

Abstract
In the past few years, many studies have attempted to measure the strength of a single molecular bond. In general, these experiments consisted in pulling on the bond and measuring the force necessary to dissociate the molecules. However, seemingly contradictory experimental results led to draw the intriguing conclusion that the strength of the bond could depend on the experiment even if the pulling conditions are similar: this paradox was first observed on the widely used streptavidin-biotin bond. Here, by doing supplementary measurements and by reanalyzing the controversial experimental results using Kramers' theory, we show that they can be conciliated. This allows us to show that the strength of a bond is very sensitive to the history of its formation, which is the key to the paradox.
INTRODUCTION
The advent of new techniques such as atomic force microscopy (AFM), the biomembrane force probe (BFP), optical tweezers, flow chambers, etc., has led to a fast-growing number of experimental articles on single molecular bonds (1–6). In these experiments a microscopic but optically visible event, like the bending of a spring or the displacement of a bead, is detected in order to measure a force. The authors usually try to interpret their results at the molecular level in terms of intrinsic parameters of the bonds such as association and dissociation constants, binding energy, or energy landscape. Such interpretations in which the observation of a visible event is supposed to directly give information on properties at the nanometric level may sometimes be presumptuous. Subtle molecular mechanisms can render the problem much more complicated than expected and lead to erroneous conclusions with the usual analysis. Recent efforts have been made to refine this analysis (7–11).
Many measurements have been reported on the streptavidin-biotin bond (1,2,6,12). A comparative reading of these results shows that some of them, among the major ones, seem to be in complete contradiction: the force of the bond under given pulling conditions depends on the technique used. By doing an in-depth analysis of these measurements and conducting complementary experiments on functionalized DNA strands with a BFP, we show that this contradiction is due to the slow molecular rearrangement of the bond that takes time to reach its most stable state. This phenomenon had never been experimentally demonstrated at the single-molecule level before.
MATERIALS AND METHODS
Force measurements with a biomembrane force probe
The biomembrane force probe (13) uses a force transducer made of a biotinylated red blood cell maintained by a glass micropipette and with a streptavidin-coated glass microbead attached on its top. The red blood cell is used as a spring of known stiffness k, which is tuned by the controlled aspiration pressure applied by the holding micropipette. The streptavidin-coated glass bead can be decorated with molecules of interest (here biotinylated DNA strands) which will be displayed on the tip of the force transducer (see Fig. 1). The glass bead also enables precise video tracking, because when observed with a slightly unfocused optical microscope, it displays a light spot with Gaussian intensity profile on its center.
FIGURE 1.

Description of the BFP experiment with DNA strands. The spring is a red blood cell whose tension is controlled by the aspiration in the pipette holding it (left pipette). A glass bead coated with DNA strands is attached to this red cell. The DNA is bound to the bead by single streptavidin-biotin bonds. The other pipette holds aggregates of latex particles that nonspecifically bind to the DNA strands. When the glass bead and the latex particles are brought in contact, the DNA strongly attaches to the latex particles. Upon separation, the streptavidin-biotin bond is the first one to unbind. This protocol allows the measurements of rupture forces of bonds that have been given several hours to form.
The force measurements consist in approach-contact-retraction automatized cycles of the BFP force transducer. During the approach phase, the force transducer is translated with constant speed into contact with target latex microbeads maintained by another micropipette facing the first one. The contact is ensured by a 20-pN compression force exerted on the force transducer (i.e., a compression of the red blood cell). The contact is held during 100 ms, and then the retraction phase is initiated. If a bond has been formed between the BFP tip (the DNA-coated bead) and the facing latex bead surface during the contact, a force will be exerted on the force transducer. Reciprocally the bond between the BFP tip and the latex beads will experience a force, until it breaks. The force exerted on the bond between the BFP tip and the target latex bead during the retraction phase is a ramp, with a slope being the loading rate r. The force exerted on the bond is then F = r × t where t is the time, starting from the beginning of the retraction phase. The force-extension curves are directly visualized on computer screen during experiments, and are also recorded and further analyzed by a homemade analysis program.
For BFP force-transducer and target latex-microbeads manipulations, micromanipulators were mounted on the stage of a Leica inverted microscope (DMIRB type, Leica, Solms, Germany). Glass micropipettes with inner diameter of 1.5–2.5 μm were attached and connected to homemade water manometers for pressure adjustment. The red blood cell membrane tension is set by the pipette aspiration pressure, and the spring constant of the cell is obtained at the beginning of each experiment for each cell by multiplying this pressure by a geometrical factor measured for each red blood cell with a calibrated program used with a video device coupled to the microscope (63× Leica objective with 1.5× supplementary lens, camera purchased from JAI, Yokohama, Japan). The micropipette holding the force transducer is coupled to a linear piezoelectric translator (Physik Instrumente, Karlsruhe, Germany) connected to a digital-analog converter connected to a personal computer. The DNA-coated bead on the top of the red blood cell is tracked by video processing with the camera connected to the microscope. The tracking procedure was kindly provided by V. Croquette and adapted to our device. Video tracking gives the bead position, whereas the piezoelectric translator provides the position of the extremity of the micropipette holding the red blood cell. The difference of these two positions gives the elongation of the red blood cell with an accuracy of a few nanometers. When multiplying it by the spring constant k of the red blood cell, the force exerted on the force transducer is obtained with an accuracy of a few picoNewtons. The desired loading rate is given by r = k × v, where v is the constant retraction speed set by the piezoelectric. This speed is adapted for each red blood cell. Effective loading rate was afterward verified on the force-extension curves.
Micropipettes were first obtained by elongating borosilicate glass capillaries (1-mm outer diameter, 0.78-mm inner diameter, Harvard Apparatus, Holliston, MA) with a micropipette puller (P-2000, Sutter Instrument, Novato, CA). Next, a custom-made microforge allowed opening the extremity of the micropipettes at the desired diameter.
Experiments were conducted in a chamber made of two glass coverslips facing each other where ∼200 μL of fluid was held by capillary forces. Micropipettes could access to the chamber from its sides. Before red blood cells and beads introduction, the chamber was incubated for 1 h in phosphate-buffered saline (PBS 0.01 M, 150 mM NaCl, 290 mOsm, pH 7.4) with 5% bovine serum albumin (BSA, Sigma-Aldrich, Lyon, France). The chamber was next washed several times in PBS, and all experiments were conducted in PBS, pH 7.4, at room temperature.
Biocytin (Sigma-Aldrich) at a final concentration of 0.1 mg/ml was used to block the empty streptavidin sites.
Red blood cells were covalently linked with PEG-biotin polymers, following the protocol kindly provided by E. Evans. More details about this protocol can be read in Merkel et al. (2).
DNA-coated silica and latex microbeads (Fig. 2)
Amino silane groups (n-(2-Aminoethyl)-3-aminopropylmethydimethoxysilane, ABCR GmbH, Karlsruhe, Germany) were covalently bound to glass microbeads (uniform silica microspheres, mean diameter 3 μm, Bangs Laboratories, Fishers, IN). A mixture of Amine-reactive polyethylene oxide polyethylene glycol with biotin (NHS-PEG3400-biotin, Interchim, Montluçon, France) and Sulfo-MBS (Pierce, c/o Touzard et Matignon, Les Ullis, France) was then covalently bound to the silanized microbeads. The biotinylated microbeads were finally incubated in a 2 mg/mL streptavidin solution (Jackson Immunoresearch Laboratories, West Grove, PA). Beads were finally washed several times in PBS and stored in PBS at 4°C. J.-F. Allemand kindly provided 15-kb DNA strands that were biotinylated at one end with a single biotin (biotinylated ends were purchased from Roche, Nutley, NJ). The day before each experiment, streptavidin-coated silica beads previously prepared were incubated in a 48 μL of PBS + 2 μL of 0.8 ng/μL DNA solution overnight at 4°C under low agitation. It can be noted that the attachment between these DNA-coated beads and the biotinylated red blood cells was possible because the DNA beads still exhibited free streptavidin sites on their surface.
Latex microbeads (Roche Diagnostics, Mannheim, Germany) were stored in PBS. They had the form of few micrometers aggregates under experimental conditions.
RESULTS AND DISCUSSION
The streptavidin-biotin paradox
Because of its high affinity as a noncovalent bond, the streptavidin-biotin complex is well-known (14,15) and widely studied (see for instance (1,2,6,16,17)). It is also often used to couple molecules. For instance, DNA stretching has been studied by grafting each extremity of the double strand to latex beads through a single streptavidin-biotin bond (18,19). After attachment to the beads, the DNA is stretched with a controlled pulling force. Up to 80 pN, the bonds are stable over 1 min, whereas, when the pulling force exceeds 100 pN, one of the bonds often breaks after a few seconds. This strongly contradicts another work where the rupture force of the bond was measured with a BFP (2). In the latter experiments, two glass beads, one coated with ligand (biotin) and the other one with receptor (streptavidin), are brought into contact to allow a single bond to form. An increasing traction is then immediately applied on the bond until separation of the molecules. The rate at which the traction is applied (variation of the force with time, df/dt) is called the loading rate. Because thermal fluctuations will ultimately be responsible for bond breakage when traction is applied, two rupture force measurements on the same bond will give different results. A consequence of this nonreproducibility is that the strength of the bond is not characterized by a given force but by a distribution of the rupture force that depends on the loading rate. In the BFP experiments, rupture force distribution of the streptavidin-biotin complex was obtained for several loading rates. A convenient way of visualizing the robustness of the bond is to plot the most likely rupture force as a function of the loading rate (Fig. 3). From the BFP measurements, it can be deduced that for a pulling force of 75 pN, an upper bound for the lifespan of the bond is 75 ms, which is at least two orders-of-magnitude smaller than the one obtained with the DNA experiment. Hence, one has to reach the senseless conclusion that for a given constant pulling force, the lifespan of the bond will depend on the technique used to apply the force. This is the streptavidin-biotin paradox. The rest of the article is devoted to understanding its origin.
FIGURE 3.

Experimental curve obtained by Evans' group (adapted from Merkel et al. (2)) of the most likely rupture force of a single streptavidin-biotin bond as a function of the loading rate. Two regimes can be observed as indicated by the two linear slopes.
Three energy barriers in the streptavidin-biotin landscape
To obtain hints about the origin of the paradox, we shall focus on other fundamental results related to the streptavidin-biotin complex. Firstly, molecular dynamics simulations provide relevant information about the energy landscape of the bond. By pulling a biotin out of an avidin binding pocket in half a nanosecond, Schulten's group has been able to completely follow the trajectory of the biotin being extracted (17). From this trajectory, they approximated the neighborhood of the minima in the energy landscape (Fig. 4). This study indicates that there are three minima. The two intermediate metastable states can be found at ∼0.3 nm and 1.0 nm from the deepest one. The presence of three minima indicates that there are also three barriers in the energy landscape. Their positions cannot be accurately defined from molecular dynamics simulations since they correspond to locations that are only transiently visited by the biotin during the extraction process. Even though these are not actual simulations of the extraction of a biotin from a streptavidin pocket, it can be used to approximate it for several reasons. Firstly, streptavidin and avidin structures are very similar and have very close affinities with biotin (20,21). Secondly, the presence and positions of the three barriers seems to be confirmed by Grubmüller et al. (16), who have conducted simulations on streptavidin without deriving an approximate energy landscape.
FIGURE 4.

Energy landscape of the streptavidin-biotin bond. The landscape used to obtain the probabilities in Fig. 5 with the parameters from Table 1 (shaded line) is superimposed to the one predicted by molecular dynamics (solid lines, given in Merkel et al. (2) and deduced from original data given in Izrailev et al. (17)). In the simulations, the instantaneous energy was computed over a half-nanosecond extraction from the biotin-avidin binding pocket. The denser regions with rapid fluctuations correspond to the minima in the energy landscape, while the heights of the barriers (maxima in the energy landscape) cannot be found. The shaded dashed line represents the inmost barrier that is seen in the DNA experiments but not in the rupture force measurements. The values xm1(0), xm2(0), xm3(0), xb1(0), xb2(0), and xb3(0), are, respectively, the positions of the first, second, and third minimums and of the first, second, and third barriers under zero force. These positions will move when a force is applied.
The presence of three barriers is corroborated by an independent experiment in which streptavidin-coated beads rolled on a biotinylated surface in a flow chamber (6). Transitions from the deepest metastable state (i.e., the second deepest energy minimum) outward to the second metastable state as well as forward to the deepest energy minimum were observed, indicating the presence of three energy barriers. The transfer rates were 5.3 s−1 and 1.3 s−1, respectively. From these two studies, it is clear that three main energy barriers can be observed during the rupture of the streptavidin-biotin complex.
Can the history of the bond be at the origin of the paradox?
It is commonly admitted that each linear regime in the curve of the most likely rupture force as a function of the logarithm of the loading rate corresponds to a given barrier in the energy landscape (3–5,22,23). Even though this assumption has been somewhat shaken recently (7,24), it can be claimed that n different regimes correspond at most to n different barriers (8). Two linear regimes can be found in Fig. 3. Thus, in the BFP experiments, only two barriers are observed in the energy landscape of the streptavidin-biotin bond. As molecular dynamics simulations and flow chamber data showed that three barriers are present, one of these barriers is missing in the BFP measurements. To determine which one, we have reanalyzed in details the distributions of rupture forces for all the loading rates. These distributions can be theoretically predicted by applying reaction-rate theory, also known as Kramers' theory (25–27), to the energy landscape of the bond. To proceed with the analysis, it is necessary to describe this theory in the case where there are two barriers in a one-dimensional energy landscape. The probabilities of being in each of the two energy minima are given by
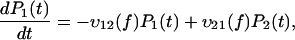 |
(1a) |
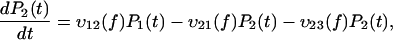 |
(1b) |
where P1(t) and P2(t) are the probability to be respectively in the first and second minimums, f is the pulling force which is related to the time t through the loading rate r by f = r × t, and υif(f) indicates the transition rates from a minimum i to a neighbor minimum j, 3 referring to the unbound state. When the landscape is locally approximated by an harmonic potential around each minimum and each maximum, υij(f) can be written as
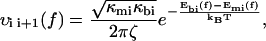 |
(2a) |
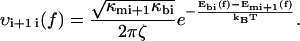 |
(2b) |
Where ζ is a damping coefficient, the subscripts mi and bi refer, respectively, to the ith metastable state and to the ith barrier, the κ-values are the local curvatures of the landscape, and E(x,f) the energy of the potential tilted by the force—i.e., E(x,0) − x × f. It is important to note that the local curvatures are necessary to predict the positions xmi(f) and xbi(f) and therefore the associated energies Emi(xmi(f),f) and Ebi(xbi(f),f). The diffusive microscopic times are usually written
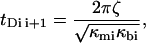 |
(3a) |
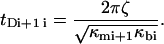 |
(3b) |
To suppress any further reference to time, the set of master equations, Eqs. 1a and 1b, can equivalently be rewritten as
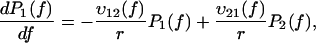 |
(4a) |
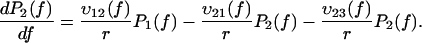 |
(4b) |
The overall evolution will be fully determined by the knowledge of the initial values of the probabilities, P1(0) and P2(0). The force distribution is then given by
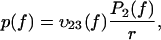 |
(5) |
which is also 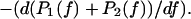
From Eqs. 2a, 2b, 4a, 4b, and 5, it is clear that the complete knowledge of the energy landscape is sufficient to predict the theoretical rupture force distribution. Since the measurements are not infinitely accurate, an experimental error has to be added, which will slightly widen the force distribution. For our purpose, we have chosen a Gaussian error with a reasonable width (see Fig. 5). We have tried to apply Kramers' theory to each possible pair of barriers of the energy landscape. In each case, we have varied the different parameters (i.e., height, position, and curvature for each minimum and each barrier) with the constraint that they should remain consistent with the molecular dynamics simulations. The only way by which the experimental rupture force distributions obtained by Merkel and co-workers (2) could be fitted was by keeping the two outer barriers from the molecular dynamics and assuming that the bond is in the second deepest minimum at the start of the separation process (values of the parameters are given in Table 1). The predicted rupture force distributions are given in Fig. 5. The agreement with the experimental histograms is almost perfect for all the loading rates. Such a good prediction could not have been achieved otherwise. Therefore, in the BFP experiment the streptavidin-biotin bond did not reach its deepest minimum. This may be the difference between these measurements and the DNA stretching technique. Intuitively, it can be understood that, in the BFP, the bond is given a fraction of second to form, whereas for the study of DNA stretching, the DNA strand has been attached to the streptavidin-coated beads for several minutes before any pulling force was applied. Hence, we can assume that the history of the bond is at the origin of the streptavidin-biotin paradox.
FIGURE 5.

Probability density of the rupture force at different loading rates superimposed to the experimental rupture force frequency obtained from Merkel et al. (2). They are deduced by the numerical resolution of the set of master equations, Eqs. 4a and 4b (see text), in the energy landscape given in Fig. 4 and taking P1(0) = 1 and P2(0) = 0. For the curves in solid line, experimental error is taken into account by changing a given rupture force probability density p(f) to an effective rupture force probability density peff(f) by using the relationship 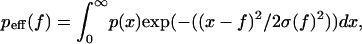 where the Gaussian error has a width σ(f), which is inspired by the experiments. Briefly, the force f is equal to kx, where k is the spring constant and x the spring extension. Therefore, df = kdx + xdk = kdx + fdk/k. The value dx is a constant due to the thermal fluctuations and the accuracy on the detection of the position of the bead in the BFP experiments; thus kdx is of the order of 100 pN/μm × 10 nm = 1 pN. The error on k is mainly due to the poor accuracy on various length measurements (inner diameter of the pipette, diameter of the red blood cell, and diameter of the contact between the red cell and the bead, all of the order of 1 μm); it can be estimated to be between 10 and 30%. Following these constraints, we chose σ(f) = Max[10, 0.20 × f] (in pN). (The curves in dashed line represent p(x), meaning that the experimental error is not taken into account. The difference between the solid and dashed lines demonstrates the importance of the experimental error in the analysis.)
where the Gaussian error has a width σ(f), which is inspired by the experiments. Briefly, the force f is equal to kx, where k is the spring constant and x the spring extension. Therefore, df = kdx + xdk = kdx + fdk/k. The value dx is a constant due to the thermal fluctuations and the accuracy on the detection of the position of the bead in the BFP experiments; thus kdx is of the order of 100 pN/μm × 10 nm = 1 pN. The error on k is mainly due to the poor accuracy on various length measurements (inner diameter of the pipette, diameter of the red blood cell, and diameter of the contact between the red cell and the bead, all of the order of 1 μm); it can be estimated to be between 10 and 30%. Following these constraints, we chose σ(f) = Max[10, 0.20 × f] (in pN). (The curves in dashed line represent p(x), meaning that the experimental error is not taken into account. The difference between the solid and dashed lines demonstrates the importance of the experimental error in the analysis.)
TABLE 1.
Parameters taken in the energy landscape of the streptavidin-biotin bond
| Wells | Barriers | |||||
|---|---|---|---|---|---|---|
| Number of the extremum | 1 | 2 | 3 | 1 | 2 | 3 |
| Position (nm) | 0 | 0.39 | 1.09 | 0.31 | 0.89 | 1.31 |
| Energy (kBT) | 0 | 5 | 13 | 32 | 26 | 37 |
| Curvature (kBT · nm−2) | 590 | 1000 | 600 | 750 | 94 | 2660 |
| Diffusive microscopic time (s), outward | tD12 | tD23 | tD34 | |||
| 2.1 10−11 | 9.3 10−11 | 2.3 10−11 | ||||
| Diffusive microscopic time (s), inward | tD21 | tD32 | ||||
| 3.2 10−12 | 4.3 10−11 | |||||
The damping coefficient ζ (compare to Eqs. 2a, 2b, 3a, and 3b) was taken equal to 4.10−11 Ns m−1, which is a typical value deduced from molecular dynamics simulations. The diffusive microscopic times were obtained from the curvatures of the landscape κm and κb around the metastable states and barriers through Eqs. 3a and 3b. No curvature of the first minimum and the first barrier could be deduced from the experiments, only diffusive microscopic time (tD12 and tD21).
Experimental validation and complete description of the energy landscape
To test this assumption we have conducted experiments in which both experimental approaches were combined: we have used the BFP technique with streptavidin-coated beads that had previously been incubated with DNA strands biotinylated on one end (see Materials and Methods and Fig. 1). As the streptavidin-biotin bonds have been formed a long time before the pulling process starts, the rupture forces should be larger than the ones obtained previously with the BFP. However, as it is important to make a large number of measurements (a least 100 per loading rate) to obtain good statistics and smooth distributions, it is necessary to keep the same bead over several approaching-separation cycles. Thus, it can happen that a DNA strand that had previously been detached from the bead reattaches through a newly formed streptavidin-biotin bond. Consequently, the expected distribution should present two peaks: one corresponding to the old bonds, like in the DNA stretching studies, and one corresponding to the new bonds, like in the previous BFP measurements. This is exactly what we have observed (Fig. 6). By adding biocytin (0.1 mg/ml) in the solution to block all the available streptavidin sites, the first peak disappears, confirming that it was due to the formation of new streptavidin/biotin bonds during the measurements (Fig. 7). The experimental distributions of Fig. 6 can be predicted using the complete energy landscape of Fig. 4 with initial conditions in which the probability to be in the deepest minimum is ∼0.5. These predictions are obtained using Kramers' theory with an extra minimum and the corresponding probability P3(f) in Eqs. 4a and 4b,
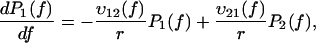 |
(6a) |
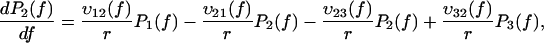 |
(6b) |
 |
(6c) |
The most likely rupture forces of the peak corresponding to the deepest minimum can also be accurately predicted with the same energy landscape (see Fig. 4 and Table 1). Thus, we have been able to probe experimentally unambiguously the presence of the three barriers with our system and completely find the energy landscape of the streptavidin-biotin bond. We now have to check that this energy landscape is consistent with the other experimental observations, namely:
In the BFP, only the second deepest minimum is reached whereas in the DNA stretching experiments, equilibrium is attained.
The lifespan of the bond in the DNA stretching experiments is at least of 10 s at 75 pN and a few seconds at 100 pN.
The transfer rates are consistent with the ones measured in the flow chamber.
FIGURE 6.

Experimental distributions of the rupture force obtained with the DNA coated beads for two loading rates: 12 pN/s and 2400 pN/s. The corresponding probability density of the rupture forces predicted from the energy landscape given in Fig. 4 and Kramers' equations (Eq. 6a–e) is superimposed. The initial conditions are P1(0) = λ, P2(0) = 1–λ, and P3(0) = 0. Because of small variations in the experiments, λ had to be adjusted with the loading rate. Here, λ = 0.3 for 12 pN/s and 0.6 for 2400 pN/s. Nevertheless, it was always of the order of 0.5. The fit at 12 pN/s is not perfect because of the presence of nonspecific forces that slightly merge with the first peak and artificially decrease the height of the second one after normalization. The experimental error, σ(f), is the same as that used in Fig. 5.
FIGURE 7.

Experimental distributions of the rupture force obtained with the DNA coated beads in a solution containing biocytin at 0.1 mg/ml to prevent formation of any streptavidin/biotin bond during the measurement. The corresponding probability density of the rupture forces predicted from the energy landscape given in Fig. 4 and Kramers' equations (Eq. 6) is superimposed. The initial conditions are P1(0) = 1, P2(0) = 0, and P3(0) = 0. The experimental error, σ(f), is the same as that used in Fig. 5.
The filling up of the different minima can be directly obtained by applying the master equations (Eqs. 6a–c) to the energy landscape with the initial conditions P1(0) = P2(0) = 0 and P3(0) = 1. The corresponding curves are given in Fig. 8. They show that it takes >10 s to fill up the deepest state. This result confirms that in the BFP, when the bonds are just formed, they are only in the second deepest minimum. However, we are not able to understand one subtle detail at small loading rates (typically <100 pN/s). In these cases, the potential is not tilted too quickly by the pulling force, which is applied sufficiently slowly. Then, the bond still has a chance to reach the deepest minimum even after the separation process has started. One explanation can be suggested to understand this problem. It is related to the geometry of the system, in which both the streptavidin and the biotin were grafted on glass beads. It is possible that because of geometrical constraints, such as lever effects (28), random pulling forces were applied to the bond. The time required to reach the deepest minimum was then increased, preventing the bond from reaching the deepest minimum during the pulling phase.
FIGURE 8.

Filling up of the energy landscape wells. Probability for a bond that is initially in the outermost minimum (minimum number 3 in Fig. 4) to be in a well as a function of time. For each well, there is a sharp transition from fully occupied to empty and from empty to fully occupied at times that are orders-of-magnitude different. These curves are obtained through the master equations, Eqs. 6a–c.
Nevertheless, this remark about the possibility of obtaining strong forces at low loading rates brings the counterintuitive result that the average rupture force at small loading rate may decrease with the loading rate before reincreasing at larger loading rates (Fig. 9). Usually, the most likely rupture force of a single bond increases continuously with the loading rate (27).
FIGURE 9.

Average rupture force as a function of the loading rate for a bond that is given 0, 100, or 500 ms before any pulling force is applied (respectively solid, dotted, and dash-dotted lines). Three regimes are observed: 1), Above typically 100 pN/s, the bond is in the second metastable state, the average rupture force increases with the loading rate and the rupture force distributions are the ones given in Fig. 3. 2), Between 5 and 100 pN/s, the lifetime of the bond is longer, more time is given to reach the most favorable state, and therefore, the average rupture force decreases with increasing loading rate. 3), Below 5 pN/s, the bond always reaches the most stable state during the pulling phase; the intermediary metastable state is never observed.
The lifespan of the bond under a constant force can also be obtained from Kramers' equations. When a 100-pN pulling force is applied, the bond breaks within a few seconds (Fig. 10), while for a pulling force of 75 pN, its lifespan is much longer (of the order of 10 s, and 1 min for a pulling force of 50 pN). This is exactly what is experimentally observed in the DNA stretching experiments.
FIGURE 10.

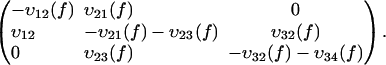
Next, the transfer rate from the second minimum to the deepest one (respectively, the third one) was 0.4 s−1 (respectively, 5.3 s−1) in the energy landscape we obtained, which is coherent with 1.3 s−1 (respectively, 5.3 s−1) in the flow chamber experiments (6).
Finally, rupture forces stronger than the ones observed in Merkel et al. (2) have been measured with an AFM (12). In this latter case, their values are comparable to the ones we obtained for the second peaks in Fig. 6 (see Fig. 11 for a comparison of the forces). Therefore, in these AFM experiments they seem to have reached the deepest minimum. As we do not know their precise experimental protocol, we cannot explain how they managed to reach it.
FIGURE 11.

Most likely rupture forces as a function of the loading rate, for both states: the first (higher force, solid triangles) and second deepest (lower force, open triangles) minima. The two observed peaks with the DNA-coated beads experiments correspond to the ones observed by Merkel et al. (2) (second deepest minimum, open squares) and by Yuan et al. (12) (deepest minimum, crosses). They are very well adjusted by the most likely forces predicted from the energy landscape given in Fig. 4 and Eq. 6 (thick and thin solid lines).
Therefore, with DNA-coated beads, we have been able to demonstrate that the history of the bond is at the origin of the streptavidin-biotin paradox: in the two contradictory experiments, the bond did not reach the same state before a pulling force was applied on it. This influence of the history of the bond had already been mentioned during the pulling phase (29,30). Here, we show that the history of the bond during the bond formation phase is also crucial. Our results also indicate that the time a system takes to reach equilibrium can be relevant on experimental timescales and may be too often neglected in many fields such as chemistry or biology where association constants are commonly used. Therefore, there should be not just one association constant per molecular complex, but one per metastable state, relevant in the timescales of the considered process. Of course, the higher the barriers, the longer it takes to fill up the minima.
FIGURE 2.

Glass beads coated with DNA strands. The left picture shows glass beads in direct illumination. These beads are coated with DNA fluorescently labeled strands. The label is yoyo1. The right picture shows the same beads observed by fluorescence, indicating that the DNA completely covers the beads.
Acknowledgments
The authors are grateful to J.-F. Allemand, A. Crut, C. Gourier, F. Heslot, E. Perez, and D. Tareste for fruitful discussions.
This work was partly funded by a University Paris 7 BQR contract.
References
- 1.Florin, E. L., V. T. Moy, and H. E. Gaub. 1994. Adhesion forces between individual ligand-receptor pairs. Science. 264:415–417. [DOI] [PubMed] [Google Scholar]
- 2.Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 397:50–53. [DOI] [PubMed] [Google Scholar]
- 3.Chen, S. Q., and T. A. Springer. 2001. Selectin receptor-ligand bonds: formation limited by shear rate and dissociation governed by the Bell model. Proc. Natl. Acad. Sci. USA. 98:950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanley, W., O. McCarty, S. Jadhav, Y. Tseng, D. Wirtz, and K. Konstantopoulos. 2003. Single molecule characterization of P-selectin/ligand binding. J. Biol. Chem. 278:10556–10561. [DOI] [PubMed] [Google Scholar]
- 5.Li, F. Y., S. D. Redick, H. P. Erickson, and V. T. Moy. 2003. Force measurements of the α5β1 integrin-fibronectin interaction. Biophys. J. 84:1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pierres, A., D. Touchard, A.-M. Benoliel, and P. Bongrand. 2002. Dissecting streptavidin-biotin interaction with a laminar flow chamber. Biophys. J. 82:3214–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hummer, G., and A. Szabo. 2003. Kinetics from nonequilibrium single-molecule pulling experiments. Biophys. J. 85:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derenyi, I., D. Bartolo, and A. Ajdari. 2004. Effects of intermediate bound states in dynamic force spectroscopy. Biophys. J. 86:1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raible, M., M. Evstigneev, P. Reimann, F. W. Bartels, and R. Ros. 2004. Theoretical analysis of dynamic force spectroscopy experiments on ligand-receptor complexes. J. Biotechnol. 112:13–23. [DOI] [PubMed] [Google Scholar]
- 10.Friedsam, C., A. K. Wehle, F. Kuhner, and H. E. Gaub. 2003. Dynamic single-molecule force spectroscopy: bond rupture analysis with variable spacer length. J. Phys. Condens. Mat. 15:S1709–S1723. [Google Scholar]
- 11.Kuhner, F., L. T. Costa, P. M. Bisch, S. Thalhammer, W. M. Heckl, and H. E. Gaub. 2004. LexA-DNA bond strength by single molecule force spectroscopy. Biophys. J. 87:2683–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan, C. B., A. Chen, P. Kolb, and V. T. Moy. 2000. Energy landscape of streptavidin-biotin complexes measured by atomic force microscopy. Biochemistry. 39:10219–10223. [DOI] [PubMed] [Google Scholar]
- 13.Evans, E., K. Ritchie, and R. Merkel. 1995. Sensitive force technique to probe molecular adhesion and structural linkages at biological interfaces. Biophys. J. 68:2580–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green, N. M. 1975. Avidin. Adv. Protein Chem. 29:85–133. [DOI] [PubMed] [Google Scholar]
- 15.Chilkoti, A., and P. S. Stayton. 1995. Molecular origins of the slow streptavidin-biotin dissociation kinetics. J. Am. Chem. Soc. 117:10622–10628. [Google Scholar]
- 16.Grubmüller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- 17.Izrailev, S., S. Stepaniants, M. Balsera, Y. Oono, and K. Schulten. 1997. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72:1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith, S. B., Y. J. Cui, and C. Bustamante. 1996. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 271:795–799. [DOI] [PubMed] [Google Scholar]
- 19.Baumann, C. G., S. B. Smith, V. A. Bloomfield, and C. Bustamante. 1997. Ionic effects on the elasticity of single DNA molecules. Proc. Natl. Acad. Sci. USA. 94:6185–6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livnah, O., E. A. Bayer, M. Wilchek, and J. L. Sussman. 1993. Three-dimensional structures of avidin and the avidin-biotin complex. Proc. Natl. Acad. Sci. USA. 90:5076–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freitag, S., I. LeTrong, L. Klumb, P. S. Stayton, and R. E. Stenkamp. 1997. Structural studies of the streptavidin binding loop. Protein Sci. 6:1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans, E., and P. M. Williams. 2002. Dynamic force spectroscopy. In Physics of Bio-Molecules and Cells. F. Julicher, P. Ormos, F. David, and H. Flyvbjerg, editors. Springer Verlag, Berlin, Germany. 145–204.
- 23.Evans, E., and F. Ludwig. 2000. Dynamic strengths of molecular anchoring and material cohesion in fluid biomembranes. J. Phys. Condens. Mat. 12:A315–A320. [Google Scholar]
- 24.Bartolo, D., I. Derenyi, and A. Ajdari. 2002. Dynamic response of adhesion complexes: beyond the single-path picture. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 65:051910. [DOI] [PubMed] [Google Scholar]
- 25.Kramers, H. A. 1940. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica (Utrecht). 7:284–304. [Google Scholar]
- 26.Hanggi, P., P. Talkner, and M. Borkovec. 1990. Reaction-rate theory—50 years after Kramers. Rev. Mod. Phys. 62:251–341. [Google Scholar]
- 27.Evans, E., and K. Ritchie. 1997. Dynamic strength of molecular adhesion bonds. Biophys. J. 72:1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stout, A. L. 2001. Detection and characterization of individual intermolecular bonds using optical tweezers. Biophys. J. 80:2976–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans, E., A. Leung, V. Heinrich, and C. Zhu. 2004. Mechanical switching and coupling between two dissociation pathways in a P-selectin adhesion bond. Proc. Natl. Acad. Sci. USA. 101:11281–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marshall, B. T., K. K. Sarangapani, J. Lou, R. P. McEver, and C. Zhu. 2005. Force history dependence of receptor-ligand dissociation. Biophys. J. 88:1458–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]