

Abstract
We demonstrate that fluorescence resonance energy transfer spectroscopy is a powerful tool for in situ structural analysis of multimeric membrane proteins by measuring the conformational changes involved in gating the mechanosensitive ion channel of large conductance. Ensemble analysis is used to analyze the intensity of light emitted by AlexaFluor-labeled cysteine mutants reconstituted into artificial liposomes before and after acceptor photobleaching. The diameter of the protein is found to increase by 16 Å upon channel activation.
Transmembrane channels facilitate the movement of small molecules and ions across cell membranes, a process responsible for such diverse roles as nerve conduction and the regulation of cell volumes. Our understanding of these proteins recently took a major step forward with the availability of structures from x-ray crystallography (1,2). A key difficulty remains, however, in that membrane channels adopt multiple conformations with at least two stable states: conductive and nonconductive. Understanding how these channels work is thus difficult from static crystal structures (3). Here we use fluorescence spectroscopy to determine the structural changes involved in gating the mechanosensitive channel of large conductance (MscL) from bacteria in situ. MscL channels act as safety valves in bacterial cells, opening a wide pore to release pressure during hypo-osmotic stress (4–6). To measure structural changes we label specific sites with fluorescent probes and use fluorescence resonance energy transfer (FRET) to measure the distance between them.
The MscL protein contains no cysteine residues. We create sites that can be uniquely labeled with fluorescent probes using site-directed mutagenesis to introduce the substitution M42C, a site located on the outer edge of the extracellular side of the protein. A crystal structure of the MscL protein in a closed state (2) shows it to be comprised of five identical subunits surrounding a central pore. Thus, this mutation introduces five identical cysteine sites. The mutants were cloned into a pQE-32 expression vector as BamHI-SalI fragments, expressed in Escherichia coli and purified using Ni-NTA affinity chromatography.
The protein is labeled with a racemic mixture of AlexaFluor488 (AF488) and AlexaFluor568 (AF568) maleimide that specifically binds to the cysteine residues. Thus, each channel protein has five fluorophores attached, with an equal probability of any site being occupied by AF488 or AF568. Excess dye was removed using dialysis and the labeled protein was reconstituted into artificial phosphatidylcholine (PC) liposomes (7). All results described here were made with a protein/lipid ratio of 1:250 by weight; however, similar results were obtained using a ratio of 1:500. The liposomes were imaged on a Biorad MRC1000/1024 UV laser scanning confocal microscope (Bio-Rad, Hercules, CA) using a Nikon 60× NA 1.2 water-immersion objective lens (Nikon, Tokyo, Japan). AF488 was excited at 488 nm with an argon laser (∼60 μW at sample) and detected through a 522/35-nm bandpass filter, and AF568 was excited at 543 nm using a green helium neon laser (∼60 μW at sample) and detected through a 585-nm long-pass filter.
In the first and third columns of Fig. 1 A, we show representative images of four separate liposomes incorporating the labeled MscL channels. After taking these images, AF568 was bleached using the 543-nm laser and the intensity of the AF568 emission is reduced to <5% of its original value, as pictured in the second column of Fig. 1 A. The intensity of the AF488, however, increases significantly, as seen in the last column. At rest (first two rows of Fig. 1 A) the MscL channels remain in a closed state. We can open the channels, however, by adding lysophosphatidylcholine (LPC) to the sample, as depicted in Fig. 1 B, which shows patch-clamp recordings of WT channels in artificial liposomes and channel activation analogous to that obtained by membrane pressure. As described previously (8), at the levels of concentration applied to our liposomes (25% molar ratio to PC), LPC induces membrane curvature and/or a change in the transbilayer pressure profiles sufficient to activate the channel for prolonged periods. An example of images of a liposome with open channels is shown in row 3 of Fig. 1 A.
FIGURE 1.

(a) Confocal microscope images of liposomes containing MscL protein labeled with both AF488 and AF568. The rows show different liposomes, whereas the columns represent the emission of the AF568 before and after bleaching, and AF488 before and after bleaching, respectively. Nine millimolar (25% molar ratio) LPC has been added in the last two rows. (b) Activation of MscL by LPC. The activation of MscL was recorded during suction to a patch pipette (left) or addition of 2 μM LPC in the absence of applied pressure (right). Pipette voltage was +30 mV.
In FRET, energy absorbed by one molecule known as the donor (AF488 in this case) can be transferred to another, the acceptor (AF568), through a nonradiative dipole-dipole interaction. That the donor intensity increased after acceptor bleaching is a sure sign that FRET had been taking place beforehand. The fraction of energy transferred, termed the efficiency E, is dependent on the distance between the molecules. We determine this from 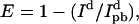 where Id and
where Id and 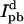 are the donor emission intensities before and after acceptor photobleaching. We use the average intensity of the membrane, avoiding regions with bright blebs. Since the bleaching of AF568 is never 100% complete, we correct
are the donor emission intensities before and after acceptor photobleaching. We use the average intensity of the membrane, avoiding regions with bright blebs. Since the bleaching of AF568 is never 100% complete, we correct 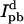 by multiplying the change in donor intensity by
by multiplying the change in donor intensity by 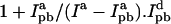 is increased by a further 8% to account for the measured bleaching of AF488 by the 543-nm laser.
is increased by a further 8% to account for the measured bleaching of AF488 by the 543-nm laser.
In the inset of Fig. 2 A, we plot the measured transfer efficiency against the bleaching of AF568. We find efficiencies of 0.61 ± 0.01 and 0.44 ± 0.01 for the closed and open states of the channel.
FIGURE 2.

Measured FRET efficiency and channel radius. (a) FRET efficiency for individual liposomes in the closed channels (solid circles) and open channels (open circles) and average values (blue) are shown in the inset. The curve relating pentamer size to transfer efficiency is plotted with the data from our experiments shown by blue lines with error margins in red. (b) Schematic diagram indicating the scale of conformational change involved in channel gating.
Although the relationship between the fluorophore separation and transfer efficiency is well known for single donor-acceptor pairs, the situation is more complicated in our case, since each protein contains a random mix of five fluorophores, and given that transfer could arise between fluorophores attached to different channels. We use a Monte Carlo scheme described in an accompanying article (9) to relate the efficiency of energy transfer to the radius of the pentamers of fluorophores as shown in Fig. 2 A.
Using an orientation factor of κ2 = 2/3, we determined the radii of the pentamer of fluorophores to be 45 ± 1 Å for the closed channels and 53 ± 1 Å for the open channels. This radius refers to the positions of the fluorophores and is likely to be somewhat larger than the radius of the protein itself. Therefore, a more interesting measure is the change of radius upon channel activation, 8 ± 2 Å. This is less likely to be influenced by any systematic errors in the FRET measurements and is less sensitive to the value of the κ2 (and R0) than is the absolute radius of the channel. By measuring the intensity of the liposomes illuminated with linearly polarized light, we are able to set limits on the value of the κ2 (B. Corry, D. Jayatilaka, B. Martinac, and P. Rigby, unpublished data), and apply different values for transfer between neighboring and next closest fluorophores. Taking this into account, we find the change in radius of the channel to lie between 8.0 and 8.5 Å.
In Fig. 2 B we illustrate schematically the change in protein size created by an 8 Å radius change. The left-hand image shows the crystal structure of the channel with models of the fluorophores attached to site M42C. The right-hand image shows the same structure to-scale, after an 8 Å radius increase. As we expect the total protein volume to stay constant upon channel activation, this implies the opening of a large pore whose likely size is depicted. This represents one of the largest conformational changes known in membrane proteins and creates a large pore consistent with models derived from studies of spin-labeled MscL (10).
The images shown in row 4 of Fig. 1 A are particularly interesting. One liposome resides within another, and although the images were taken after adding LPC this is unlikely to have reached the inner liposome. Thus, channels in the outer liposome can be expected to be in the open state, whereas those in the inner liposome should remain closed. This is supported by the measured transfer efficiencies of 0.44 and 0.59 for the outer and inner liposomes.
The calculated limits on the fluorophore orientation are the same for the open and closed channels, suggesting that the 8 Å radius change is a result of a reconfiguration of the protein rather than of the fluorophores. We have examined a site M42 on the exterior of the protein because it is unlikely to affect channel function, in a protein that undergoes large conformational changes. This has enabled us to overcome a number of technical challenges and demonstrate the utility of spectroscopic techniques in probing the structure of membrane proteins in situ. With this study we have set a stage for the exciting possibility of gaining a complete insight into gating of multimeric proteins such as MscL, by analyzing the motions of many more sites within reconstituted proteins.
Acknowledgments
We thank Dr. Shankar Balasubramanian for his inspiring lecture that gave rise to this research, A. Prof. Dylan Jayatilaka for critically reading the manuscript, and Albert Raso for technical assistance.
This work was funded by the Australian Research Council, The National Health and Medical Research Council of Australia, and The University of Western Australia. The Biomedical Imaging and Analysis Facility is supported by LotteryWest.
Zhen-Wei Liu's and Boris Martinac's present address is School of Biomedical Sciences, University of Queensland, Brisbane, Australia.
References
- 1.Doyle, D. A., J. Morais Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 2.Chang, G., R. H. Spencer, A. T. Lee, M. T. Barclay, and D. C. Rees. 1998. Structure of the MscL homologue from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 282:2220–2226. [DOI] [PubMed] [Google Scholar]
- 3.Selvin, P. R. 2003. Lighting up single ion channels. Biophys. J. 84:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinac, B. 2004. Mechanosensitive ion channels: molecules of mechanotransduction. J. Cell Sci. 117:2449–2460. [DOI] [PubMed] [Google Scholar]
- 5.Hamill, O. P., and B. Martinac. 2001. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 81:685–740. [DOI] [PubMed] [Google Scholar]
- 6.Levina, N., S. Totemeyer, N. R. Stokes, P. Louis, M. A. Jones, and I. R. Booth. 1999. Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: identification of genes required for MscS activity. EMBO J. 18:1730–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Häse, C. C., A. C. LeDain, and B. Martinac. 1995. Purification and functional reconstitution of the recombinant large mechanosensitive ion channel (MscL) of Escherichia coli. J. Biol. Chem. 270:18329–18334. [DOI] [PubMed] [Google Scholar]
- 8.Perozo, E., A. Kloda, D. M. Cortes, and B. Martinac. 2002. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 9:696–703. [DOI] [PubMed] [Google Scholar]
- 9.Corry, B., D. Jayatilaka, and P. Rigby. 2005. A flexible approach to the calculation of resonance energy transfer efficiency between multiple donors and acceptors in complex geometries. Biophys. J. 89:3822–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perozo, E., D. M. Cortes, P. Sompornpisut, A. Kloda, and B. Martinac. 2002. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418:942–948. [DOI] [PubMed] [Google Scholar]