

Abstract
A fundamental attribute of cell membranes is transmembrane asymmetry, specifically the formation of ordered phase domains in one leaflet that are compositionally different from the opposing leaflet of the bilayer. Using model membrane systems, many previous studies have demonstrated the formation of ordered phase domains that display complete transmembrane symmetry; but there have been few reports on the more biologically relevant asymmetric membrane structures. Here we report on a combined atomic force microscopy and fluorescence microscopy study whereby we observe three different states of transmembrane symmetry in phase-separated supported lipid bilayers formed by vesicle fusion. We find that if the leaflets differ in gel-phase area fraction, then the smaller domains in one leaflet are in registry with the larger domains in the other leaflet and the system is dynamic. In a presumed lipid flip-flop process similar to Ostwald ripening, the smaller domains in one leaflet erode away whereas the large domains in the other leaflet grow until complete compositional asymmetry is reached and remains stable. We have quantified this evolution and determined that the lipid flip-flop event happens most frequently at the interface between symmetric and asymmetric DSPC domains. If both leaflets have identical area fraction of gel-phase, gel-phase domains are in registry and are static in comparison to the first state. The stability of these three DSPC domain distributions, the degree of registry observed, and the domain immobility have biological significance with regards to maintenance of lipid asymmetry in living cell membranes, communication between inner leaflet and outer leaflet, membrane adhesion, and raft mobility.
INTRODUCTION
Transmembrane asymmetry and the formation of ordered phase domains in the plasma membrane have elicited extensive attention for more than 30 years. It has been shown that many important functions of cellular membranes are closely associated with their compositional and structural heterogeneity (1,2). For example, it is well know that in the plasma membrane of eukaryotic cells, phosphatidylserine and phosphatidylethanolamine are the predominant lipid species in the intracellular leaflet whereas phosphatidylcholine and sphingomyelin are generally located in the extracellular leaflet (3). The maintenance of this asymmetric distribution is a result of a continuous transfer of lipids between the two monolayers. Several types of proteins such as flippases, floppases, and scramblases are involved in the active transbilayer movement which occurs on the timescale of minutes (4). However spontaneous transbilayer diffusion of lipids is usually very slow (hours to days) (5) and is thought to have little contribution to the maintenance of transmembrane lipid asymmetry. Nevertheless, it has been shown that introducing transient defects into the membrane can greatly increase the spontaneous transbilayer flip-flop rate from hours to minutes (6,7), demonstrating that nonactive transport may still play a significant role in transmembrane asymmetry. Another important factor for maintaining lipid asymmetry is the interaction between the lipids of the intracellular leaflet and proteins of the cytoskeleton. Although the existence of these interactions is not questioned (6,8), the necessity of cytoskeleton-lipid interactions for lipid asymmetry has not been determined.
In addition to the asymmetric lipid distribution across the bilayer of the plasma membrane, lateral segregation, or lipid ‘raft’ formation, within each monolayer is proposed to play an important role in membrane protein sorting, signal transduction, and pathogen binding (9,10). In the raft theory, rafts are believed to be enriched in long-chained glycolipids, sphingolipids, and cholesterol. Due to the asymmetric distribution of these various lipids and the different viscosities measured between the intracellular and extracellular leaflets (11,12), it is unlikely that raft domains maintain a stable symmetric distribution between the two leaflets of the bilayer (2).
Model membrane systems such as giant unilamellar vesicles (GUVs) and supported lipid bilayers have been extensively used in understanding the fundamental properties of heterogeneity in biological membranes (13–15). These model systems have successfully demonstrated the coexistence of ordered and disordered phases for a variety of different lipid compositions. The physical properties of ordered phase lipid domains in model membranes (e.g., lipid density, chain dynamics, and lateral mobility) bear a striking resemblance to plasma membrane rafts described in the raft theory. However, there are several discrepancies between the structural organization of proposed rafts in biological membranes and phase-separated domains in model membrane systems. For example, in nearly all giant vesicle and supported lipid bilayer (formed through vesicle fusion) studies, lipid domains are transmembrane symmetric. Despite the advances made in our understanding of ordered phase domains using model membrane systems, very little work has been done to study transmembrane asymmetric distributions in these systems (16).
In this work, we report an atomic force microscopy (AFM) and fluorescence microscopy study of the distribution of gel-phase domains in supported lipid bilayers. Unlike most previous studies, the distribution of gel-phase lipid ranges from completely symmetric to completely asymmetric. Supported lipid bilayers were formed of dilauroylphosphatidylcholine/distearoylphosphatidylcholine (DLPC/DSPC) mixtures through the method of vesicle fusion. The thermal history of the vesicles was varied as well as the substrate-vesicle suspension temperature differential during deposition—making three distinct conditions for formation of the DLPC/DSPC-supported lipid bilayers. AFM, qualitative fluorescence recovery after photobleaching (FRAP), and cobalt-quenching were used to characterize the supported lipid bilayers. Using the information obtained by each of these characterization techniques, it was deduced that the three distinct sample preparation techniques gave three distinct DSPC domain distributions: symmetric, asymmetric, and symmetric/asymmetric. Technically DSPC domains should be regarded as DSPC rich since DLPC has a limited solubility in DSPC domains (<15 mol %) (17). Observations were made with respect to domain stability, lateral mobility, and the registry of DSPC domains in each leaflet of the bilayer. We statistically analyzed the temporal distribution of DSPC within the individual leaflets for bilayers containing symmetric/asymmetric bilayers. We discuss the significance of our results with regard to maintenance of lipid asymmetry in living cell membranes, communication between inner leaflet and outer leaflet, membrane adhesion, and raft mobility.
MATERIALS AND METHODS
Materials
1,2-Dilauroyl-sn-glycero-3-phosphocholine (DLPC), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and 1-oleoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphocholine (18:1–06:0 NBD-PC) were purchased in chloroform from Avanti Polar Lipids (Birmingham, AL) and used without further purification. Cobalt (II) chloride was purchased from Fisher Scientific (Pittsburgh, PA) and used without further purification. All water used in these experiments was purified in a Barnstead Nanopure System (Barnstead Thermolyne, Dubuque, IA), with resistivity ≥17.9 MΩ and pH 5.5.
Preparation of large multilamellar vesicles
Vesicles were prepared from mixtures of DLPC/DSPC and doped with a fluorescent probe NBD-PC when necessary. Because the tail-labeled fluorescent probe partitions in the fluid DLPC areas (18), the concentration of the probe was calculated only relative to the fluid phase. A mixture of lipid in chloroform was dried in a clean glass reaction vial under a small stream of N2. Purified water was added to the vial to resuspend the lipids to a final lipid concentration of 0.5 mg/mL. The suspension was then incubated in a 65°C water bath for 5 min with votexing periods of 15 s. The milky lipid suspension (containing multilamellar vesicles, MLVs) was then transferred into a plastic tube at room temperature before further treatments.
Small vesicle preparation methods
Method A
The MLV suspension was pushed through a polycarbonate membrane of defined pore size (50 nm in diameter) using gas-tight glass syringes. The center part of the extruder (i.e., the polycarbonate membrane and the membrane holder) (Model LiposoFast-Basic, Avestin, Ottawa, Canada) was encircled by a heating blanket and heated up to 65°C, whereas the syringes were kept at room temperature (20°C). Using the syringes the lipid suspension was pushed slowly through the center part of the extruder 20 times. During the extrusion process the vesicle suspension was thermally annealed, i.e., as the suspension passed through the center of the extruder, it was heated above the phase transition temperature of DSPC 55°C and then cooled in the room temperature syringe. The SUV suspension was then incubated in 65°C hot water bath for 2 min and used immediately to make a supported lipid bilayer (summarized in Fig. 1).
FIGURE 1.

Flow chart for methods of formation of DLPC/DSPC-supported lipid bilayers. Resulting domain symmetry for each method is given at the bottom.
Method B and B′
In this method we used either the tip sonification or extrusion method to make SUVs. In the tip sonification method, the MLV suspension was sonicated using a tip sonifier Model 250 (Branson Ultrasonics, Danbury, CT) equipped with a double stepped microtip (101-063-212, Branson Ultrasonics) in new condition at the lowest power for 30 s twice with a 20 s pause in between. In the extrusion method, the MLV suspension was pushed through a polycarbonate membrane of defined pore size (50 nm in diameter) 20 times using an extruder with gas-tight glass syringes. The whole extrusion process was performed in a 65°C water bath. In Method B the SUVs were cooled to room temperature and used immediately after cooling to make a supported lipid bilayer. Alternatively, in Method B′ the SUVs were incubated at 65°C in a hot water bath for 2 min and used immediately to make a supported lipid bilayer (summarized in Fig. 1).
Supported bilayer deposition conditions
A 150 μl droplet of the SUV suspension was added to a freshly cleaved room temperature mica disk which was glued to a small metal puck as described previously (19). When heated SUV suspensions (prepared by Method A and Method B′) were used, formation of the supported lipid bilayer occurred during a thermal quench from slightly above the Tm of DSPC to room temperature 20°C. For both the quenched and nonquenched vesicle deposition methods, the vesicle droplet was incubated on the mica disk for 30 min and then rinsed 40 times with purified water to remove excess vesicles (summarized in Fig. 1).
AFM imaging
Samples were imaged with a Digital Instruments NanoScope IIIa (Santa Barbara, CA) in contact mode with a J scan head. Experimental detail is described elsewhere (20). A public domain software package, Imagetool (University of Texas Health Science Center, San Antonio, TX), was used to analyze the size, perimeter, and area fraction of the solid phase domains in the AFM images of our samples.
Fluorescence recovery after photobleaching
Supported lipid bilayers that had been scanned by AFM were transferred into a Petri dish containing purified water. The samples were managed carefully so that the surfaces were hydrated at all times during the transfer. FRAP experiments were carried out on a Nikon Eclipse 400 fluorescence microscope (Nikon, Melville NY) equipped with a fluorescence filter cube (EF-4 FITC HYQ, Nikon) that matches the excitation and emission spectrum of NBD-PC. Images were captured with a high resolution Orca digital camera (Hamamatsu, Japan) at varying periods of time after a 5 s photobleaching. The excitation light was attenuated at least 400 times while observing the fluorescence recovery. Capture times were adjusted from 0.1 s to 1 s, depending on the sample, to get better imaging quality.
RESULTS
Supported lipid bilayers containing fluid-phase DLPC and gel-phase DSPC were formed by several different methods in an attempt to control the distribution of DSPC domains within the individual leaflets of the bilayer. Fig. 1 summarizes the different supported lipid bilayer preparation methods used in this study. In the following text, we initially focus on determining the distribution of DSPC domains in each leaflet for each preparation method. Then, we will characterize the time-dependent redistribution of DSPC in the case where we observed an initial uneven distribution of DSPC between the two leaflets. In the discussion, we will relate our results to their possible biological significance.
AFM section analysis of DLPC/DSPC-supported bilayers
Method A
AFM imaging revealed that supported lipid bilayers prepared in this manner contained DSPC domains extending ∼1.8 nm from the DLPC fluid-phase matrix (Fig. 2 A). The domains were immobile and were relatively centrosymmetric in shape. Individual domains of 1.8 nm in height remained completely unchanged over a 4 h observation period.
FIGURE 2.

AFM images and section analyses (the dotted lines denote the location of the sections) of phase-separated supported lipid bilayers. Lighter shading represent higher surfaces. (A) Supported lipid bilayer made by Method A. The measured domain heights extend ∼1.8 nm above the surrounding DLPC fluid-phase matrix. (B) Supported lipid bilayer made by Method B′. The bilayers prepared in this manner contained domains with areas extending 1.8 nm and 1.1 nm above the surrounding DLPC matrix. (Inset) 1.8 nm domains convert into 1.1 nm domains after ∼4 h; time after supported lipid bilayer formation: left, 30 min, middle, 1.5 h, and right, 4 h. (C) Supported lipid bilayer made by Method B. The domains are ∼1.1 nm higher than the fluid-phase region.
Method B
AFM imaging revealed that supported lipid bilayers prepared in this manner contained immobile DSPC domains extending only 1.1 nm above the surrounding DLPC matrix (Fig. 2 C). These bilayers remained unchanged over 4 h time.
Method B′
In comparison to the AFM images of supported lipid bilayers prepared by Method A or B where one domain height (1.8 nm or 1.1 nm, respectively) was observed, the supported lipid bilayers prepared in this manner contained domains with areas extending 1.8 nm and 1.1 nm above the surrounding DLPC matrix (Fig. 2 B). In this case, the domains were noticeably unstable and, after a period of several hours, converted to the lower height (1.1 nm) while the apparent domain area increased (Fig. 2 B, inset). After the conversion to the lower height, the bilayers remained stable for another 4 h observation period.
AFM section analysis of Langmuir-Blodgett deposited DLPC/DSPC-supported lipid bilayers
It has been shown previously that when Langmuir-Blodgett (L-B) deposition is used to form phase-separated supported lipid bilayers, the phases of each monolayer do not perfectly line up with each other (21,22). Therefore, a supported lipid bilayer containing DLPC and DSPC in both leaflets formed using L-B deposition should contain regions of symmetric fluid-phase DLPC (opposing monolayers of DLPC), symmetric gel-phase DSPC (opposing monolayers of DSPC), and asymmetric gel-phase DSPC (DSPC monolayer opposing a DLPC monolayer). AFM images of supported lipid bilayers formed by L-B deposition (for example, see Fig. 3) revealed that domains extended above the fluid-phase DLPC matrix at two heights, 1.1 nm, corresponding to asymmetric gel-phase DSPC, and 1.8 nm, corresponding to symmetric gel-phase DSPC. These are the same two heights that we observed in the supported lipid bilayers that were formed by vesicle fusion methods.
FIGURE 3.

AFM image and section analysis of an L-B-deposited supported lipid bilayer.
Therefore we can deduce that symmetric DSPC domains were formed by Method A (i.e., 1.8 nm height) (Fig. 1, bottom left). This deduction also is supported by x-ray diffraction measurements in which the bilayer thickness for DSPC and DLPC are 4.7 nm and 3.0 nm, respectively (23). By Method B, asymmetric DSPC domains were formed (i.e., 1.1 nm height) in only one monolayer (Fig. 1, bottom right) or in both monolayers but not in registry. By Method B′, symmetric DSPC domains were directly adjacent to asymmetric DSPC domains (i.e., both 1.8 nm and 1.1 nm heights) (Fig. 1, bottom middle). By their appearance (Fig. 2 B), each of these structures formed by Method B′ seemed to comprise DSPC domain(s) in one leaflet in registry with a larger DSPC domain in the opposing leaflet.
Fluorescence recovery of DLPC/DSPC-supported lipid bilayers in comparison to DLPC- and DSPC-supported lipid bilayers
It has been shown that nanometer size symmetric gel-phase DSPC domains can greatly obstruct lateral diffusion of surrounding fluid lipids (20,24). These works demonstrated a decrease in fluid-phase diffusion at increasing symmetric DSPC gel-phase domain area fraction. Therefore, we intentionally increased the concentration of DSPC in the DLPC/DSPC mixture to form supported lipid bilayers containing a very high domain area fraction (>0.7) to further investigate the location (in either one or both leaflets) of DSPC domains. We included 1 mol % NBD-PC (2 mol % for DSPC bilayer) to trace the diffusion of the fluid DLPC lipids in the bilayers; as previously stated, NBD-PC partitions in less ordered phases (18). We performed qualitative FRAP experiments on supported lipid bilayers prepared by Method A and B. Each sample was imaged by AFM before FRAP measurements. An octagonal spot (∼50 μm in size) on the bilayer was photobleached for 5 s. The excitation light was attenuated at least 400 times while observing the fluorescence recovery.
The supported lipid bilayer formed by Method A containing symmetric DSPC domains of height 1.8 nm recovered much more slowly (Fig. 4 B for AFM image and B1 for FRAP images) than the supported lipid bilayer formed by Method B containing asymmetric DSPC domains of height 1.1 nm (Fig. 4 C for AFM image and C1 for FRAP images). In fact, the recovery time of the bilayer containing 1.1 nm DSPC domain heights was very close to that of a supported lipid bilayer consisting of only fluid-phase DLPC (Fig. 4 D for AFM image and D1 for FRAP images), and after 90 s, merely a faint remnant of the original bleached spot was visible. In comparison, the fluorescence recovery of the supported lipid bilayer containing DSPC domains of height 1.8 nm resembled that of a pure DSPC-supported lipid bilayer (Fig. 4 A for AFM image and A1 for FRAP images), indicating a long-range diffusion coefficient several orders of magnitude lower than for a pure fluid bilayer. The slow recovery of the DLPC/DSPC bilayers formed by Method A (symmetric DSPC domains) indicates that the fluid phase in both monolayers was highly obstructed. Therefore, the DSPC domains spanned across the lipid bilayer, almost completely obstructing long-range diffusion of the probe in the fluid phase (illustration in Fig. 4 B). The much faster recovery of the DLPC/DSPC bilayers formed by Method B (asymmetric DSPC domains) indicates that there existed a large proportion of the fluorescent probe which was completely unobstructed. The fact that we observed a faint photobleached spot after the initial recovery is consistent with this notion and indicates that the domains were confined completely to one of the monolayers in which recovery was highly obstructed (illustration in Fig. 4 C). This also excludes the possibility that the intermediate height difference of 1.1 nm was due to an interdigitated version of the bilayer, because this would have led to slow diffusion (i.e., obstructed diffusion).
FIGURE 4.

AFM images (A–D), fluorescent images from FRAP experiments (A1–D1), and cobalt-quenching experiments (A2–D2) for supported lipid bilayers prepared by different methods. (A) Supported DSPC lipid bilayer doped with 2 mol % NBD-PC. (B) A supported lipid bilayer made by Method A doped with 1 mol % NBD-PC. The area fraction of gel-phase region is ∼0.79 (DSPC/DLPC molar ratio ∼70:20). (C) A supported lipid bilayer made by Method B doped with 1 mol % NBD-PC. The area fraction of the gel-phase region is ∼0.75 (DSPC/DLPC molar ratio ∼40:60). (D) Supported DLPC bilayer doped with 1 mol % NBD-PC. The top illustrates the type of supported lipid bilayer. In each FRAP experiment, images were taken after photobleaching. The original bleached spot is ∼50 μm in diameter. In the cobalt-quenching experiments, fluorescent images were taken before (left) and after (right) addition of 50 mM cobalt chloride ions in the water subphase. The measured intensity is labeled on each image. The scale bar is 10 μm unless specified.
Fluorescence quenching of DLPC/DSPC-supported lipid bilayers
To determine if DSPC occupies the monolayer proximal or distal to the mica substrate in DLPC/DSPC-supported lipid bilayers prepared by Method B (asymmetric DSPC domains), fluorescence-quenching experiments were performed using cobalt ions as quenchers. It has been observed that these cations do not penetrate the lipid bilayer at concentrations lower than 100 mM (25). Therefore, upon addition of CoCl2 to the water subphase of the NBD-PC-labeled supported lipid bilayer, we would expect to observe a decrease in the fluorescence signal due to the static quenching of the distal monolayer. If we assume NBD-PC partitions evenly in the fluid-phase DLPC, then the fraction of the fluorescence signal remaining after quenching is roughly in proportion to the fraction of DLPC in the proximal monolayer. The same bilayers that were used to perform FRAP experiments were used for cobalt-quenching experiments, and a calculated amount of CoCl2 was added to the water subphase to achieve a final CoCl2 concentration of 50 mM. Fluorescent images were taken before and 15 min after addition of CoCl2. For supported lipid bilayers containing DSPC (formed by Method A), DLPC/DSPC (formed by Method A—symmetric DSPC domains), and DLPC, we observed a nearly 50% decrease in the fluorescence signal (Fig. 4, A2, B2, and D2, respectively) after adding cobalt ions, which indicates a generally even distribution of NBD-PC in the proximal and distal leaflets in these bilayers. For the supported lipid bilayer formed by Method B (asymmetric DSPC domains), we obtained only an 18% decrease of the fluorescence signal (Fig. 4, C2). Based on this reduced fluorescent signal, we calculated the relative area covered by fluid phase in the distal leaflet to be 22%, i.e., the remaining 78% of the area was covered by nonfluorescent regions, presumably the DSPC domains. Analysis of the AFM images for the same bilayer resulted in a DSPC domain area fraction of 0.75, matching the determined DSPC domain area fraction from the cobalt-quenching data. Therefore we conclude that the asymmetric domains formed by Method B exclusively partitioned in the distal monolayer. It is worth noting that in each experiment after rinsing the cobalt ion from the subphase, we observed an almost complete recovery of fluorescence (∼99%), indicating that indeed this ion was not significantly penetrating the bilayer at the experimental concentrations. The illustrations at the bottom of Fig. 1 summarize our observations with regard to the dependence of DSPC distribution on preparation method presented thus far.
Quantifying lipid flip-flop in bilayers with uneven distribution of DSPC
As mentioned previously, we observed that the supported lipid bilayers formed by Method B′ were unstable: the domains that extended 1.8 nm (symmetric) above the DLPC matrix converted over a period of hours to the lower (asymmetric) height (1.1 nm), whereas the apparent domain area increased (Fig. 2 B, inset). By acquiring images over several hours, we were able to characterize changes in the area and perimeter of the two different domain regions (symmetric and asymmetric) throughout the conversion process. We believe the AFM scanning, i.e., tip-sample contact, did not affect this conversion since the conversion speed did not change with the number of scans that were performed. Fig. 5 illustrates the change in domain area as a function of time for a DLPC/DSPC bilayer prepared by Method B′. The total area occupied by DSPC was calculated by adding the domain area of the 1.1 nm region to two times the domain area of the 1.8 nm region. We found that during the conversion, the total area occupied by DSPC remained roughly constant (Fig. 5, solid squares). Combining this with the fact that we did not observe any vesicle budding or fusion phenomena during the experiment, we believe that DSPC molecules transferred, or flipped, from the proximal leaflet to the distal leaflet during the conversion process. This flip-flop process did not occur evenly throughout the DSPC domains. It could be seen that the height converted rapidly from 1.8 nm to 1.1 nm at the interface between symmetric and asymmetric DSPC gel-phase domains (Fig. 6 A, white dashed arrows), whereas the interface between symmetric gel-phase DSPC domains and fluid-phase DLPC was relatively stable (Fig. 6 A, white solid arrows). The most rapid conversion occurred toward pools of asymmetric fluid-phase DLPC trapped inside the domain. These were not visible until the area around the pool had converted, revealing a small region with a 1.1 nm step height (Fig. 6 A, black arrows). We plotted the distribution of the interface movement toward the center of the domain for each case and fitted with Gaussian curves (Fig. 6 B). The center of the Gaussian peaks for symmetric DLPC-symmetric DSPC interface, symmetric DSPC-asymmetric DSPC interface, and interface associated with DLPC pools located at −1.7 (i.e., ∼0), 36.5, and 85.3 nm/h, respectively.
FIGURE 5.

Change in domain area of a DLPC/DSPC-supported lipid bilayer formed by Method B′ as a function of time. Open circles represent the area of DSPC symmetric domains; open squares represent the area of DSPC asymmetric domains. The total area occupied by DSPC (solid squares) is the summation of asymmetric domains and two times the symmetric domains. The dashed line represents the average value. Data shown here are average results from five 10 μm × 10 μm AFM bilayer scans.
FIGURE 6.

Interface movement due to lipid flip-flop in supported lipid bilayers. (A) Time sequence images of a bilayer made by Method B′ showing the evolution of the domain structure after bilayer formation. The unit of time is an hour. We observed a fast interface movement at the interface between 1.1 nm height and 1.8 nm height (white dashed arrow; symmetric DSPC-asymmetric DSPC interface) and a slow interface movement at the interface between 0 nm height and 1.8 nm height (white solid arrow; symmetric DLPC-symmetric DSPC interface). In addition, the fast interface movement also happened when there was a fluid DLPC pool trapped within the gel domain (black arrow). (B) A histogram of interface movement for each case. Dashed lines represent the Gaussian fit of each group. The center of the Gaussian peaks located at 1.7, 36.5, and 85.3 nm/h for symmetric DLPC-symmetric DSPC interface, symmetric DSPC-asymmetric DSPC interface, and interface associated with DLPC pools, respectively.
We examined two possible models for this conversion process: lipids flipping uniformly throughout the symmetric domain region or exclusively at the domain perimeter (i.e., the symmetric DSPC-asymmetric DSPC interface). To determine where the lipid flip-flop event happened most frequently, area and perimeter information was analyzed as a function of time for domain structures only containing a symmetric DSPC-asymmetric DSPC interface (for example, see Fig. 7 B). The distribution of the interface movement for these domain structures has its Gaussian peak located at 45.5 nm/h. If the lipid flip-flop happened evenly throughout the entire symmetric DSPC portion of the domain structure, a simple two-compartment model can be used to describe the event (7). The time rate of change of DSPC molecules in the proximal leaflet, which can be calculated as change of area of the symmetric DSPC domain region (i.e., 1.8 nm in height) A1 (assuming area per lipid molecule is a constant), is given by 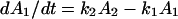 where A2 is the DSPC domain area in the distal leaflet and k1 and k2 are the rate constants of lipid flipping from the proximal to the distal monolayer and from the distal to the proximal layer, respectively. With the experimental condition that the total area occupied by DSPC was constant (A1 + A2 = constant), we would expect the solution, A1(t), to have the form of exp[−(k1 + k2)t]. Interestingly, we did not observe this trend in our data analysis. When fitted with an exponential equation, the mean R-squared value among the 42 domains that were analyzed was only 0.901 (data not shown).
where A2 is the DSPC domain area in the distal leaflet and k1 and k2 are the rate constants of lipid flipping from the proximal to the distal monolayer and from the distal to the proximal layer, respectively. With the experimental condition that the total area occupied by DSPC was constant (A1 + A2 = constant), we would expect the solution, A1(t), to have the form of exp[−(k1 + k2)t]. Interestingly, we did not observe this trend in our data analysis. When fitted with an exponential equation, the mean R-squared value among the 42 domains that were analyzed was only 0.901 (data not shown).
FIGURE 7.

Quantifying lipid flip-flop in supported lipid bilayers formed by Method B′. (A) Measured perimeter of four symmetric DSPC domains (in asymmetric DSPC/symmetric DSPC domain structures) as a function of time. The solid line represents the polynomial fit of the data. (B) Time sequence of the type of domain used for this analysis. (C) Distribution of the rate constant K. The average value of K is 77 ± 17 h−1.
On the other hand, if the flipping event happened most frequently at the symmetric DSPC-asymmetric DSPC interface, the time rate of change of DSPC molecules in the proximal leaflet would be directly proportional to the number of DSPC molecules present at the interface:
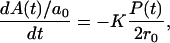 |
(1) |
where A is the area of DSPC in the proximal leaflet, P(t) is the perimeter of the symmetric DSPC domain-asymmetric DSPC domain interface, 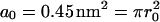 is the area of an individual DSPC molecule, and K is the rate constant for the flipping event. After integration, Eq. 1 becomes
is the area of an individual DSPC molecule, and K is the rate constant for the flipping event. After integration, Eq. 1 becomes
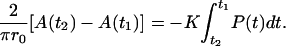 |
(2) |
Fig. 7 A shows the domain perimeter data points as a function of time for four individual domains and the polynomial fits (in solid line) that have R-squared values >0.998. The resulting P(t) along with the measured area of DSPC in the proximal leaflet allows us to solve for the rate constant K. Despite the wide range of sizes of DSPC domain structures, we obtained a narrow distribution of the rate constant K with the average of 76 ± 17 h−1 (Fig. 7 C). Thus, it is likely that the flip-flop process occurred mainly at the interface between symmetric DSPC and asymmetric DSPC.
DISCUSSION
We developed three methods involving vesicle preparation and deposition temperature which have allowed us to control the initial distribution of gel-phase DSPC in a DLPC/DSPC-supported lipid bilayer. Bilayers prepared by Method A resulted in an even distribution of gel-phase DSPC in both leaflets, whereas bilayers formed by Method B resulted in gel-phase DSPC domains exclusively partitioned in the distal monolayer relative to the mica substrate. In contrast, Method B′ resulted in an uneven distribution of DSPC between the two leaflets that was highly unstable. We believe that the initial DSPC distribution in the supported lipid bilayer was controlled mainly by the bilayer characteristics of the vesicles (e.g., lateral mixing, leaflet asymmetry) used for vesicle fusion. For example, although it is known that GUVs are transbilayer symmetric, it has been reported that in SUVs, due to the highly curved surface, the lipid distribution can be transbilayer asymmetric and the partitioning depends on the overall molecular shape. In general, in mixed lipid vesicles, those lipids which have larger area per molecule tend to distribute preferentially in the outer leaflet (26) and lipids located in the inner leaflet usually pack tighter than the same species located in the outer leaflet (27). Therefore it is likely that DSPC has the tendency to locate at the inner leaflet of DLPC/DSPC SUVs at room temperature. It follows that supported bilayers formed by Method B, where SUV deposition occurred at room temperature, should contain DSPC domains partitioned to the distal leaflet as we observed. This general area will be a subject of future investigation by our group. However, our main goal was to investigate the biophysical properties of supported lipid bilayers with symmetric and asymmetric distributions of gel-phase lipids to gain insight into the consequences of lipid asymmetry in living cell membranes.
We find that supported lipid bilayers containing DSPC domains in either one or both leaflets are always laterally immobile regardless of the area fraction of the domains. This was not necessarily expected in the case of asymmetric DSPC domains since they were not in contact with the substrate (i.e., we found the domains to be distal to the substrate). In the case of the symmetric DSPC domains, the immobility of symmetric gel-phase domains has been observed before in supported lipid bilayers of DLPC/DSPC (20), DLPC/DPPC (15), and DOPC/DPPC (28). It is generally believed that the immobility of objects that extend toward the substrate (such as proteins and symmetric lipid domains) in supported lipid bilayers arise from an attractive interaction between the closely positioned substrate and the object (29,30). The lack of mobility for the asymmetric DSPC domain cannot be readily explained by an extension of the bilayer toward the substrate since the asymmetric DSPC domain extends from the distal side of the bilayer not the proximal side and chain extension (i.e., ordering) of the DLPC in the neighboring leaflet is precluded, as discussed below. One can argue that the diffusion coefficient of a large object (i.e., asymmetric DSPC domains) in the bilayer would be lowered since it is related to the radius of the object (31): a DSPC domain of 250 nm in diameter would have a diffusion coefficient ∼50 times lower than that of a fluid lipid (assuming 63 Å2/molecule). However even such a slow diffusion would have given us more than 50 μm root mean-square displacement in an hour, which we did not observe over the several hours of imaging.
A possible explanation comes from the impact of the asymmetric DSPC domain on the mechanical properties of the bilayer. The total interaction energy between the substrate and the supported bilayer is a balance of attractive van der Waals and repulsive steric forces. Among the three main types of steric forces, the hydration force, the undulation force, and the peristaltic force, the latter two forces are inversely proportional to the mechanical properties of the bilayer, bending modulus, and area expansion modulus, respectively (32,33). It is known that the bending and area expansion modulus of gel-phase lipid bilayers is nearly 10-fold higher than that of the fluid-phase lipid bilayers (34). Consequently gel-phase-supported lipid bilayers will have much stronger interactions with the mica substrate than fluid bilayers as a result of 10-fold reduction in steric repulsive forces. Therefore, symmetric DSPC domains will exhibit strong adhesive interactions to the mica substrate. The lack of mobility of the asymmetric DSPC domains in the distal leaflet suggests a mechanical coupling of the stiff DSPC distal monolayer with the proximal DLPC monolayer. This mechanical coupling would result in flattening the thermal fluctuations of the proximal DLPC monolayer. As a result, the DSPC asymmetric bilayer unit behaves mechanically similar to the gel-phase symmetric bilayer unit, i.e., a strong adhesion to the mica substrate and asymmetric domain immobilization (Fig. 1, sketch). This mechanism may apply to biological membranes resulting in an additional role of rafts in cellular membranes. The mechanical coupling observed for asymmetric DSPC domains in this model membrane system suggests that rafts or ordered phased domains in one leaflet are able to locally decrease the membrane undulation and lead to a strong adhesion and close contact between the other leaflet and a substrate, which can be cytoskeleton or another membrane. This novel mechanism may play an important role in exocytosis pathways and intracellular trafficking, which can be regulated by lipid rafts, cholesterol, and sphingolipid-rich domains that are enriched in the extracellular membrane (35,36). Our data suggest that, besides docking essential proteins for intracellular membrane fusion (SNARE and SNAREs, for example), ordered lipid domains in the extracellular membrane can also provide an environment that promotes close contact on the other side of the membrane that then leads to membrane fusion.
Our results do not indicate that the asymmetric DSPC domain is causing a significant ordering effect on the proximal DLPC monolayer. If there were an ordering effect, we would expect a significantly slow fluorescence recovery in the case of a high domain area fraction of asymmetric DSPC domains, since ordered domains would have existed in both leaflets of the bilayer (ordered DSPC domains in the distal leaflet and corresponding ordered DLPC domains in the proximal leaflet). In addition, if asymmetric DSPC domains induced an ordered phase in the proximal monolayer, we would expect NBD-PC to have a significantly lower partitioning in this phase. Therefore, we would have obtained similar results for both symmetric and asymmetric DSPC domains in the cobalt-quenching experiments, i.e., the gel-phase asymmetric bilayer unit would behave structurally similar to the gel-phase symmetric bilayer unit. It has been reported that, in DOPC/sphingomyelin/cholesterol-supported bilayer, ordered domains in the proximal leaflet may induce ordering in the distal leaflet (22). This ordering effect is likely due to interdigitation of cholesterol across the bilayer since cholesterol vibrates perpendicular to the bilayer continuously and penetrates into the opposing monolayer by 5–11 Å (37,38). Combining these results, we conclude that gel-phase domains consisting of long-chain saturated lipids in one leaflet do not seem to be capable of any strong ordering effect in the neighboring leaflet when made of a short-chain saturated lipid. These conclusions in combination with the mechanical coupling discussed above indicate that the mechanical properties transferred to the proximal monolayer from asymmetric DSPC domains do not dramatically alter the phase of the proximal monolayer. Therefore, we observe no strong ordering effect, but the mechanical effect is not negligible and can lead to strong and stable adhesive contact of the neighboring monolayer with a substrate.
We find that when domains exist in each monolayer, the DSPC gel-phase regions tend to register across the two leaflets as much as possible. When there is an even distribution of DSPC between the two leaflets, we observe complete registry (symmetric DSPC domains). This is in agreement with previous work involving GUVs, which always seem to display a symmetric distribution of gel-phase lipids in mixed fluid-gel bilayers. In those cases, the gel domains in each leaflet always superimpose upon each other. It has been proposed that the origin of the superimposed phases observed in model membranes is strong intermonolayer coupling between similar phases (13,39). It has been suggested that the source of this increased coupling results from increased interactions between the tails of gel-phase lipids relative to the tails of gel-phase and fluid-phase lipids at the bilayer midplane (40). These increased interactions may drive gel-phase lipids to align in opposing monolayers. The results from supported lipid bilayers containing an uneven DSPC distribution strongly support these conclusions. Under these conditions we always observe maximal overlap of DSPC domains within the two leaflets, indicating a strong gel-phase tail-tail interaction. Several functions of rafts involve transient communication between ordered phase domains in the outer leaflet and ordered phase domains in the inner leaflet. It has been postulated that this communication may arise from transient interleaflet interactions (41). Our results suggest that ordered phase domains in one leaflet will indeed align with ordered phase domains in the neighboring leaflet. The transience of this interleaflet association may result in part from rapid flip-flop, as discussed below.
Qualitatively, we observed that each symmetric DSPC domain formed from two same-sized monolayer domains that are in perfect registry have a long time stability (hours). This is similar to the case of most previous studies, especially involving giant vesicles (13). These domains do not seem to reflect the situation in living cell membranes where domains are mainly in one leaflet and are not coupled for long periods of time to domains in the neighboring leaflet. The question arises—if we see strong coupling in model systems, why do we not see the same thing in living membranes. Our study may provide a partial answer to this question. By forming bilayers of uneven domain distributions between the two leaflets of the bilayer, we were able to observe that although the symmetric DSPC-symmetric DLPC interface is extremely stable, the symmetric DSPC-asymmetric DSPC interface which occurs when domains of uneven sizes are superimposed is extremely unstable.
Quantitatively, we found that a rapid one-way flip-flop process occurs at those interfaces, which moves the DSPC to the leaflet rich in DSPC, resulting in domain growth in the distal leaflet. The process is most rapid in the case of a pool of trapped DLPC, such that, theoretically, symmetric-ordered regions of biological size (∼50 nm) can be converted to asymmetric domains within minutes. Most likely this is an Ostwald ripening process, as it results in more DSPC being moved away from the perimeter of the domain where there is a very unfavorable hydrophobic mismatch due to the six-carbon difference in acyl-chain length between the two lipid species. We do not know why the symmetric DSPC-symmetric DLPC interface is so stable, and we are beginning computer simulations to investigate. Our results suggest that only domains that are of exactly equal size and in perfect registry will be completely unchanged and stable on the order of hours. Although this can be the case for giant vesicles or supported lipid bilayers, it is clearly not the case in living membranes that the compositions of domains and their size would be the same in each leaflet. Therefore, the drive will always be toward enrichment of long-chained/ordered phase lipids in one leaflet of the bilayer.
CONCLUSION
In this work, we controlled the distribution of DSPC domains within the individual leaflets of a DLPC/DSPC-supported lipid bilayer to gain insight into the consequences of lipid asymmetry in living cell membranes. When DSPC domains were in both leaflets but in unequal proportions, symmetric DSPC domains (i.e., DSPC in both leaflets) were unstable and converted, through lipid flip-flop, to the stable asymmetric distribution (i.e., DSPC domains were exclusively in the leaflet distal to the substrate). Since it is highly unlikely that the cell membrane exists in a symmetric state, this instability suggests a passive mechanism in which cell membranes maintain asymmetric lipid distributions. Asymmetric domains remained completely immobile even though they were found to exist in the distal leaflet. This indicates a strong mechanical coupling between gel-phase domains in one leaflet and fluid-phase lipids in the adjacent leaflet. These results suggest that ordered phase domains in cellular membranes may be able to locally modulate membrane stiffness, which can increase the strength and lifetime of adhesion events on either side of the bilayer. We did not observe any significant ordering effect induced by asymmetric gel-phase domains in one leaflet onto the opposing fluid-phase monolayer in contrast to previous studies of the ordering effect of cholesterol-containing domains. We also observed maximal alignment of gel-phase domains across the leaflets of the bilayer indicating a strong gel-phase tail-tail interaction. Our work suggests a mechanism by which ordered phase domains in the two leaflets of the cellular membrane may transiently communicate: ordered phase alignment accompanied by a rapid one-way flip-flop of the ordered lipids from one leaflet into the other leaflet. The results we have obtained have lead to several novel mechanisms by which ordered phase domains in cellular membranes may be able to locally alter membrane mechanical properties, contribute to a passive process of lipid asymmetry, and transiently communicate between the two leaflets.
Acknowledgments
We thank Dr. Steven Boxer, Stanford University, and his laboratory group for the cobalt-quenching protocol. M.L.L. acknowledges funding by the Center for Polymeric Interfaces and Macromolecular Assemblies (National Science Foundation grant DMR 0213618), the Nanoscale Interdisciplinary Research Teams Program of the National Science Foundation (under award numbers CHE 0210807 and BES 0506602), the Materials Research Institute at Lawrence Livermore National Laboratory (MI-03-117), and a generous endowment from Joe and Essie Smith. C.D.B. acknowledges funding from the National Institutes of Health Biotechnology Training Grant of the University of California, Davis. This work was performed under the auspices of the U.S. Dept. of Energy by the University of California/Lawrence Livermore National Laboratory under contract No. W-7405-Eng-48.
References
- 1.Brown, D. A., and E. London. 2000. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275:17221–17224 [Review]. [DOI] [PubMed] [Google Scholar]
- 2.Devaux, P. F., and R. Morris. 2004. Transmembrane asymmetry and lateral domains in biological membranes. Traffic. 5:241–246. [DOI] [PubMed] [Google Scholar]
- 3.Devaux, P. F. 1991. Static and dynamic lipid asymmetry in cell membranes. Biochemistry. 30:1163–1173. [DOI] [PubMed] [Google Scholar]
- 4.Daleke, D. L. 2003. Regulation of transbilayer plasma membrane phospholipid asymmetry. J. Lipid Res. 44:233–242. [DOI] [PubMed] [Google Scholar]
- 5.Kornberg, R. D., and H. M. McConnell. 1971. Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry. 10:1111–1120. [DOI] [PubMed] [Google Scholar]
- 6.Boon, J. M., and B. D. Smith. 2002. Chemical control of phospholipid distribution across bilayer membranes. Med. Res. Rev. 22:251–281. [DOI] [PubMed] [Google Scholar]
- 7.John, K., S. Schreiber, J. Kubelt, A. Herrmann, and P. Muller. 2002. Transbilayer movement of phospholipids at the main phase transition of lipid membranes: implications for rapid flip-flop in biological membranes. Biophys. J. 83:3315–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Middelkoop, E., B. H. Lubin, E. M. Bevers, J. A. F. O. Denkamp, P. Comfurius, D. T. Y. Chiu, R. F. A. Zwaal, L. L. M. Vandeenen, and B. Roelofsen. 1988. Studies on sickled erythrocytes provide evidence that the asymmetric distribution of phosphatidylserine in the red-cell membrane is maintained by both ATP-dependent translocation and interaction with membrane skeletal proteins. Biochim. Biophys. Acta. 937:281–288. [DOI] [PubMed] [Google Scholar]
- 9.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- 10.Fantini, J., M. Maresca, D. Hammache, N. Yahi, and O. Delezay. 2000. Glycosphingolipid (GSL) microdomains as attachment platforms for host pathogens and their toxins on intestinal epithelial cells: activation of signal transduction pathways and perturbations of intestinal absorption and secretion. Glycoconj. J. 17:173–179. [DOI] [PubMed] [Google Scholar]
- 11.Seigneuret, M., A. Zachowski, A. Hermann, and P. F. Devaux. 1984. Asymmetric lipid fluidity in human erythrocyte membrane: new spin-label evidence. Biochemistry. 23:4271–4275. [DOI] [PubMed] [Google Scholar]
- 12.Chahine, J. M. E. H., S. Cribier, and P. F. Devaux. 1993. Phospholipid transmembrane domains and lateral diffusion in fibroblasts. Proc. Natl. Acad. Sci. USA. 90:447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagatolli, L. A., and E. Gratton. 2000. A correlation between lipid domain shape and binary phospholipid mixture composition in free standing bilayers: a two-photon fluorescence microscopy study. Biophys. J. 79:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Almeida, R. F. M., L. M. S. Loura, A. Fedorov, and M. Prieto. 2002. Nonequilibrium phenomena in the phase separation of a two-component lipid bilayer. Biophys. J. 82:823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokumasu, F., A. J. Jin, G. W. Feigenson, and J. A. Dvorak. 2003. Nanoscopic lipid domain dynamics revealed by atomic force microscopy. Biophys. J. 84:2609–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rinia, H. A., M. M. E. Snel, J. P. J. M. van der Eerden, and B. de Kruijff. 2001. Visualizing detergent resistant domains in model membranes with atomic force microscopy. FEBS Lett. 501:92–96. [DOI] [PubMed] [Google Scholar]
- 17.Mabrey, S., and J. M. Sturtevant. 1976. Investigation of phase transitions of lipids and lipid mixtures by high sensitivity differential scanning calorimetry. Proc. Natl. Acad. Sci. USA. 73:3862–3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mesquita, R., E. Melo, T. E. Thompson, and W. L. C. Vaz. 2000. Partitioning of amphiphiles between coexisting ordered and disordered phases in two-phase lipid bilayer membranes. Biophys. J. 78:3019–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKiernan, A. E., T. V. Ratto, and M. L. Longo. 2000. Domain growth, shapes, and topology in cationic lipid bilayers on mica by fluorescence and atomic force microscopy. Biophys. J. 79:2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ratto, T. V., and M. L. Longo. 2002. Obstructed diffusion in phase-separated supported lipid bilayers: a combined atomic force microscopy and fluorescence recovery after photobleaching approach. Biophys. J. 83:3380–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollars, C. W., and R. C. Dunn. 1998. Submicron structure in L-alpha-dipalmitoylphosphatidylcholine monolayers and bilayers probed with confocal, atomic force, and near-field microscopy. Biophys. J. 75:342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stottrup, B. L., S. L. Veatch, and S. L. Keller. 2004. Nonequilibrium behavior in supported lipid membranes containing cholesterol. Biophys. J. 86:2942–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marsh, D. 1990. CRC Handbook of Lipid Bilayers. CRC Press, Boca Raton, FL.
- 24.Almeida, P. F. F., W. L. C. Vaz, and T. E. Thompson. 1992. Lateral diffusion and percolation in two-phase two-component lipid bilayers topology of the solid-phase domains in-plane and across the lipid bilayer. Biochemistry. 31:7198–7210. [DOI] [PubMed] [Google Scholar]
- 25.Bayerl, T. M., and M. Bloom. 1990. Physical properties of single phospholipid bilayers adsorbed to micro glass beads. A new vesicular model system studied by 2H-nuclear magnetic resonance. Biophys. J. 58:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Israelachvili, J. N., D. J. Mitchell, and B. W. Ninham. 1977. Theory of self-assembly of lipid bilayers and vesicles. Biochim. Biophys. Acta. 470:185–201. [DOI] [PubMed] [Google Scholar]
- 27.Huang, C., and J. T. Mason. 1978. Geometric packing constraints in egg phosphatidylcholine vesicles. Proc. Natl. Acad. Sci. USA. 75:308–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giocondi, M. C., V. Vie, E. Lesniewska, P. E. Milhiet, M. Zinke-Allmang, and C. Le Grimellec. 2001. Phase topology and growth of single domains in lipid bilayers. Langmuir. 17:1653–1659. [Google Scholar]
- 29.Brian, A. A., and H. M. McConnell. 1984. Allogeneic stimulation of cyto-toxic T-cells by supported planar membranes. Proc. Natl. Acad. Sci. USA. 81:6159–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan, P. Y., M. B. Lawrence, M. L. Dustin, L. M. Ferguson, D. E. Golan, and T. A. Springer. 1991. Influence of receptor lateral mobility on adhesion strengthening between membranes containing LFA-3 and CD2. J. Cell Biol. 115:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans, E., and E. Sackmann. 1988. Translational and rotational drag coefficients for a disk moving in a liquid membrane associated with a rigid substrate. J. Fluid Mech. 194:553–561. [Google Scholar]
- 32.Israelachvili, J. N. 1992. Intermolecular & Surface Forces, 2nd ed. Academic Press, London.
- 33.Radler, J. O., T. J. Feder, H. H. Strey, and E. Sackmann. 1995. Fluctuation analysis of tension-controlled undulation forces between giant vesicles and solid substrates. Phys. Rev. E. 51:4526–4536. [DOI] [PubMed] [Google Scholar]
- 34.Lee, C.-H., W.-C. Lin, and J. Wang. 2001. All-optical measurements of the bending rigidity of lipid-vesicle membranes across structural phase transitions. Phys. Rev. E. 64:020901. [DOI] [PubMed] [Google Scholar]
- 35.Salaun, C., D. J. James, and L. H. Chamberlain. 2004. Lipid rafts and the regulation of exocytosis. Traffic. 5:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helms, J. B., and C. Zurzolo. 2004. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic. 5:247–254. [DOI] [PubMed] [Google Scholar]
- 37.Endress, E., H. Heller, H. Casalta, M. F. Brown, and T. M. Bayerl. 2002. Anisotropic motion and molecular dynamics of cholesterol, lanosterol, and ergosterol in lecithin bilayers studied by quasi-elastic neutron scattering. Biochemistry. 41:13078–13086. [DOI] [PubMed] [Google Scholar]
- 38.Hildenbrand, M. F., and T. M. Bayerl. 2005. Differences in the modulation of collective membrane motions by ergosterol, lanosterol, and cholesterol: a dynamic light scattering study. Biophys. J. 88:3360–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korlach, J., P. Schwille, W. W. Webb, and G. W. Feigenson. 1999. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 96:8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bagatolli, L. A., and E. Gratton. 2000. Two photon fluorescence microscopy of coexisting lipid domains in giant unilamellar vesicles of binary phospholipid mixtures. Biophys. J. 78:290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subczynski, W. K., and A. Kusumi. 2003. Dynamics of raft molecules in the cell and artificial membranes: approaches by pulse EPR spin labeling and single molecule optical microscopy. Biochim. Biophys. Acta. 1610:231–243. [DOI] [PubMed] [Google Scholar]