

Abstract
Phosphate binding to the sarcoplasmic reticulum Ca2+-ATPase was studied by time-resolved Fourier transform infrared spectroscopy with ATP and isotopically labeled ATP ([β-18O2, βγ-18O]ATP and [γ-18O3]ATP). Isotopic substitution identified several bands that can be assigned to phosphate groups of bound ATP: bands at 1260, 1207, 1145, 1110, and 1085 cm−1 are affected by labeling of the β-phosphate, bands likely near 1154, and 1098–1089 cm−1 are affected by γ-phosphate labeling. The findings indicate that the strength of interactions of β- and γ- phosphate with the protein are similar to those in aqueous solution. Two bands, at 1175 and 1113 cm−1, were identified for the phosphate group of the ADP-sensitive phosphoenzyme Ca2E1P. They indicate terminal and bridging P-O bond strengths that are intermediate between those of ADP-insensitive phosphoenzyme E2P and the model compound acetyl phosphate in water. The bridging bond of Ca2E1P is weaker than for acetyl phosphate, which will facilitate phosphate transfer to ADP, but is stronger than for E2P, which will make the Ca2E1P phosphate less susceptible to attack by water.
INTRODUCTION
P-type ATPases are a large family of membrane proteins that are responsible for active transport of cations across biological membranes to keep the cationic cellular milieu in correct balance for cellular function. A fundamental property of P-type ATPases is their ability to bind the substrate ATP with high affinity and to catalyze phosphorylation (hence the name P-type ATPase) of a conserved aspartic acid residue in the presence of activating ions. The SR Ca2+-ATPase of muscle cells (SERCA1a) (1–9) is a typical P-type ATPase with a single polypeptide chain and 110-kDa molecular mass. The crystal structure of presumably the Ca2+ form Ca2E1 (6) shows that the Ca2+-ATPase consists of a transmembrane region with 10 transmembrane α-helices, a small lumenal region and a cytoplasmic region comprising the nucleotide binding domain (N domain), the phosphorylation domain (P domain), actuator or anchor domain (A domain), and hinge regions.
The Ca2+-ATPase (1) catalyzes Ca2+ transport against a concentration gradient from the cytoplasm of muscle cells into the SR lumen for muscle relaxation (7–9). The energy required to transfer Ca2+ ions against the electrochemical potential gradient is derived from hydrolysis of the terminal phosphate of ATP. The reaction cycle modified from de Meis and Vianna (10) is shown in Scheme 1. After binding of Ca2+ from the cytoplasmic side of the SR and binding of ATP, the γ-phosphate of ATP is transferred to Asp351 and the first phosphoenzyme intermediate Ca2E1P is formed. The subsequent conversion to the E2P phosphoenzyme intermediate is associated with Ca2+ release to the lumenal side of the SR membrane. The two phosphoenzyme intermediates Ca2E1P and E2P have different catalytic properties: Ca2E1P dephosphorylates with ADP to reform ATP, whereas E2P hydrolyzes and does not react with ADP. The formation of Ca2E1ATP, as well as the further processing of phosphoenzyme intermediates is associated with protein conformational changes that couple Ca2+ transport in the transmembrane region with ATP hydrolysis in the cytoplasmic region.
SCHEME 1.

Reaction cycle of the SR Ca2+-ATPase.
Crystal structures of ATPase with a bound ATP analog AMPPCP (Ca2E1AMPPCP) have been published recently (11,12). They show that all phosphate groups of AMPPCP are engaged in interactions with the protein and reveal an uncommon zigzag conformation of the triphosphate chain that results in a hydrogen bond between the ribose 3′-OH and the β-phosphate group.
Previously we studied binding of ATP (13–16) to the Ca2+-ATPase and the phosphate group of the phosphoenzymes (17–19) by time-resolved FTIR difference spectroscopy (20–28). Modifications at the adenine ring and the ribose ring of the ATP molecule affect ATP-ATPase interactions upon binding and phosphorylation, as indicated by effects on phosphorylation rates and on the fingerprint signals of conformational change in the amide I region (15,29). The three stretching vibrations of the terminal P-O bonds of the E2P phosphate group could be identified in an isotope exchange experiment which detected a 20% weaker bridging P-O bond as compared to the model compound acetyl phosphate in aqueous solution (19). In these studies, the small infrared absorbance changes associated with nucleotide binding and ATPase phosphorylation could be detected because these reactions were triggered directly in the infrared cuvette using photolytic release of nucleotides from inactive photolabile derivatives, i.e., P3-1-(2-nitrophenyl)ethyl nucleotides (NPE caged nucleotides) (30).
Here, time-resolved FTIR difference spectroscopy was applied to study the phosphate groups of ATP in the complex with the Ca2+-ATPase and of the Ca2E1P phosphoenzyme phosphate group with the help of isotopic labeling. Two isotopomers of ATP were used, one labeled at the three terminal oxygens of the γ-phosphate ([γ-18O3]ATP) and one labeled at the two terminal oxygens of the β-phosphate and at the bridging oxygen between β- and γ- phosphate ([β-18O2, βγ-18O]ATP). Their structures are shown in Scheme 2. Isotopic labeling was used to assign bands in the difference spectrum of ATP binding to phosphate groups of ATP in analogy to previous studies of free GTP and GTP bound to H-Ras (31–35) and of free ATP (36–38).
SCHEME 2.

Structures of ATP and isotopically labeled ATP. The labeled atoms are highlighted in bold. (Top) [β-18O2, βγ-18O]ATP. (Bottom) [γ-18O3]ATP.
EXPERIMENTAL PROCEDURES
Materials
Caged ATP
Labeled caged ATPs were a gift from M. Webb and J. E. T. Corrie (National Institute for Medical Research, London).
Sample preparation
SR vesicles were prepared as described (39). After a 60-min dialysis of 50 μl SR vesicles in 100 ml buffer (10 mM methylimidazole/HCl, pH 7.5, 200 μM CaCl2, and 10 mM KCl) at 4°C, infrared samples were prepared as before (15,16), containing ∼1.2 mM Ca2+-ATPase, 0.5 mg/ml A23187, 1 mg/ml adenylate kinase, 150 mM methylimidazole, 150 mM KCl, 10 mM CaCl2, 10 mM dithiothreitol, and 10 mM caged ATP.
Control samples in the absence of ATPase were made for each caged ATP isotopomer in the same way as described above but without SR vesicles. Instead, 15 μl buffer were added to maintain the same pH value and ion concentrations.
In all samples, 10 mM Ca2+ was used instead of the physiological cosubstrate Mg2+ to inhibit the Ca2E1P-to-E2P transition to achieve a maximum steady-state level of Ca2E1P and to slow down the phosphorylation reaction (40–43). This enables longer observation times for Ca2E1ATP and Ca2E1P, resulting in a better signal/noise ratio of difference spectra.
Supplementary experiments were done with [15N]caged ATP. Because of labeling at the nitro position of the (2-nitrophenyl)ethyl group, the photolysis band at 1526 cm−1 shifts to 1499 cm−1 (13,16,38), which allows the identification of protein bands in the 1526 cm−1 region. Since ATP released from [15N]caged ATP is identical to that released from unlabeled [14N]caged ATP, its interactions with the protein are expected to be the same, and thus the spectrum obtained with [15N]caged ATP can be used as a reference for a complete compensation of the 1526 cm−1 photolysis band when the spectrum associated with the photolysis reaction is subtracted from our raw data (see below).
Methods
FTIR measurements
Time-resolved FTIR measurements of Ca2+-ATPase partial reactions were performed at 1°C with a Bruker IFS 66 spectrometer (Bruker Optics, Ettlingen, Germany), as described previously (29,43). Photolytic release of ATP was triggered by a xenon flash tube (N-185C, Xenon, Woburn, MA). A Schott UG11 filter was used to block the flash light of the xenon tube in the visible and infrared spectral region. This setup released ∼3 mM ATP from 10 mM caged ATP. These conditions gave saturating ATP binding signals as described (15).
A series of time-resolved spectra with a maximum time resolution of 65 ms was recorded after the photolysis flash. They were converted into difference spectra with respect to a reference spectrum recorded before photolysis. With each isotopomer, 19–37 experiments were repeated for averaging.
Photolysis spectra
Spectra obtained with control samples in the absence of ATPase were used to subtract photolysis signals in the spectra of ATPase samples, as described below. The control sample spectra show only signals evoked by release of free ATP and byproduct, and by consumption of caged ATP upon photolysis. They are therefore named “photolysis spectra”.
Binding spectra
To calculate difference spectra that reflect ATP binding, the reference spectrum characterizing the absorption of the initial Ca2E1 state was subtracted from a spectrum that reflected the absorption of Ca2E1ATP. The latter spectrum was averaged in the time slot from 0.26 to 0.90 s after the photolysis flash (15). These spectra are named “uncorrected spectra of ATP binding” in the following. Apart from absorbance changes due to ATP binding, these spectra also contain signals due to the photolysis reaction.
Subtraction of photolysis spectrum
Photolysis signals were subtracted using the photolysis spectra, which reflect only the absorbance changes due to the photolysis reaction. The resulting spectra are termed “ATP binding spectra”, indicating that these spectra show only signals due to ATP binding (ATP + Ca2E1 → Ca2E1ATP). The correct subtraction factor in the interactive subtraction was determined with the help of the uncorrected spectrum obtained with [15N]caged ATP. This spectrum is shown as a thin line in Fig. 1. Isotopic substitution shifts the νas (NO2) band of caged ATP from 1526 cm−1 to 1499 cm−1 revealing underlying protein bands (13). The subtraction factor in the subtraction of photolysis signals was chosen such that the 1526 cm−1 region of the processed spectrum corresponded to that region in the spectrum obtained with [15N]caged ATP (13). A comparison of this spectrum (Fig. 1, thin line) with the binding spectra shown in Fig. 2 A demonstrates the agreement between the resulting binding spectra and the spectrum obtained with [15N]caged ATP in the region around 1526 cm−1. The ATP binding spectra with labeled ATP were normalized to that of unlabeled ATP using the spectral region above 1300 cm−1 outside phosphate absorption.
FIGURE 1.

Comparison of the uncorrected spectrum of ATP binding (bold line and small black labels), the photolysis spectrum (shaded line and shaded labels, multiplied by a factor of 0.45, which was used for subtraction to obtain the ATP binding spectrum in Fig. 2) and the uncorrected spectrum obtained upon photolysis of [15N]caged ATP (thin line and large black labels with arrows). Experimental conditions were 1°C, pH 7.5, 3 mM released ATP, and 10 mM Ca2+.
FIGURE 2.

Binding (A and B), phosphorylation (C and D), and isotope effect (E) spectra obtained with labeled and unlabeled ATP. (A–D) Spectra obtained upon release of 3 mM ATP (thin line), [β-18O2, βγ-18O]ATP (shaded line), or [γ-18O3]ATP (bold line). (A) Binding spectra shown from 1750 to 980 cm−1. (B) Binding spectra shown from 1350 to 1000 cm−1. In A and B, positive bands show the absorption of Ca2E1ATP, negative bands that of free ATP and Ca2E1. (C) Phosphorylation spectra shown from 1750 to 980 cm−1. (D) Phosphorylation spectra shown from 1350 to 1000 cm−1. In C and D, positive bands show the absorption of Ca2E1P and ADP, negative bands that of Ca2E1ATP. (E) IE binding spectra (thin lines) and IE phosphorylation spectra (bold lines). IE spectra were calculated by subtraction between spectra obtained with labeled ATP and those obtained with unlabeled ATP (IE binding spectra from binding spectra shown in A and B, and IE phosphorylation spectra from phosphorylation spectra shown in C and D, see Methods). The sign convention is according to Scheme 3. Arrows with broken lines indicate band shifts of bound ATP, and arrows with solid lines in the lower panel indicate band shifts of the phosphoenzyme phosphate. (Top) Isotope effects of labeling the β-phosphate. (Bottom) Isotope effects of labeling the γ-phosphate.
Phosphorylation spectra
Phosphorylation spectra with ATP and labeled ATP (reflecting absorbance changes upon phosphorylation, Ca2E1ATP → Ca2E1P) were calculated from the same series of time-resolved spectra as binding spectra using different time slots. They were obtained by subtracting a spectrum that reflected the absorption of a mixture of Ca2E1ATP and Ca2E1P (0.26–1.9 or 3.2 s after photolysis flash) from a spectrum that reflected solely Ca2E1P absorption and which was averaged in the time slot from 3.25 to 16.2 s after the photolysis flash. The time interval for the first spectrum was longer than that used for the nucleotide binding spectra. Thus, this spectrum contains a contribution of partially formed Ca2E1P. However, due to the longer acquisition time, the quality of this spectrum is better than that of a spectrum obtained in a shorter time interval, which results in a better phosphorylation spectrum. Since this spectrum covers only part of the phosphorylation reaction, its amplitude needs to be corrected to obtain the spectrum of the complete reaction. This was done for the spectrum with unlabeled ATP by comparison with a difference spectrum that covered the full reaction (time interval for first spectrum, 0.26–0.90 s). The spectra with labeled ATP were normalized to the phosphorylation spectrum obtained with unlabeled ATP, as described above for the ATP binding spectra. Phosphorylation spectra with unlabeled and γ-labeled ATP shown in Fig. 2 were calculated with a longer time interval for the first spectrum, 0.26–3.2 s, and those with β-labeled ATP with the shorter time interval, 0.26–1.9 s, for reasons explained below. The phosphorylation spectrum with unlabeled ATP, to which those with the ATP isotopomers are compared, is virtually identical after normalization to equal band amplitudes for the two time intervals.
Phosphorylation spectra are not disturbed by photolysis signals because photolysis is complete before recording of the first spectrum used in the subtraction. Thus, they are not affected by manual subtraction of photolysis signals employed in the calculation of binding spectra. It should be noted that phosphorylation spectra are different from Ca2E1P formation spectra, described by us in previous articles (15,29,43,44), which reflect absorbance changes due to the reaction Ca2E1 → Ca2E1P.
IE
IE spectra were used to study band shifts due to isotopic labeling of ATP's phosphate groups. IE binding spectra were obtained by subtracting the binding spectrum of unlabeled ATP from that of labeled ATP. IE phosphorylation spectra were obtained by subtracting the phosphorylation spectrum of labeled ATP from that of unlabeled ATP to get the same sign as in IE binding spectra for bands of bound labeled and unlabeled ATP. The time interval for the early spectrum used to calculate the phosphorylation spectrum was chosen so that the best signal/noise ratio in the IE phosphorylation spectra was obtained. For the IE phosphorylation spectrum with γ-labeled ATP the time interval 0.26–3.2 s was used for two phosphorylation spectra used in the calculation of the IE phosphorylation spectrum. For the IE phosphorylation spectrum with β-labeled ATP the shorter time interval 0.26–1.9 s was used for the two respective spectra. This difference in time intervals did not alter the bands observed in the IE phosphorylation spectra.
Scheme 3 shows how IE spectra were obtained and how band shifts were symbolized. IE binding spectra have contributions from bound and free ATP. IE phosphorylation spectra show contributions of bound ATP (Ca2E1ATP), phosphoenzyme phosphate (Ca2E1P), and ADP (free or bound to Ca2E1P). In both types of IE spectra, a downshift of a band for bound ATP due to isotopic labeling gives rise to a minimum at higher wavenumber close to the spectral band position of the unlabeled compound and a maximum at lower wavenumber close to that of the labeled compound. We symbolize this kind of shift with “∪∩”. A downshift of a band for free ATP due to isotopic labeling will give rise to the opposite “∩∪” band profile in the IE binding spectra. The same band profile is expected for band downshifts of ADP and of the phosphoenzyme phosphate group in IE phosphorylation spectra. It should be noted that the band position in an IE spectrum will not generally correspond to the peak position in an absorbance spectrum. This is because a small downshift of a band will decrease the absorbance most on the high-wavenumber side of the original band and increase the absorbance most on the low-wavenumber side of the shifted band. Thus, the minima and maxima in IE spectra are often further apart than the two peak positions in absorbance spectra.
SCHEME 3.

Illustration of the band shifts in the difference spectra upon isotopic labeling (top panels) and in the IE spectra (bottom panels). The wavenumber scale runs according to convention from high wavenumbers on the left-hand side to low wavenumbers on the right-hand side. Illustrated are downshifts of positive and negative bands upon isotopic labeling. (A) Binding spectra and IE binding spectra, with downshift of a positive band, characteristic of bound ATP (left), and downshift of a negative band, characteristic of free ATP (right). (B) Phosphorylation and IE phosphorylation spectra, with shift of a positive band, characteristic of the phosphoenzyme phosphate or ADP (left), and shift of a negative band, characteristic of bound ATP (right). Note that the sign convention in IE binding and IE phosphorylation spectra is different, to obtain the same band profile for band shifts of bound ATP.
Subtraction of flash artifact
After replacing our flash tube we noted a “new” band at 1282 cm−1. This is a flash artifact and is observed also when the flash is ignited in the empty spectrometer. The flash artifact was subtracted from the IE spectra shown in Fig. 2 E, but is evident in the phosphorylation spectra shown in Fig. 2 D.
RESULTS
Overview
We studied the interactions of the Ca2+-ATPase with phosphate groups of ATP and with the aspartylphosphate of Ca2E1P by monitoring the reaction sequence Ca2E1 → Ca2E1ATP → Ca2E1P with time-resolved infrared spectroscopy. The reactions were triggered by photolytic release of ATP from caged ATP. We will focus on the spectral region of phosphate group absorption below 1300 cm−1.
Fig. 1 shows the uncorrected spectrum of ATP binding (bold line), which reflects absorbance changes due to ATP binding and due to photolysis of caged ATP (Ca2E1 + caged ATP → Ca2E1ATP + photolysis byproducts). Positive bands are due to the absorption of ATPase in complex with ATP, of byproducts, and of both bound and extra free ATP because the ATPase/ATP ratio was 1:2.5 to saturate binding of ATP. Negative bands are characteristic of caged ATP and the ATP-free ATPase Ca2E1. The uncorrected spectrum of ATP binding reflects conformational changes of the protein backbone in the amide I (1700–1610 cm−1) and amide II (1580–1500 cm−1) regions. Absorbance changes of phosphate groups are expected below 1300 cm−1. In addition, environmental and structural changes of side chains and ATP contribute in the entire spectral region shown. Groups or structures not involved in the conformational change or in the interaction do not show in difference spectra.
The photolysis spectrum (caged ATP → ATP + byproducts) is shown as a shaded line in Fig. 1. It was obtained with samples in the absence of ATPase. Positive bands show the absorption of free ATP and photolysis byproducts, and negative bands are characteristic of caged ATP. As described in Experimental Procedures, the photolysis spectrum was used to subtract the photolysis signals from the uncorrected spectrum of ATP binding with the help of the uncorrected spectrum obtained with [15N]caged ATP, which is shown as a thin line in Fig. 1. The result of the subtraction of photolysis signals are ATP binding spectra, which are shown in Fig. 2, A and B, for ATP and labeled ATP. Here, ATP bands are positive for bound ATP and negative for free ATP. (This is because the extra free ATP contribution is over-subtracted in the subtraction of the photolysis spectrum. Consider the following conditions as an example: 1 Ca2E1 + 2 caged ATP → 1 free ATP + 1 Ca2E1ATP. Thus, in the uncorrected spectrum we have positive bands of free and bound ATP. However, if the photolysis spectrum of the reaction 2 caged ATP → 2 free ATP is subtracted, the absorption of 1 free ATP appears negative in the resulting binding spectrum.) No significant absorbance changes were observed in control samples that contained the inhibitor thapsigargin (16), which selectively inhibits the Ca2+-ATPase (45–47); 20 mM EGTA (48), which converts the ATPase to a Ca2+-free state; or fluorescein isothiocyanate-labeled ATPase (48), which blocks the ATP binding site (49,50).
We checked the robustness of our subtraction procedures in several ways:
Compared to the spectra shown in Fig. 1, spectra obtained with 30% less released ATP (data not shown) showed essentially the same bands in the region of phosphate absorption before and after subtraction of the photolysis signals, in particular the bands at 1147, 1107, and 1093 cm−1 (see Fig. 1) that will be assigned below to ATP phosphates bound to the ATPase.
Subtraction of photolysis signals from spectra obtained with a threefold higher concentration of released ATP produced a very similar binding spectrum (not shown), again showing the above mentioned bands.
A photolysis spectrum in the presence of ATPase was obtained by subtraction between a spectrum obtained with high concentration of released ATP and the spectrum under our standard conditions. The subtraction of this spectrum (not shown) gave a spectrum very similar to the subtraction of a photolysis spectrum obtained in the absence of ATPase (see Fig. 1).
The IE phosphorylation spectrum for γ-labeled ATP was calculated with independent subsets of our experiments (see Fig. S1 in Supplementary Material) to assess the variation in the spectra between different experiments. The spectra obtained with the subsets are very similar to each other as well as to the spectrum obtained with the full set.
Fig. 2 shows spectra obtained with ATP and labeled ATP that were used to assign difference bands to phosphate groups based on band shifts observed with isotopically labeled ATP. The spectra used for assignment comprise binding spectra, phosphorylation spectra, and isotope effect spectra to all of which bound ATP contributes. The assignments are listed in Table 1. They will be useful for future studies of the phosphate binding of ATP analogs.
TABLE 1.
Band assignments to phosphate groups of bound ATP (Ca2E1ATP), bound labeled ATP, free ATP, phosphoenzyme phosphate moiety (Ca2E1P), and bound ADP (to Ca2E1P)
| Band position/cm−1
|
|||
|---|---|---|---|
| ATP | [β-18O2, βγ-18O] ATP | [γ-18O3]ATP | Assignments |
| ∼1250 |
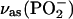 of free ATP of free ATP |
||
| 1196 |
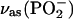 of β-phosphate of free ATP of β-phosphate of free ATP |
||
| ∼1260 |
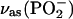 of bound ATP of bound ATP |
||
| 1207 | 1174 |
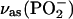 of bound ATP of bound ATP |
|
| 1154–1145 | 1128 | 1130 | β- and likely γ-phosphate of bound ATP |
| 1110–1106* | 1103 | 1106* | Predominantly α-phosphate of bound ATP |
| 1098–1085 | ∼1070 | 1072–1047 | β- and γ-phosphate of bound ATP |
| 1175 | 1147 | Phosphate of Ca2E1P | |
| 1130 | Possibly phosphate of Ca2E1P | ||
| 1113 | 1098–1089 | Phosphate of Ca2E1P | |
Band positions correspond to Fig. 2 E, except for those marked with an asterisk.
Band position from binding spectrum.
Binding spectra shown in Fig. 2, A and B, reflect absorbance differences between the initial ATP-free state Ca2E1 and free ATP in solution on the one hand (negative bands) and Ca2E1ATP on the other hand (positive bands). Above 1300 cm−1, outside the absorption region of phosphate groups, the same band positions and very similar relative band amplitudes were obtained with ATP and labeled ATP. This demonstrates the excellent reproducibility of our infrared spectra. Fig. 2 B shows the binding spectra on a larger scale below 1350 cm−1 to display band shifts upon isotopic substitution more clearly. It is evident that bands are observed at different positions in the three spectra, which is due to band shifts caused by labeling of oxygen atoms on the β- or γ-phosphate (discussed below).
Phosphorylation spectra with ATP or labeled ATP (Fig. 2, C and D) were obtained by subtracting a spectrum characteristic of Ca2E1ATP from a spectrum characteristic of Ca2E1P. Therefore, phosphorylation spectra reflect absorbance differences between the ATP-bound state Ca2E1ATP (negative bands) and the ADP-sensitive phosphoenzyme Ca2E1P with ADP (partially) bound (positive bands).
Fig. 2 E shows IE spectra, which are obtained by a subtraction of spectra obtained with unlabeled and labeled ATP. They clearly reveal differences due to the different absorption of labeled and unlabeled ATP. The IE spectra shown are robust in the sense that they are only slightly affected by changes in the time intervals used for spectra averaging and by the subtraction factor used in the calculation of IE spectra (interactive subtraction of spectra for labeled and unlabeled ATP). Nevertheless, it is obvious from an inspection of the IE spectra outside the region of phosphate absorption (>1300 cm−1) that a complete compensation of isotope unrelated bands is impossible in IE spectra. The level of confidence of these spectra was assessed by evaluation of independent subsets of our experiments, as shown in Fig. S1 of Supplementary Material.
Expected isotope shifts
Upon 18O labeling of phosphate groups, their vibrations are expected to shift downward by up to 40 cm−1 for an isolated P-O oscillator. For the coupled P-O vibrations of phosphate groups, shifts of ∼40 cm−1 are indeed observed for the symmetric stretching vibrations 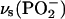 and
and 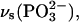 whereas shifts of the respective anti- or asymmetric vibrations, νas, are ∼30 cm−1 (51). Infrared spectra of labeled and unlabeled ATP (36,52), GTP, and guanosine diphosphate (32) in aqueous solution have been reported before. The pattern of labeling is different for the published spectra of ATP (36) compared with our isotopes in that different bridging oxygen atoms were labeled in the phosphate groups. This different labeling pattern will affect differently those vibrations with a contribution of bridging P-O bonds. However, vibrations of terminal P-O bonds that are discussed in the following will experience largely similar shifts with both labeling patterns.
whereas shifts of the respective anti- or asymmetric vibrations, νas, are ∼30 cm−1 (51). Infrared spectra of labeled and unlabeled ATP (36,52), GTP, and guanosine diphosphate (32) in aqueous solution have been reported before. The pattern of labeling is different for the published spectra of ATP (36) compared with our isotopes in that different bridging oxygen atoms were labeled in the phosphate groups. This different labeling pattern will affect differently those vibrations with a contribution of bridging P-O bonds. However, vibrations of terminal P-O bonds that are discussed in the following will experience largely similar shifts with both labeling patterns.
Strategy for band assignments
In the following discussion of band shifts due to isotopic labeling, we will focus on IE spectra that are shown in Fig. 2 E and reveal isotopic band shifts most clearly. The isotope shifts manifest in IE spectra as ∪∩ and ∩∪ band profiles as explained in “Methods” and shown in Scheme 3. The high-wavenumber component of a band profile indicates the absorption of unlabeled compound, and the low-wavenumber component that of the labeled compound. As a general approach we attributed bands that appear similar in both IE spectra to bound ATP. Additionally, we checked whether or not a band in an IE spectrum corresponds to a band in the respective binding and phosphorylation spectra. Bands of bound ATP contribute to both the binding and phosphorylation spectra as positive and negative bands, respectively. Therefore, they are best assigned by a comprehensive discussion of band shifts observed in binding, phosphorylation, and IE spectra. This discussion is provided in Supplementary Material. Here, the main results are summarized.
β-phosphate bands of bound ATP
The IE spectra of binding and phosphorylation that we obtained upon β-labeling are shown in the top panel of Fig. 2 E. The expected downshifts of bands upon isotopic labeling will give rise to ∪∩ band profiles in both IE spectra for bound ATP, and to ∩∪ band profiles for free ATP in the IE binding spectrum and for ADP in the IE phosphorylation spectrum.
For free ATP or GTP, the band of the antisymmetric stretching vibration of α- and 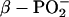 groups,
groups, 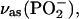 consists of a main band near 1234 cm−1 and a shoulder near 1216 cm−1 (32,36). Upon labeling of the β-phosphate, the
consists of a main band near 1234 cm−1 and a shoulder near 1216 cm−1 (32,36). Upon labeling of the β-phosphate, the 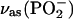 band loses intensity and becomes a clear double band with component bands at 1220–1226 cm−1 and 1178–1190 cm−1 (32,36). Accordingly, we assign a ∩∪ band profile in the IE spectrum of binding (Fig. 2 E, top, thin line) with a positive band at 1248 cm−1 and a negative band at 1196 cm−1 to the downshift of the
band loses intensity and becomes a clear double band with component bands at 1220–1226 cm−1 and 1178–1190 cm−1 (32,36). Accordingly, we assign a ∩∪ band profile in the IE spectrum of binding (Fig. 2 E, top, thin line) with a positive band at 1248 cm−1 and a negative band at 1196 cm−1 to the downshift of the 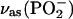 band of free ATP upon labeling. For bound ATP, a small negative band at 1260 cm−1 was observed in the IE phosphorylation spectrum (Fig. 2 E, top, bold line), which we attribute to the corresponding intensity decrease of the main
band of free ATP upon labeling. For bound ATP, a small negative band at 1260 cm−1 was observed in the IE phosphorylation spectrum (Fig. 2 E, top, bold line), which we attribute to the corresponding intensity decrease of the main 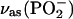 band of bound ATP. In addition, a ∪∩ band profile is evident with the negative band at 1207 cm−1 and the positive band at 1174 cm−1, which we assign to the isotope shift of the shoulder of the
band of bound ATP. In addition, a ∪∩ band profile is evident with the negative band at 1207 cm−1 and the positive band at 1174 cm−1, which we assign to the isotope shift of the shoulder of the 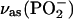 band.
band.
Between 1150 and 1100 cm−1, in the spectral region of the in-phase symmetric stretching vibration of α- and 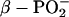 groups,
groups, 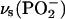 (32,36), four bands at 1145 (−), 1128 (+), 1110 (−), and 1103 (+) cm−1 are observed in the IE binding spectrum and the IE phosphorylation spectrum. We attribute them to two ∪∩ band profiles of bound ATP and one ∩∪ band profile of free ATP. The first ∪∩ band profile of bound ATP exhibits its negative band at 1145 cm−1 and its positive band at 1128 cm−1, the second has its negative band at 1110 cm−1 and the positive band at 1103 cm−1. Overlapped is the expected ∩∪ band profile of free ATP with the positive band contributing at 1128 cm−1.
(32,36), four bands at 1145 (−), 1128 (+), 1110 (−), and 1103 (+) cm−1 are observed in the IE binding spectrum and the IE phosphorylation spectrum. We attribute them to two ∪∩ band profiles of bound ATP and one ∩∪ band profile of free ATP. The first ∪∩ band profile of bound ATP exhibits its negative band at 1145 cm−1 and its positive band at 1128 cm−1, the second has its negative band at 1110 cm−1 and the positive band at 1103 cm−1. Overlapped is the expected ∩∪ band profile of free ATP with the positive band contributing at 1128 cm−1.
The absorption spectrum of free ATP shows a shoulder at 1087 cm−1, which is assigned to the out-of-phase 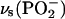 vibration (36). The isotope shift of this band of bound ATP gives rise to a ∪∩ band profile that is observed in both the IE binding spectrum and the IE phosphorylation spectrum. It consists of the negative band at 1085 cm−1, with a shoulder at 1094 cm−1, and a positive band near 1070 cm−1.
vibration (36). The isotope shift of this band of bound ATP gives rise to a ∪∩ band profile that is observed in both the IE binding spectrum and the IE phosphorylation spectrum. It consists of the negative band at 1085 cm−1, with a shoulder at 1094 cm−1, and a positive band near 1070 cm−1.
Summarizing the isotope effects upon β-labeling, we have identified several bands for bound ATP that involve the β-phosphate: those at 1260, 1207, 1144, 1110, and 1085 cm−1 for unlabeled ATP, and those at 1174, 1128, 1103, and 1070 cm−1 for β-labeled ATP. Several of these bands are obvious also in the ATP binding spectra: the positive bands at 1200, 1149, 1106, and 1093 cm−1 for unlabeled ATP (Fig. 2 B, thin line), and at 1128, 1100, and 1069 cm−1 for β-labeled ATP (Fig. 2 B, shaded line). The bound ATP bands identified are also present as negative bands in the phosphorylation spectra: at 1209, 1148, 1096 (shoulder), and 1087 cm−1 for unlabeled ATP (Fig. 2 D, thin line) and at 1178, 1103, and 1069 cm−1 for β-labeled ATP (Fig. 2 D, shaded line).
γ-Phosphate bands of bound ATP
IE spectra upon γ-labeling are shown in the bottom panel of Fig. 2 E. Downshifts because of isotopic substitution will give rise to ∪∩ band profiles for bound ATP in both types of IE spectra, and to ∩∪ band profiles for free ATP in the IE binding spectrum and for the phosphoenzyme phosphate group in the IE phosphorylation spectrum. Very similar IE phosphorylation spectra (see Fig. S1 in Supplementary Material) were obtained from independent experiments at slightly different conditions of which phosphorylation spectra were published previously (17).
The positive band at 1249 cm−1 in the IE binding spectrum (Fig. 2 E, bottom, thin line) is assigned to an intensity decrease of the 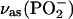 band of free ATP upon γ-phosphate labeling, in line with published (32,36) and our unpublished absorbance spectra.
band of free ATP upon γ-phosphate labeling, in line with published (32,36) and our unpublished absorbance spectra.
The band at 1130 cm−1 is observed in the IE phosphorylation spectrum and in the IE binding spectrum. It is therefore assigned to a γ-phosphate vibration of bound labeled ATP because bound ATP is the only species that contributes to both spectra and a positive band is expected for the labeled isotopomer. The corresponding negative band of unlabeled bound ATP is likely found at 1154 cm−1 in the IE binding spectrum and contributes to the negative band at 1147 cm−1 in the IE phosphorylation spectrum.
Another ∩∪ band profile for bound ATP is evident in both IE binding and phosphorylation spectra of γ-labeled ATP, with a negative band between 1098 and 1089 cm−1 indicating absorption of unlabeled ATP, and positive bands between 1070 and 1047 cm−1 indicating absorption of γ-labeled ATP.
In summary, we have identified two bands for bound ATP that involve the γ-phosphate: presumably one near 1154 cm−1 shifting to 1130 cm−1 upon γ-labeling, and another between 1098 and 1089 cm−1 shifting to 1070–1047 cm−1. Most of these bands are observed at similar positions as positive bands in the binding spectra of unlabeled ATP (1093 cm−1 and shoulder near 1086 cm−1; Fig. 2 B, thin line) and of γ-labeled ATP (1131 cm−1; Fig. 2 B, bold line). They are also found in the phosphorylation spectrum with unlabeled ATP as negative bands at 1148 and 1087 cm−1, and as a shoulder at 1096 cm−1 (Fig. 2 D, thin line).
Bands of the Ca2E1P phosphate
Bands that are present in the IE phosphorylation spectrum but not in the IE binding spectrum upon γ-labeling (Fig. 2 E, bottom) can be assigned to the Ca2E1P phosphate group. They are found at 1175 (+), 1147 (−), and 1113 cm−1 (+). The former two represent a ∩∪ profile expected for the phosphoenzyme phosphate with the positive 1175 cm−1 band indicating absorption of the unlabeled phosphate group and the negative 1147 cm−1 band that of the labeled group. The positive 1113 cm−1 band represents the absorption of the unlabeled phosphate group. The shifted corresponding band of the labeled phosphate group is expected to overlap with negative bands of unlabeled bound ATP in the 1098–1089 cm−1 region.
These assignments are at variance with our previous study (17), in which we tentatively assigned a band near 1131 cm−1 to the phosphoenzyme phosphate (Ca2E1P). Our more complete study here does not exclude this assignment, but suggests an alternative or additional assignment of the 1131 cm−1 band to γ-labeled bound ATP.
Other bands
In Fig. 2 B, three bands at 1175, 1164, and ∼1150 cm−1 in the binding spectra are always observed with ATP or labeled ATP. The insensitivity to labeling indicates that these bands cannot be assigned to β- and γ-phosphate. Instead they might be induced by conformational or environmental changes of side chains of ATPase. In general, this spectral region corresponds to the absorption region of the stretching vibration of single bonds. In particular ν(C-O) of protonated Asp and Glu could contribute, as could the bending vibration δ(C-OH) of Ser and Tyr, which is sensitive to changes in hydrogen bonding (53,54). Another possibility is an assignment of these bands to the stretching vibrations of the 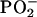 and C-O moieties of phospholipids.
and C-O moieties of phospholipids.
DISCUSSION
Interactions between phosphate groups and ATPase: β-phosphate
We have identified five bands of unlabeled bound ATP that are sensitive to labeling of the β-phosphate (see Fig. 2 E, top, and Table 1) at 1260, 1207, 1145, 1110, and 1085 cm−1. The former two represent 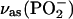 vibrations, the latter three
vibrations, the latter three 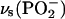 vibrations that are coupled to α- and γ-phosphate vibrations. Compared with bands of free ATP, the bands at 1260 and 1145 cm−1 are found at higher wavenumber, the bands at 1207 and 1110 cm−1 at lower wavenumber, and the band at 1085 cm−1 at similar wavenumber. The different absorbance in the absence/presence of the ATPase (Fig. 1) indicates the existence of interactions between the phosphates and ATPase resulting in altered P-O bond strengths and/or an altered phosphate chain conformation, both of which will change coupling between the vibrations. Coupling of vibrations between β-phosphate and γ-phosphate explains why we observe three bands for bound ATP in the
vibrations that are coupled to α- and γ-phosphate vibrations. Compared with bands of free ATP, the bands at 1260 and 1145 cm−1 are found at higher wavenumber, the bands at 1207 and 1110 cm−1 at lower wavenumber, and the band at 1085 cm−1 at similar wavenumber. The different absorbance in the absence/presence of the ATPase (Fig. 1) indicates the existence of interactions between the phosphates and ATPase resulting in altered P-O bond strengths and/or an altered phosphate chain conformation, both of which will change coupling between the vibrations. Coupling of vibrations between β-phosphate and γ-phosphate explains why we observe three bands for bound ATP in the 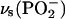 region that are sensitive to β-phosphate labeling as opposed to only two bands observed for free ATP (36).
region that are sensitive to β-phosphate labeling as opposed to only two bands observed for free ATP (36).
Higher wavenumbers of stretching vibration bands indicate stronger bonds and lower wavenumbers weaker bonds. However, because of coupling it is difficult to draw conclusions regarding the β-phosphate P-O bond strengths. Probably the best local reporters of β-phosphate bond strengths are bands of labeled β-phosphate, because labeling will reduce the coupling between α and β-phosphates. They are found at 1174 and ∼1070 cm−1 at positions similar to the corresponding bands of free β-labeled ATP (1182 and 1060 cm−1, according to our IE absorption spectrum). This indicates that P-O bond strengths of bound β-phosphate are similar to those of free ATP, i.e., that hydrogen bonds of free ATP to water are largely replaced by interactions with the protein for bound ATP. In line with this, both β-phosphate oxygens are close to Arg560 (2.6 and 3.0 Å), the closer oxygen might interact additionally with water and the more distant one seems to form a hydrogen bond with the ribose 3′-OH in the crystal structures of Ca2E1AMPPCP (11,12).
Interactions between phosphate groups and the ATPase: γ-phosphate
Two bands were found to be sensitive to γ-phosphate labeling (Fig. 2 E, bottom): likely at 1154, and one between 1098 and 1089 cm−1. The vibrations corresponding to the 1154 cm−1 band and the band between 1098 and 1089 cm−1 seem also to be coupled to the β-phosphate. These bands are in the region of of the absorption of the asymmetric stretching vibration of the 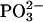 group,
group, 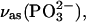 where free ATP shows only one band near 1120 cm−1. Thus, this band seems to split into two components upon binding of ATP to the ATPase. The average wavenumber is similar to the wavenumber of the free ATP band, indicating a similar bond strength of bound and free ATP.
where free ATP shows only one band near 1120 cm−1. Thus, this band seems to split into two components upon binding of ATP to the ATPase. The average wavenumber is similar to the wavenumber of the free ATP band, indicating a similar bond strength of bound and free ATP.
Comparison to Ras
Similar studies with isotopically labeled phosphates have been performed previously on GTP binding to Ras (31,33–35), and have identified bound GTP bands near 1263 (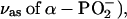 1216 (
1216 (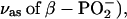 1158 and 1142 (
1158 and 1142 (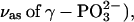 as well as 1124 and 1090 cm−1
as well as 1124 and 1090 cm−1 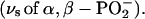 These bands are generally found at similar positions as for ATP bound to the ATPase but the extent of coupling between the phosphate vibrations seems to be different: the α-, β-, and γ-phosphate vibrations of GTP bound to Ras seem to be largely decoupled, whereas they are coupled for ATP bound to the ATPase.
These bands are generally found at similar positions as for ATP bound to the ATPase but the extent of coupling between the phosphate vibrations seems to be different: the α-, β-, and γ-phosphate vibrations of GTP bound to Ras seem to be largely decoupled, whereas they are coupled for ATP bound to the ATPase.
The Ca2E1P phosphoenzyme
This study has identified two bands of the Ca2E1P phosphate group that originate from the P-O stretching vibrations of the terminal P-O bonds, namely, the bands at 1175 and 1113 cm−1. Up to three bands are expected for the P-O stretching vibrations in an asymmetric protein environment and the third band could be located at 1130 cm−1, according to our previous assignment (17), but an additional or alternative assignment of this band is possible. The model compound acetyl phosphate in the symmetric environment of an aqueous solution shows two bands, that of the degenerated asymmetric stretching vibration at 1132 cm−1 and that of the symmetric stretching vibration at 982 cm−1 (17). For the E2P phosphate, bands at 1194, 1137, and 1115 cm−1 were found (19).
The Ca2E1P band at 1175 cm−1 indicates that the average terminal P-O bond is stronger than that of acetyl phosphate in water. Theoretically, a second explanation is possible: that the upshift is caused by a change in bond geometry. However, an increase in O-P-O bond angle of ∼15° is required to shift the antisymmetric stretching vibration from 1132 cm−1 to 1175 cm−1 (51). This required bond angle change seems to be unrealistically high and in the wrong direction, given that our estimate for the E2P phosphate is a decrease in bond angle by 4° from the value for acetyl phosphate (19). Thus, we consider it unlikely that the shift from 1132 cm−1 to 1175 cm−1 is caused solely by a geometry change.
The highest wavenumber band observed for the Ca2E1P phosphate at 1175 cm−1 is intermediate in position between those observed for E2P (1194 cm−1) and acetyl phosphate (1132 cm−1) (19). This indicates that the average terminal P-O bond strength of Ca2E1P is closer to that of E2P than to that of acetyl phosphate. Because there is some doubt regarding the assignment of the 1130 cm−1 to the phosphate group we could assign only two of the maximal three bands of the Ca2E1P phosphate group, making it impossible to calculate bond strengths of the terminal and the bridging P-O bonds for Ca2E1P as previously done for E2P (19). However, they can be estimated by regarding the highest wavenumber band as a measure of the average bond strength of the terminal P-O bonds. This estimation is based on the observation of only one of the expected three P-O bands of Ca2E1P and thus may lead to wrong estimates if the other P-O vibrations of Ca2E1P have frequencies that are very different from those of the E2P P-O vibrations. However, this is not expected because the crystal structures of Ca2E1P and E2P analogs show very similar phosphate environments. The subtle differences described below between Ca2E1P and E2P will not be detectable with x-ray crystallography because the underlying bond length changes are much smaller than the resolution of the x-ray structures.
In this estimation of P-O bond strengths in Ca2E1P we assume that the bond strength is proportional to the wavenumber of the highest wavenumber band. This approximation can be validated for the comparison between E2P and acetyl phosphate, for which a more sophisticated calculation of bond strengths is available based on the observation of all terminal P-O vibrations. This calculation gave 4.9% stronger P-O bonds for E2P in terms of bond valence (19) and the estimation assuming proportionality between bond strength and wavenumber of the highest wavenumber band gives 5.5% stronger P-O bonds for E2P. Thus, the simple approximation intended for Ca2E1P yields realistic estimates when applied to E2P.
Applied to Ca2E1P, 1.6% (1175/1194 cm−1 = 0.984) weaker terminal P-O bonds are inferred for Ca2E1P as compared to E2P, or 3.8% (1175/1132 cm−1 = 1.038) stronger bonds as compared to acetyl phosphate in water. By comparison with the known P-O bond valences of E2P and acetyl phosphate (19), the bond valence of the terminal P-O bonds of Ca2E1P is estimated to be close to 1.39 vu and that of the bridging P-O bond 0.83 vu (atomic valence of phosphorus minus three times the bond valence of the terminal P-O bonds). The corresponding values for E2P are 1.41 and 0.77 vu, and for acetyl phosphate 1.34 and 0.97 vu (19). Thus, the bond strength of the bridging P-O bond is ∼15% lower than for acetyl phosphate but ∼8% higher than for E2P.
A reduced bond strength results in a reduced bond energy. The bond energy of the bridging P-O bond of E2P is ∼18% lower than that of acetyl phosphate using the average value obtained from four relationships between bond energy and other bond parameters (19). Assuming a typical single bond energy for the bridging P-O bond of acetyl phosphate of 420 kJ/mol, the bond energy of the bridging P-O bond of E2P is thus reduced by 77 kJ/mol (range 64–90 kJ/mol) with respect to that of acetyl phosphate (19). Assuming linearity between bond valence and bond energy, the corresponding reduction of bond energy for Ca2E1P is ∼62 kJ/mol (range 51–72 kJ/mol, calculated from the relative reductions in bond order: ∼15% for Ca2E1P/∼18% for E2P ≈ 14.61/18.25% = 0.80 times the reduction in E2P). Such a linear relationship between bond valence and bond energy gives similar values as correlations between bond energy and other bond parameters (19).
A weaker bridging P-O bond and stronger terminal P-O bonds are expected for a transition state in a dissociative phosphate transfer reaction. For the Ca2E1P intermediate, the phosphate transfer is to ADP under ATP synthesis. Thus the bond strength changes observed in going from aqueous environment to the Ca2E1P environment of the phosphate group seem to indicate that the enzyme environment prepares the phosphate group for a transfer reaction with dissociative character, as also found for E2P (19). Recently, x-ray structures have been interpreted as giving evidence for strongly associative phosphate transfer reactions of both phosphoenzyme intermediates (11,55). Al3−, the mimic of phosphorus in the x-ray studies, has a distance to the leaving Asp351 oxygen atom of 1.9–2.1 Å and to the attacking oxygen of 1.8–2.0 Å in the Ca2E1P transition-state analog (11,56), and the respective distances in the E2P transition-state analog are both 2.1 Å (55). From these Al-O bond lengths in the phosphoenzyme transition state analogs, P-O bond strengths can be calculated using the bond length L versus bond valence s relationship s = exp{(162 pm − L)/37 pm} (57), giving bond valences of 0.27–0.58 vu for bonds between phosphorus and leaving or attacking oxygen (a single bond would have a bond valence of 1 vu). Thus, evaluation of the x-ray structures reveals that the transition states of Ca2E1P and E2P seem to have considerable associative and dissociative character, which is in agreement with the dissociative character inferred from our infrared experiments.
In a 100% dissociative phosphate transfer reaction, the bond energy of the P-O bond aspartyl or acetyl oxygen has to be overcome to reach the transition state. In a simplistic view and in the absence of stabilizing interactions in the transition state, the activation energy will be the bond energy of the breaking bond. Thus, the lower P-O bond energies in Ca2E1P and E2P, as compared to acetyl phosphate, will reduce the activation energy by the bond energy difference. This enhances the rate of Ca2E1P dephosphorylation by a factor of 109–1012 compared to acetyl phosphate. Compared to E2P, the bond energy in Ca2E1P is ∼27 kJ/mol higher (≈ {420 − 77}kJ/mol × 1.08), corresponding to a slowing down of phosphate transfer by a factor of 104–105 in Ca2E1P with respect to E2P. This will inhibit the dephosphorylation of Ca2E1P by water, which is an “unwanted” reaction that would lead to uncoupling of ATP hydrolysis and Ca2+ transport. In reality, the bond energy of the breaking P-O bond will be one of many energy contributions that change in the approach to the transition state and the effects on the reaction rate will be less than outlined above in the presence of some associative character of the transition state.
In summary, the estimated P-O bond strengths of Ca2E1P indicate stronger terminal P-O bonds and a weaker bridging P-O bond due to weaker interactions with the environment, as discussed previously for E2P (19). The effect is, however, less than for E2P. The weaker bridging P-O bond as compared to acetyl phosphate will reduce the activation energy required for bond breakage upon phosphate transfer, which will be one cause for the fast transfer of the aspartyl phosphate to ADP (58-60). On the other hand, the bridging P-O bond of Ca2E1P is stronger than that of E2P, which will make the aspartyl phosphate of Ca2E1P less susceptible to attack by water.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We gratefully acknowledge W. Mäntele (Institut für Biophysik, Johann Wolfgang Goethe-Universität) for provision of facilities in the initial phase of this project. The authors thank W. Hasselbach (Max-Planck-Institut, Heidelberg) for the gift of Ca2+-ATPase, J. E. T. Corrie (National Institute for Medical Research, London) for the preparation of caged compounds, and M. Webb (National Institute for Medical Research, London) for isotopic labeling of ATP.
This work was supported by Deutsche Forschungsgemeinschaft grants Ba1887/2-1 and Ma1054/18-3, Vetenskapsrådet projektbidrag 621-2002-5884, and Knut och Alice Wallenbergs Stiftelse bidrag 2002.0115.
Abbreviations used: ATP, adenosine 5′-triphosphate; ADP, adenosine 5′-diphosphate; GTP, guanosine triphosphate; AMPPCP, (β,γ-methyl)adenosine 5′-triphosphate; Ca2E1, Ca2+ bound form of Ca2+-ATPase; Ca2E1AMPPCP, AMPPCP-ATPase complex; Ca2E1ATP, ATP-ATPase complex; Ca2E1P, ADP-sensitive phosphoenzyme; E2P, ADP-insensitive phosphoenzyme; FTIR, Fourier transform infrared; IE, isotope effect; SR, sarcoplasmic reticulum; vu, valence units.
References
- 1.Hasselbach, W., and M. Makinose. 1961. Die Calciumpumpe der “Erschlaffungsgrana” des Muskels und ihre Abhängigkeit von der ATP-Spaltung. Biochem. Z. 333:518–528. [PubMed] [Google Scholar]
- 2.Andersen, J. P. 1989. Monomer-oligomer equilibrium of sarcoplasmic reticulum Ca-ATPase and the role of subunit interaction in the Ca2+ pump mechanism. Biochim. Biophys. Acta. 988:47–72. [DOI] [PubMed] [Google Scholar]
- 3.MacLennan, D. H., and N. M. Green. 2000. Pumping ions. Nature. 405:633–634. [DOI] [PubMed] [Google Scholar]
- 4.Lee, A., and J. East. 2001. What the structure of a calcium pump tells us about its mechanism. Biochem. J. 356:665–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toyoshima, C., and G. Inesi. 2004. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 73:269–292. [DOI] [PubMed] [Google Scholar]
- 6.Toyoshima, C., M. Nakasako, H. Nomura, and H. Ogawa. 2000. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature. 405:647–655. [DOI] [PubMed] [Google Scholar]
- 7.Hasselbach, W. 1979. The sarcoplasmic calcium pump. Top. Curr. Chem. 78:1–56. [PubMed] [Google Scholar]
- 8.Hasselbach, W. 1983. Energetics and electrogenicity of the SR Ca-Pump. Annu. Rev. Physiol. 45:325–339. [DOI] [PubMed] [Google Scholar]
- 9.Martonosi, A. N., and S. Pikula. 2003. The network of calcium regulation in muscle. Acta Biochim. Pol. 50:1–30. [PubMed] [Google Scholar]
- 10.De Meis, L., and A. Vianna. 1979. Energy interconversion by the Ca2+-dependent ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 48:275–292. [DOI] [PubMed] [Google Scholar]
- 11.Sørensen, T. L.-M., J. V. Møller, and P. Nissen. 2004. Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science. 304:1672–1675. [DOI] [PubMed] [Google Scholar]
- 12.Toyoshima, C., and T. Mizutani. 2004. Crystal structure of the calcium pump with a bound ATP analogue. Nature. 430:529–535. [DOI] [PubMed] [Google Scholar]
- 13.Von Germar, F., A. Barth, and W. Mäntele. 2000. Structural changes of the sarcoplasmic reticulum Ca2+-ATPase upon nucleotide binding studied by Fourier transform infrared spectroscopy. Biophys. J. 78:1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu, M., and A. Barth. 2002. Mapping nucleotide binding site of calcium ATPase with IR spectroscopy: effects of ATP γ-phosphate binding. Biospectroscopy. 67:267–270. [DOI] [PubMed] [Google Scholar]
- 15.Liu, M., and A. Barth. 2003. Mapping interactions between the Ca2+-ATPase and its substrate ATP with infrared spectroscopy. J. Biol. Chem. 278:10112–10118. [DOI] [PubMed] [Google Scholar]
- 16.Liu, M., and A. Barth. 2003. TNP-AMP binding to the sarcoplasmic reticulum Ca2+-ATPase studied by infrared spectroscopy. Biophys. J. 85:3262–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barth, A., and W. Mäntele. 1998. ATP-induced phosphorylation of the sarcoplasmic reticulum Ca2+ ATPase: Molecular interpretation of infrared difference spectra. Biophys. J. 75:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barth, A. 1999. Phosphoenzyme conversion of the sarcoplasmic reticulum Ca2+ ATPase. Molecular interpretation of infrared difference spectra. J. Biol. Chem. 274:22170–22175. [DOI] [PubMed] [Google Scholar]
- 19.Barth, A., and N. Bezlyepkina. 2004. P-O bond destabilisation accelerates phosphoenzyme hydrolysis of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 279:51888–51896. [DOI] [PubMed] [Google Scholar]
- 20.Gerwert, K. 1993. Molecular reaction mechanisms of proteins as monitored by time-resolved FTIR spectroscopy. Curr. Opin. Struct. Biol. 3:769–773. [DOI] [PubMed] [Google Scholar]
- 21.Mäntele, W. 1996. Infrared and Fourier-transform infrared spectroscopy. In Biophysical Techniques in Photosynthesis. J. Amesz and A. J. Hoff, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 137–160.
- 22.Barth, A., and C. Zscherp. 2000. Substrate binding and enzyme function investigated by infrared spectroscopy. FEBS Lett. 477:151–156. [DOI] [PubMed] [Google Scholar]
- 23.Barth, A., and C. Zscherp. 2002. What vibrations tell us about proteins. Q. Rev. Biophys. 35:369–430. [DOI] [PubMed] [Google Scholar]
- 24.Jung, C. 2000. Insight into protein structure and protein-ligand recognition by Fourier transform infrared spectroscopy. J. Mol. Recognit. 13:325–351. [DOI] [PubMed] [Google Scholar]
- 25.Vogel, R., and F. Siebert. 2000. Vibrational spectroscopy as a tool for probing protein function. Curr. Opin. Chem. Biol. 4:518–523. [DOI] [PubMed] [Google Scholar]
- 26.Wharton, C. W. 2000. Infrared spectroscopy of enzyme reaction intermediates. Nat. Prod. Rep. 17:447–453. [DOI] [PubMed] [Google Scholar]
- 27.Kim, S., and B. A. Barry. 2001. Reaction-induced FT-IR spectroscopic studies of biological energy conversion in oxygenic photosynthesis and transport. J. Phys. Chem. 105:4072–4083. [Google Scholar]
- 28.Zscherp, C., and A. Barth. 2001. Reaction-induced infrared difference spectroscopy of the study of protein reaction mechanisms. Biochemistry. 40:1875–1883. [DOI] [PubMed] [Google Scholar]
- 29.Liu, M., and A. Barth. 2004. Phosphorylation of the sarcoplasmic reticulum Ca2+-ATPase from ATP and ATP analogs studied by infrared spectroscopy. J. Biol. Chem. 279:49902–49909. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan, J. H., B. Forbush, and J. F. Hoffman. 1978. Rapid photolytic release of ATP from a protected analogue: utilization by the Na:K pump of human red blood cell ghosts. Biochemistry. 17:1929–1935. [DOI] [PubMed] [Google Scholar]
- 31.Cepus, V., A. J. Scheidig, R. S. Goody, and K. Gerwert. 1998. Time-resolved FTIR studies of the GTPase reaction of H-ras p21 reveal a key role for the β-phosphate. Biochemistry. 37:10263–10271. [DOI] [PubMed] [Google Scholar]
- 32.Wang, J. H., D. G. Xiao, H. Deng, R. Callender, and M. R. Webb. 1998. Vibrational study of phosphate modes of GDP and GTP and their interaction with magnesium in aqueous solution. Biospectroscopy. 4:219–227. [DOI] [PubMed] [Google Scholar]
- 33.Du, X., H. Frei, and S.-H. Kim. 2000. The mechanism of GTP hydrolysis by Ras probed by Fourier transform infrared spectroscopy. J. Biol. Chem. 275:8492–8500. [DOI] [PubMed] [Google Scholar]
- 34.Allin, C., and K. Gerwert. 2001. Ras catalyzes GTP hydrolysis by shifting negative charges from γ- to β-phosphate as revealed by time-resolved FTIR difference spectroscopy. Biochemistry. 40:3037–3046. [DOI] [PubMed] [Google Scholar]
- 35.Cheng, H., S. Sukal, R. Callender, and T. S. Leyh. 2001. γ-phosphate protonation and pH-dependent unfolding of the Ras·GTP·Mg2+ complex. J. Biol. Chem. 276:9931–9935. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi, H., H. Murata, and I. Harada. 1988. Interaction of adenosine 5′-triphosphate with Mg2+: vibrational study of coordination sites by use of 18O-labeled triphosphates. J. Am. Chem. Soc. 110:392–397. [Google Scholar]
- 37.Barth, A., K. Hauser, W. Mäntele, J. E. T. Corrie, and D. R. Trentham. 1995. Photochemical release of ATP from ‘caged ATP’ studied by time-resolved infrared spectroscopy. J. Am. Chem. Soc. 117:10311–10316. [Google Scholar]
- 38.Barth, A., J. E. T. Corrie, M. J. Gradwell, Y. Maeda, W. Mäntele, T. Meier, and D. R. Trentham. 1997. Time-resolved infrared spectroscopy of intermediates and products from photolysis of 1-(2-nitrophenyl)ethyl phosphates: reaction of the 2-nitrosoacetophenone byproduct with thiols. J. Am. Chem. Soc. 119:4149–4159. [Google Scholar]
- 39.De Meis, L., and W. Hasselbach. 1971. Acetyl phosphate as substrate for calcium uptake in skeletal muscle microsomes. J. Biol. Chem. 246:4759–4763. [PubMed] [Google Scholar]
- 40.Shigekawa, M., S. Wakabayashi, and H. Nakamura. 1983. Reaction mechanism of calcium-dependent ATPase of SR. J. Biol. Chem. 258:8698–8707. [PubMed] [Google Scholar]
- 41.Lacapere, J.-J., and F. Guillain. 1990. Reaction mechanism of Ca-ATPase of SR. Equilibrium and transient study of phosphorylation with Ca.ATP. J. Biol. Chem. 265:8583–8589. [PubMed] [Google Scholar]
- 42.Suzuki, H., S. Nakamura, and T. Kanazawa. 1994. Effects of divalent cations bound to the catalytic site on ATP-induced conformational changes in the sarcoplasmic reticulum Ca2+-ATPase: Stopped- flow analysis of the fluorescence of N-acetyl-N′-(5-sulfo-1-naphthyl)ethylenediamine attached to cysteine-674. Biochemistry. 33:8240–8246. [DOI] [PubMed] [Google Scholar]
- 43.Barth, A., F. von Germar, W. Kreutz, and W. Mäntele. 1996. Time-resolved infrared spectroscopy of the Ca2+-ATPase. The enzyme at work. J. Biol. Chem. 271:30637–30646. [DOI] [PubMed] [Google Scholar]
- 44.Barth, A., W. Kreutz, and W. Mäntele. 1994. Changes of protein structure, nucleotide microenvironment, and Ca2+ binding states in the catalytic cycle of sarcoplasmic reticulum Ca2+ ATPase: investigation of nucleotide binding, phosphorylation, and phosphoenzyme conversion by FTIR difference spectroscopy. Biochim. Biophys. Acta. 1194:75–91. [DOI] [PubMed] [Google Scholar]
- 45.Kijima, Y., E. Ogunbunmi, and S. Fleischer. 1991. Drug action of thapsigargin on the Ca2+ pump protein of sarcoplasmic reticulum. J. Biol. Chem. 266:22912–22918. [PubMed] [Google Scholar]
- 46.Sagara, Y., F. Fernandez-Belda, L. De Meis, and G. Inesi. 1992. Characterization of the inhibition of intracellular Ca2+ transport ATPases by thapsigargin. J. Biol. Chem. 267:12606–12613. [PubMed] [Google Scholar]
- 47.Fortea, M. I., F. Soler, and F. Fernandez-Belda. 2001. Unravelling the interaction of thapsigargin with the conformational states of Ca2+-ATPase from skeletal sarcoplasmic reticulum. J. Biol. Chem. 276:37266–37272. [DOI] [PubMed] [Google Scholar]
- 48.Barth, A., W. Mäntele, and W. Kreutz. 1991. Infrared spectroscopic signals arising from ligand binding and conformational changes in the catalytic cycle of sarcoplasmic reticulum Ca2+ ATPase. Biochim. Biophys. Acta. 1057:115–123. [DOI] [PubMed] [Google Scholar]
- 49.Pick, U., and S. Bassilian. 1981. Modification of the ATP binding site of the Ca2+-ATPase from sarcoplasmic reticulum by fluorescein isothiocyanate. FEBS Lett. 123:127–130. [DOI] [PubMed] [Google Scholar]
- 50.Highsmith, S. 1984. Evidence that the ATP binding site of SR Ca-ATPase has a Mg ion binding subsite. Biochem. Biophys. Res. Commun. 124:183–189. [DOI] [PubMed] [Google Scholar]
- 51.Deng, H., J. Wang, R. Callender, and W. J. Ray. 1998. Relationship between bond stretching frequencies and internal bonding for [16O4]- and [18O4]phosphates in aqueous solution. J. Phys. Chem. B. 1022:3617–3623. [Google Scholar]
- 52.Brintzinger, H. 1963. The structures of adenosine triphosphate metal ion complexes in aqueous solution. Biochim. Biophys. Acta. 77:343–345. [DOI] [PubMed] [Google Scholar]
- 53.Madec, C., J. Lauransan, and C. Garrigou-Lagrange. 1978. Etude du spectre de vibration de la dl-serine et des ses dérivés deuteries. Can. J. Spectrosc. 23:166–172. [Google Scholar]
- 54.Hienerwadel, R., A. Boussac, J. Breton, B. Diner, and C. Berthomieu. 1997. Fourier transform infrared difference spectroscopy of photosystem II tyrosine D using site-directed mutagenesis and specific isotope labeling. Biochemistry. 36:14712–14723. [DOI] [PubMed] [Google Scholar]
- 55.Olesen, C., T. L.-M. Sørensen, R. C. Nielsen, J. V. Møller, and P. Nissen. 2004. Dephosphorylation of the calcium pump coupled to counterion occlusion. Science. 306:2251–2255. [DOI] [PubMed] [Google Scholar]
- 56.Toyoshima, C., H. Nomura, and T. Tsuda. 2004. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 432:361–368. [DOI] [PubMed] [Google Scholar]
- 57.Brown, I. D. 2002. The Chemical Bond in Inorganic Chemistry. The Bond Valence Model. Oxford University Press, Oxford, UK.
- 58.Pickart, C. M., and W. P. Jencks. 1984. Energetics of the calcium-transporting ATPase. J. Biol. Chem. 259:1629–1643. [PubMed] [Google Scholar]
- 59.Inesi, G., and L. De Meis. 1985. Kinetic regulation of catalytic and transport activities in sarcoplasmic reticulum ATPase. In The Enzymes of Biological Membranes, 2nd ed. A. Martonosi, editor. Plenum Press, New York, London. 157–191.
- 60.Inesi, G., M. Kurzmack, and D. Lewis. 1988. Kinetic and equilibrium characterization of an energy-transducing enzyme and its partial reactions. Methods Enzymol. 157:154–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.