coli protein HypF aggregates under conditions in which the protein is still folded, a result that is apparently similar to that described here for AcPDro2 (37). Yet, HypF-N was found to be energetically destabilized under conditions promoting aggregation, and the kinetic analysis revealed that aggregation can indeed occur from a partially folded conformation, such as that accumulating during folding, which is at equilibrium scarcely populated but still in rapid equilibrium with the native structure (37). By contrast, AcPDro2 appears to be as stable as under nonaggregating conditions and the kinetic analysis shows that the a mechanism of aggregation via partial unfolding is largely inconsistent with the experimental data. An alternative model, in which the protein is not viewed to unfold before self-assembly, needs to be invoked to outline the aggregation mechanism of AcPDro2 in these conditions. One possibility is that native-like molecules of AcPDro2 interact through their edge β-strands.
The question naturally arises as to why AcPDro2 requires small concentrations of TFE to aggregate. In its native state, human muscle acylphosphatase, a protein sharing 36% sequence identity and belonging to the same structural family as AcPDro2 (49), undergoes limited proteolysis by trypsin, chymotrypsin, elastase, and subtilisin at the same sites along the sequence in the presence and absence of 10% TFE. However, the rate of proteolysis appears to be more rapid at all sites in 10% TFE, indicating that the native protein has gained flexibility in the presence of TFE (49). It is reasonable that in the presence of small concentrations of TFE, the native fold of a protein such as AcPDro2 becomes flexible and plastic enough so that more opportunities exist for intermolecular interactions to occur within the context of native or native-like structures.
Although this hypothesis awaits further experimental investigation, the most important message that emerges from this study is that folded proteins can aggregate into fibrillar aggregates under conditions that do not promote their unfolding or destabilization. In the highly crowded environment of a cell, or in the extracellular space where macromolecules still overcrowd the available space, albeit to a lower extent, native proteins have to face a considerable number of events that can possibly alter and distort their fold or simply increase their flexibility. Specific interactions with ligands and partner proteins, nonspecific interactions with a variety of alcohols or lipids that are present in vivo, and translocation between organelles across phospholipid membranes are just a few examples in which this can occur. Perhaps the evolutionary adaptations that all-β proteins have designed to prevent aggregation via interactions of the edge strands of their native β-sheets is the most intriguing indication that native or native-like aggregation is indeed possible and needs to be actively combated by living organisms (50).
CONCLUSIONS
The finding that a protein with a well defined three-dimensional fold does not need to unfold to initiate aggregation does not contradict the notion that many other proteins need to unfold, at least partially, to form amyloid-like structures. The fact remains that the fully or partially unfolded states that are populated after biosynthesis on the ribosome, stress conditions, or mutation are the conformational states that are most susceptible to initiate aggregation. Nevertheless, folded proteins spend most of their lifetime in a folded rather than an unfolded state. In some of these diseases the residual propensity of folded states to aggregate, albeit much lower than that of unfolded states, can become significant under physiological conditions, especially when considering that amyloid formation is extremely slow even in the most aggressive manifestations of protein deposition diseases.
The identification of the aggregation pathway followed in vivo by a protein that is normally folded is necessary to fully understand the pathogenesis of the disease with which it is associated and to identify possible molecular targets for drug design. More generally, the elucidation of potential aggregation pathways involving proteins that are not associated with pathological states under conditions close to physiological makes it possible to comprehend the strategies that Nature has devised during evolution to contrast protein aggregation.
Acknowledgments
We thank Giordana Marcon and Donatella Degl'Innocenti for their technical assistance. We also thank Alessandra Gliozzi and Ranieri Rolandi for helpful discussion.
This work was supported by grants from the European Union (Project HPRN-CT-2002-00241), the Italian Ministero dell'Istruzione, dell'Università e della Ricerca (FIRB project Nos. RBAU015B47_001 and RBNE01S29H; PRIN project Nos. 2003025755_003 and 2003054414_004; L. 449/97 Sector “Genomica funzionale”, project “Strutture ed interazioni molecolari di prodotti genici”), the Compagnia di San Paolo (project 2003.0727), and the Ente Cassa di Risparmio di Firenze (project Nos. 2003.437 and 2003.2029).
References
- 1.Dobson, C. M. 2003. Protein folding and misfolding. Nature. 426:884–890. [DOI] [PubMed] [Google Scholar]
- 2.Stefani, M., and C. M. Dobson. 2003. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81:678–699. [DOI] [PubMed] [Google Scholar]
- 3.Uversky, V. N., and A. L. Fink. 2004. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim. Biophys. Acta. 1698:131–153. [DOI] [PubMed] [Google Scholar]
- 4.Walsh, D. M., and D. J. Selkoe. 2004. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett. 11:213–228. [DOI] [PubMed] [Google Scholar]
- 5.Sunde, M., and C. Blake. 1997. The structure of amyloid fibrils by electron microscopy and x-ray diffraction. Adv. Protein Chem. 50:123–159. [DOI] [PubMed] [Google Scholar]
- 6.Serpell, L. C., M. Sunde, M. D. Benson, G. A. Tennent, M. B. Pepys, and P. E. Fraser. 2000. The protofilament substructure of amyloid fibrils. J. Mol. Biol. 300:1033–1039. [DOI] [PubMed] [Google Scholar]
- 7.Tycko, R. 2003. Insights into the amyloid folding problem from solid-state NMR. Biochemistry. 42:3151–3159. [DOI] [PubMed] [Google Scholar]
- 8.Klunk, W. E., R. F. Jacob, and R. P. Mason. 1999. Quantifying amyloid by Congo red spectral shift assay. Methods Enzymol. 309:285–305. [DOI] [PubMed] [Google Scholar]
- 9.LeVine 3rd, H. 1999. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309:274–284. [DOI] [PubMed] [Google Scholar]
- 10.Krebs, M. R., E. H. Bromley, and A. M. Donald. 2005. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J. Struct. Biol. 149:30–37. [DOI] [PubMed] [Google Scholar]
- 11.Khurana, R., V. N. Uversky, L. Nielsen, and A. L. Fink. 2001. Is Congo red an amyloid-specific dye? J. Biol. Chem. 276:22715–22721. [DOI] [PubMed] [Google Scholar]
- 12.Bousset, L., V. Redeker, P. Decottignies, S. Dubois, P. Le Marechal, and R. Melki. 2004. Structural characterization of the fibrillar form of the yeast Saccharomyces cerevisiae prion Ure2p. Biochemistry. 43:5022–5032. [DOI] [PubMed] [Google Scholar]
- 13.Chow, M. K., H. L. Paulson, and S. P. Bottomley. 2004. Destabilization of a non-pathological variant of ataxin-3 results in fibrillogenesis via a partially folded intermediate: a model for misfolding in polyglutamine disease. J. Mol. Biol. 335:333–341. [DOI] [PubMed] [Google Scholar]
- 14.Lindberg, M. J., L. Tibell, and M. Oliveberg. 2002. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc. Natl. Acad. Sci. USA. 99:16607–16612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canet, D., A. M. Last, P. Tito, M. Sunde, A. Spencer, D. B. Archer, C. Redfield, C. V. Robinson, and C. M. Dobson. 2002. Local cooperativity in the unfolding of an amyloidogenic variant of human lysozyme. Nat. Struct. Biol. 9:308–315. [DOI] [PubMed] [Google Scholar]
- 16.Kelly, J. W. 1998. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 8:101–106. [DOI] [PubMed] [Google Scholar]
- 17.Rochet, J. C., and P. T. Lansbury, Jr. 2000. Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 10:60–68. [DOI] [PubMed] [Google Scholar]
- 18.Chow, M. K., A. M. Ellisdon, L. D. Cabrita, and S. P. Bottomley. 2005. Polyglutamine expansion in Ataxin-3 does not affect protein stability: implications for misfolding and disease. J. Biol. Chem. 279:47643–47651. [DOI] [PubMed] [Google Scholar]
- 19.Bouchard, M., J. Zurdo, E. J. Nettleton, C. M. Dobson, and C. V. Robinson. 2000. Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 9:960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pedersen, J. S., G. Christensen, and D. E. Otzen. 2004. Modulation of S6 fibrillation by unfolding rates and gatekeeper residues. J. Mol. Biol. 341:575–588. [DOI] [PubMed] [Google Scholar]
- 21.Plakoutsi, G., N. Taddei, M. Stefani, and F. Chiti. 2004. Aggregation of the acylphosphatase from Sulfolobus solfataricus: the folded and partially unfolded states can both be precursors for amyloid formation. J. Biol. Chem. 279:14111–14119. [DOI] [PubMed] [Google Scholar]
- 22.Degl'Innocenti, D., M. Ramazzotti, R. Marzocchini, F. Chiti, G. Raugei, and G. Ramponi. 2003. Characterization of a novel Drosophila melanogaster acylphosphatase. FEBS Lett. 535:171–174. [DOI] [PubMed] [Google Scholar]
- 23.Zuccotti, S., C. Rosano, M. Ramazzotti, D. Degl'Innocenti, M. Stefani, G. Manao, and M. Bolognesi. 2004. Three-dimensional structural characterization of a novel Drosophila melanogaster acylphosphatase. Acta Crystallogr. D Biol. Crystallogr. 60:1177–1179. [DOI] [PubMed] [Google Scholar]
- 24.Camici, G., G. Manao, G. Cappugi, and G. Ramponi. 1976. A new synthesis of benzoyl phosphate: a substrate for acyl phosphatase assay. Experientia. 32:535–536. [DOI] [PubMed] [Google Scholar]
- 25.Johnson, W. C., Jr. 1990. Protein secondary structure and circular dichroism: a practical guide. Proteins. 7:205–214. [DOI] [PubMed] [Google Scholar]
- 26.Ramponi, G., C. Treves, and A. A. Guerritore. 1966. Aromatic acyl phosphates as substrates of acyl phosphatase. Arch. Biochem. Biophys. 115:129–135. [DOI] [PubMed] [Google Scholar]
- 27.Santoro, M. M., and D. W. Bolen. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenyl-methanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 27:8063–8068. [DOI] [PubMed] [Google Scholar]
- 28.Jackson, S. E., and A. R. Fersht. 1991. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 30:10428–10435. [DOI] [PubMed] [Google Scholar]
- 29.Venyaminov, S. Yu., and K. S. Vassilenko. 1994. Determination of protein tertiary structure class from circular dichroism spectra. Anal. Biochem. 222:176–184. [DOI] [PubMed] [Google Scholar]
- 30.Relini, A., C. Canale, S. Torrassa, R. Rolandi, A. Gliozzi, C. Rosano, M. Bolognesi, G. Plakoutsi, M. Bucciantini, F. Chiti, and M. Stefani. 2004. Monitoring the process of HypF fibrillization and liposome permeabilization by protofibrils. J. Mol. Biol. 338:943–957. [DOI] [PubMed] [Google Scholar]
- 31.Stryer, L. 1965. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 13:482–495. [DOI] [PubMed] [Google Scholar]
- 32.Ptitsyn, O. B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47:83–229. [DOI] [PubMed] [Google Scholar]
- 33.Matulis, D., and R. Lovrien. 1998. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys. J. 74:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stefani, M., N. Taddei, and G. Ramponi. 1997. Insights into acylphosphatase structure and catalytic mechanism. Cell. Mol. Life Sci. 53:141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamada, D., F. Chiti, J. I. Guijarro, M. Kataoka, N. Taddei, and C. M. Dobson. 2000. Evidence concerning rate-limiting steps in protein folding from the effects of trifluoroethanol. Nat. Struct. Biol. 7:58–61. [DOI] [PubMed] [Google Scholar]
- 36.Matouschek, A., J. T. Kellis, Jr., L. Serrano, M. Bycroft, and A. R. Fersht. 1990. Transient folding intermediates characterized by protein engineering. Nature. 346:440–445. [DOI] [PubMed] [Google Scholar]
- 37.Marcon, G., G. Plakoutsi, C. Canale, A. Relini, N. Taddei, C. M. Dobson, G. Ramponi, and F. Chiti. 2005. Amyloid formation from HypF-N under conditions in which the protein is initially in its native state. J. Mol. Biol. 347:323–335. [DOI] [PubMed] [Google Scholar]
- 38.DuBay, K. F., A. P. Pawar, F. Chiti, J. Zurdo, C. M. Dobson, and M. Vendruscolo. 2004. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J. Mol. Biol. 341:1317–1326. [DOI] [PubMed] [Google Scholar]
- 39.Chiti, F., M. Calamai, N. Taddei, M. Stefani, G. Ramponi, and C. M. Dobson. 2002. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. USA. 99:16419–16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobson, C. M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24:329–332. [DOI] [PubMed] [Google Scholar]
- 41.Villegas, V., J. Zurdo, V. V. Filimonov, F. X. Aviles, C. M. Dobson, and L. Serrano. 2000. Protein engineering as a strategy to avoid formation of amyloid fibrils. Protein Sci. 9:1700–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferrao-Gonzales, A. D., S. O. Souto, J. L. Silva, and D. Foguel. 2000. The preaggregated state of an amyloidogenic protein: hydrostatic pressure converts native transthyretin into the amyloidogenic state. Proc. Natl. Acad. Sci. USA. 97:6445–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McParland, V. J., N. M. Kad, A. P. Kalverda, A. Brown, P. Kirwin-Jones, M. G. Hunter, M. Sunde, and S. E. Radford. 2000. Partially unfolded states of β(2)-microglobulin and amyloid formation in vitro. Biochemistry. 39:8735–8746. [DOI] [PubMed] [Google Scholar]
- 44.Chiti, F., N. Taddei, M. Bucciantini, P. White, G. Ramponi, and C. M. Dobson. 2000. Mutational analysis of the propensity for amyloid formation by a globular protein. EMBO J. 19:1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raffen, R., L. J. Dieckman, M. Szpunar, C. Wunschl, P. R. Pokkuluri, P. Dave, P. Wilkins Stevens, X. Cai, M. Schiffer, and F. J. Stevens. 1999. Physicochemical consequences of amino acid variations that contribute to fibril formation by immunoglobulin light chains. Protein Sci. 8:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hammarstrom, P., X. Jiang, A. R. Hurshman, E. T. Powers, and J. W. Kelly. 2002. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc. Natl. Acad. Sci. USA. 99:16427–16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramirez-Alvarado, M., J. S. Merkel, and L. Regan. 2000. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. USA. 97:8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calloni, G., S. Zoffoli, M. Stefani, C. M. Dobson, and F. Chiti. 2005. Investigating the effects of mutations on protein aggregation in the cell. J. Biol. Chem. 280:10607–10613. [DOI] [PubMed] [Google Scholar]
- 49.Monti, M., B. L. Garolla di Bard, G. Calloni, F. Chiti, A. Amoresano, G. Ramponi, and P. Pucci. 2004. The regions of the sequence most exposed to the solvent within the amyloidogenic state of a protein initiate the aggregation process. J. Mol. Biol. 336:253–262. [DOI] [PubMed] [Google Scholar]
- 50.Richardson, J. S., and D. C. Richardson. 2002. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA. 99:2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]