

Abstract
Stark (electroabsorption) spectra of the M100A mutant of photoactive yellow protein reveal that the neutral, cis cofactor of the pB intermediate undergoes a strikingly large change in the static dipole moment (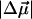 = 19 Debye) on photon absorption. The formation of this charge-separated species, in the excited state, precedes the cis → trans isomerization of the pB cofactor and the regeneration of pG. The large
= 19 Debye) on photon absorption. The formation of this charge-separated species, in the excited state, precedes the cis → trans isomerization of the pB cofactor and the regeneration of pG. The large 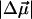 , reminiscent of that produced on the excitation of pG, we propose, induces twisting of the cis cofactor as a result of translocation of negative charge, from the hydroxyl oxygen, O1, toward the C7-C8 double bond. The biological significance of this photoinduced charge transfer reaction underlies the significantly faster regeneration of pG from pB in vitro, on the absorption of blue light.
, reminiscent of that produced on the excitation of pG, we propose, induces twisting of the cis cofactor as a result of translocation of negative charge, from the hydroxyl oxygen, O1, toward the C7-C8 double bond. The biological significance of this photoinduced charge transfer reaction underlies the significantly faster regeneration of pG from pB in vitro, on the absorption of blue light.
The physiologically important blue-light negative phototactic response of the bacterium Halorhodospira halophila has been linked to photoactive yellow protein (PYP) (1). The PYP photocycle associated with this response (Scheme 1), is initiated by the excitation of the 4-hydroxycinnamyl thioester (4-HcT) cofactor (λmax = 446 nm). In particular, excitation of the 4-HcT cofactor in the receptor state, pG, was found to produce a 26 Debye change in the static dipole moment, 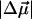 (2), thereby increasing the flexibility of the thioester tail to allow for the rotation of the cofactor in the excited state (3). Subsequently, trans → cis isomerization generates pB (λmax = 355 nm) and triggers partial unfolding of the protein (4,5). This study aims to understand the link between the excited-state electronic properties of pB and the driving force for the accelerated recovery of pG, from pB, on light absorption (see ellipse in Scheme 1). To this end, Stark spectroscopy is used to determine the change in electronic properties on photon absorption by the neutral (protonated) cis form of the 4-HcT cofactor (Scheme 2) of the pB intermediate.
(2), thereby increasing the flexibility of the thioester tail to allow for the rotation of the cofactor in the excited state (3). Subsequently, trans → cis isomerization generates pB (λmax = 355 nm) and triggers partial unfolding of the protein (4,5). This study aims to understand the link between the excited-state electronic properties of pB and the driving force for the accelerated recovery of pG, from pB, on light absorption (see ellipse in Scheme 1). To this end, Stark spectroscopy is used to determine the change in electronic properties on photon absorption by the neutral (protonated) cis form of the 4-HcT cofactor (Scheme 2) of the pB intermediate.
SCHEME 1.

Photocycle of PYP. The effect of photon absorption by pB is represented inside the ellipse.
SCHEME 2.

The cis-isomer of 4-hydroxycinnamyl thioester in the pB intermediate. The relevant atoms discussed in the text are numbered.
To facilitate the study of the pB intermediate the M100A mutant is employed, which has previously been used to characterize the ground-state recovery reaction in PYP (6). Effectively, the electron-donating capability of M100 is severely restricted due to the elimination of its electronegative sulfur atom. Consequently, the recovery of pG from pB in the M100A protein is decreased ∼1000-fold (6) and pB is therefore more easily trapped at 77 K. Note that the mutation does not change the amino acid residues surrounding the cofactor and the environmental effects exerted in the protein pocket remain similar to that in the wild type (see Supplementary Material).
The room- and low (77 K)-temperature absorption spectra of M100A in glycerol/buffer solution are presented in Fig. 1 a. The former is shown before (dotted-dashed line), and after (light solid line), illumination at 450 nm. Depending on the duration of illumination, the 77-K spectra (bold solid lines) are of two types: I, mostly enriched with pB; and II, containing a mixture of pG and pB. The absorption maxima of both pG and pB blue shift on lowering the temperature: pG to 444 ± 2 nm from 448 nm (2), and pB to 345 ± 2 nm from 355 nm. The protocol and methodology for fitting the Stark spectra for I (Fig. 1 b) and II (Fig. 1 c) are described in the Supplementary Material.
FIGURE 1.

(a) Absorption spectra of M100A in glycerol/buffer solution at 298 K: spectra before and after illumination at 450 nm are shown in dotted-dashed and light solid lines, respectively; 77-K spectra (bold solid lines) include I, with mostly pB, and II, a mixture of pG and pB. The field-normalized Stark signal (dark solid line), fit (dark dashed line), and scaled first (light dotted-dashed line) and second (light dashed line) derivatives of the absorption are shown in panel b to spectrum I and in panel c to spectrum II.
The sample containing mostly pB is fit to the absorption band between 24,500 and 32,000 cm−1 (Fig. 1 b) to yield a change in the static dipole moment, 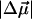 , of 19 ± 3 D, and a 350 ± 200 Å3 component corresponding to the sum of the transition moment polarizability and difference electronic polarizability. The Stark spectrum of II, containing comparable amounts of pG and pB (Fig. 1 c), is fit to pG (20,000–25,000 cm−1) and pB (>25,000 cm−1) independently, by deconvolving the absorption spectrum into two bands. This fit yields a
, of 19 ± 3 D, and a 350 ± 200 Å3 component corresponding to the sum of the transition moment polarizability and difference electronic polarizability. The Stark spectrum of II, containing comparable amounts of pG and pB (Fig. 1 c), is fit to pG (20,000–25,000 cm−1) and pB (>25,000 cm−1) independently, by deconvolving the absorption spectrum into two bands. This fit yields a 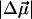 of 29 ± 2 D in the region of pG absorbance, in agreement with previous measurements (2), whereas the
of 29 ± 2 D in the region of pG absorbance, in agreement with previous measurements (2), whereas the 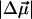 for pB, in this case, is found to be 20% larger than for I (Fig. 1 b). This discrepancy likely arises from the inherent increase in the inaccuracy of the fit for II, as the Stark signal of pB in spectrum II is significantly smaller than in I. Although the absorbances of pG and pB in II are comparable (Fig. 1 a), the Stark signal of pG is, however, much larger and overwhelms the pB Stark signal. This leads to the asymmetry of the pB signal at the red edge of II (26,000–27,000 cm−1) not evident in spectrum I. Alternatively, this asymmetry could arise due to the superposition of the Stark signal from an intermediate that potentially gives rise to the shoulder, marked with an asterisk (*) in Fig. 1 c, in the red edge of pB absorption. The presence and relative abundance of this intermediate appears to depend on pG, from which it is therefore likely to originate. Attempts to gain further insight about this species, by independently analyzing this band, lead to unstable solutions and was therefore not pursued further.
for pB, in this case, is found to be 20% larger than for I (Fig. 1 b). This discrepancy likely arises from the inherent increase in the inaccuracy of the fit for II, as the Stark signal of pB in spectrum II is significantly smaller than in I. Although the absorbances of pG and pB in II are comparable (Fig. 1 a), the Stark signal of pG is, however, much larger and overwhelms the pB Stark signal. This leads to the asymmetry of the pB signal at the red edge of II (26,000–27,000 cm−1) not evident in spectrum I. Alternatively, this asymmetry could arise due to the superposition of the Stark signal from an intermediate that potentially gives rise to the shoulder, marked with an asterisk (*) in Fig. 1 c, in the red edge of pB absorption. The presence and relative abundance of this intermediate appears to depend on pG, from which it is therefore likely to originate. Attempts to gain further insight about this species, by independently analyzing this band, lead to unstable solutions and was therefore not pursued further.
Fit I, to the sample composed mostly of pB, yields a 19 Debye 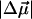 for the neutral cis isomer of 4-HcT. Such a large excited-state dipole moment for pB, heretofore unknown, would be expected to have a significant effect on the excited-state reactivity of the cofactor, as would the large local field generated in the vicinity of the cofactor, resulting from the charge separation in the excited state, on its surroundings.
for the neutral cis isomer of 4-HcT. Such a large excited-state dipole moment for pB, heretofore unknown, would be expected to have a significant effect on the excited-state reactivity of the cofactor, as would the large local field generated in the vicinity of the cofactor, resulting from the charge separation in the excited state, on its surroundings.
In pG, charge transfer from O1 to C7 produces a 26 Debye 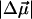 and results in an increase in the flexibility of the C4-C7-C8-C9 backbone (3). Thus, the twisting of the C7-C8-C9 moiety, and the rotation of the carbonyl group, drives the trans-cis isomerization reaction. A similar charge-transfer pathway in pB would provide added impetus for the “reverse” geometry distortion, despite the somewhat smaller
and results in an increase in the flexibility of the C4-C7-C8-C9 backbone (3). Thus, the twisting of the C7-C8-C9 moiety, and the rotation of the carbonyl group, drives the trans-cis isomerization reaction. A similar charge-transfer pathway in pB would provide added impetus for the “reverse” geometry distortion, despite the somewhat smaller 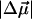 of pB compared to that of pG. Indeed, the hydroxyl oxygen, in the para position, of the neutral cofactor of pB is similar in its electron donating functionality to the oxyanion of the trans cofactor in pG. (Due to the extension of the π-conjugation, both −O− and −OH can donate an electron from the lone pair, and in general are considered to activate the phenyl ring for electrophillic substitution.)
of pB compared to that of pG. Indeed, the hydroxyl oxygen, in the para position, of the neutral cofactor of pB is similar in its electron donating functionality to the oxyanion of the trans cofactor in pG. (Due to the extension of the π-conjugation, both −O− and −OH can donate an electron from the lone pair, and in general are considered to activate the phenyl ring for electrophillic substitution.)
Therefore, electron donation from O1 to C7, as in pG, would increase the flexibility of the C4-C7-C8-C9 backbone and decrease the barrier to twist in the excited state. A neutral hydroxyl oxygen, as in pB, is however, a weaker electron donor than the −O− in pG, and could partly account for the smaller 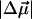 of pB (the 19 Debye value suggests that 80% of an electronic charge is transferred from O1 to C7 that are ≈5 Å apart). Interestingly, the 19 Debye
of pB (the 19 Debye value suggests that 80% of an electronic charge is transferred from O1 to C7 that are ≈5 Å apart). Interestingly, the 19 Debye 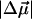 of pB is more than twice that measured for the isolated neutral all-trans 4-HcT chromophore (3), suggesting a role of the protein environment in tuning the electronic properties of the chromophore (5), beyond that induced by structural deformation. One possibility is that hydrogen bonding with Arg-52 (7) would increase the electronegativity and electron-donating capability of O1. After charge transfer, decrease in charge density on O1 would weaken the hydrogen bond with Arg-52 (Scheme 2) to disrupt the “tether” holding the cofactor, and promote twisting as well.
of pB is more than twice that measured for the isolated neutral all-trans 4-HcT chromophore (3), suggesting a role of the protein environment in tuning the electronic properties of the chromophore (5), beyond that induced by structural deformation. One possibility is that hydrogen bonding with Arg-52 (7) would increase the electronegativity and electron-donating capability of O1. After charge transfer, decrease in charge density on O1 would weaken the hydrogen bond with Arg-52 (Scheme 2) to disrupt the “tether” holding the cofactor, and promote twisting as well.
In brief, our results suggest that the photoreactivity of pB is induced by the charge-separated state produced on excitation. Furthermore, the impetus for the faster regeneration of the ground-state species, pG, would be provided by the photoinduced charge transfer reaction, and could explain the light-accelerated recovery of wild-type PYP in vitro (8).
SUPPLEMENTARY MATERIAL
An online supplement to this letter can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
L.P. and R.v.G. acknowledge the Foundation for Fundamental Research on Matter (FOM) of The Netherlands and the Dutch Science Organization (NWO) for financial support.
References
- 1.Sprenger, W. W., W. D. Hoff, J. P. Armitage, and K. J. Hellingwerf. 1993. The eubacterium Ectothiorhodospira halophila is negatively phototactic, with a wavelength dependence that fits the absorption spectrum of the photoactive yellow protein. J. Bacteriol. 175:3096–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Premvardhan, L. L., M. A. van der Horst, K. J. Hellingwerf, and R. van Grondelle. 2003. Stark spectroscopy on photoactive yellow protein, E46Q and a non-isomerising derivative, probes photo-induced charge motion. Biophys. J. 84:3226–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Premvardhan, L. L., F. Buda, M. A. van der Horst, D. C. Lührs, K. J. Hellingwerf, and R. van Grondelle. 2004. The impact of photon absorption on the electronic properties of p-coumaric acid derivatives of the photoactive yellow protein chromophore. J. Phys. Chem. B. 108:5138–5148. [Google Scholar]
- 4.Larsen, D. S., and R. van Grondelle. 2005. Initial photoinduced dynamics of the photoactive yellow protein. ChemPhysChem. 6:828–837. [DOI] [PubMed] [Google Scholar]
- 5.Mataga, N., H. Chosrowjan, and S. Taniguchi. 2004. Investigations into the dynamics and mechanisms of ultrafast photoinduced reactions taking place in photoresponsive protein nanospaces (PINS). J. Photochem. Photobiol. C. 5:155–168. [Google Scholar]
- 6.Devanathan, S., U. K. Genick, I. L. Canestrelli, T. E. Meyer, M. A. Cusanovich, E. D. Getzoff, and G. Tollin. 1998. New insights into the photocycle of Ectothiorhodospira halophila photoactive yellow protein: photorecovery of the long-lived photobleached intermediate in the Met100Ala mutant. Biochemistry. 37:11563–11568. [DOI] [PubMed] [Google Scholar]
- 7.Genick, U. K., G. E. O. Borgstahl, K. Ng, Z. Ren, C. Pradervand, P. M. Burke, V. Srajer, T. Teng, W. Schildkamp, D. E. McRee, K. Moffat, and E. D. Getzoff. 1997. Structure of a protein photocycle intermediate. Science. 275:1471–1475. [DOI] [PubMed] [Google Scholar]
- 8.Miller, A., H. Leigeber, W. D. Hoff, and K. J. Hellingwerf. 1993. A light-dependent branching reaction in the photocycle of the yellow protein from Ectothiorhodospira halophila. Biochim. Biophys. Acta. 1141:190–196. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.