

Abstract
Peridinin-chlorophyll a-protein (PCP) is a unique antenna complex in dinoflagellates that employs peridinin (a carotenoid) as its main light-harvesting pigment. Strong excitonic interactions between peridinins, as well as between peridinins and chlorophylls (Chls) a, can be expected from the short intermolecular distances revealed by the crystal structure. Different experimental approaches of nonlinear polarization spectroscopy in the frequency domain (NLPF) were used to investigate the various interactions between pigments in PCP of Amphidinium carterae at room temperature. Lineshapes of NLPF spectra indicate strong excitonic interactions between the peridinin's optically allowed S2 (1Bu+) states. A comprehensive subband analysis of the distinct NLPF spectral substructure in the peridinin region allows us to assign peridinin subbands to the two Chls a in PCP having different S1-state lifetimes. Peridinin subbands at 487, 501, and 535 nm were assigned to the longer-lived Chl, whereas a peridinin subband peaking at 515 nm was detected in both clusters. Certain peridinin(s), obviously corresponding to the subband centered at 487 nm, show(s) specific (possibly Coulombic?) interaction between the optically dark 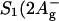 ) and/or intramolecular charge-transfer (ICT) state and S1 of Chl a. The NLPF spectrum, hence, indicates that this peridinin state is approximately isoenergetic or slightly above S1 of Chl a. A global subband analysis of absorption and NLPF spectra reveals that the Chl a Qy-band consists of two subbands (peaking at 669 and 675 nm and having different lifetimes), confirmed by NLPF spectra recorded at high pump intensities. At the highest applied pump intensities an additional band centered at ≤660 nm appears, suggesting—together with the above results—an assignment to a low-dipole moment S0 → S1/ICT transition of peridinin.
) and/or intramolecular charge-transfer (ICT) state and S1 of Chl a. The NLPF spectrum, hence, indicates that this peridinin state is approximately isoenergetic or slightly above S1 of Chl a. A global subband analysis of absorption and NLPF spectra reveals that the Chl a Qy-band consists of two subbands (peaking at 669 and 675 nm and having different lifetimes), confirmed by NLPF spectra recorded at high pump intensities. At the highest applied pump intensities an additional band centered at ≤660 nm appears, suggesting—together with the above results—an assignment to a low-dipole moment S0 → S1/ICT transition of peridinin.
INTRODUCTION
Dinoflagellates possess a unique water-soluble peripheral antenna complex, peridinin-chlorophyll a-protein (PCP). The carotenoid peridinin is the main light-harvesting pigment in PCP. The crystal structure of PCP from Amphidinium (A.) carterae reveals a trimeric organization of the complex (1). Within each monomeric subunit, pigments are organized in two clusters related by a pseudo-symmetry. Each cluster contains one chlorophyll (Chl) a surrounded (within van der Waals distances) by four peridinins (1). The closest distances between peridinins and Chls, as well as between peridinins in one cluster, are ∼4 Å. The center-to-center distance between the two Chls a in a PCP monomer is rather large (∼17 Å; see Hofmann et al. (1)). Thus, whereas pronounced peridinin-peridinin (2–6) as well as peridinin-Chl a interactions can be expected in PCP, interaction between the Chls appears to be negligible.
Peridinin is an unusual carotenoid, which has a C-37 backbone instead of the usual C-40 carbon skeleton. Peridinin also contains an allene moiety, a lactone ring in conjugation with the π-system and acetate and epoxy groups on the opposite β-rings. These features strongly influence the photophysical properties; in nonpolar solvents, peridinin shows a typical carotenoid vibrational substructure of the optically allowed S0 → S2 transition in the 350–550 nm range. (Note that the following nomenclature for carotenoid singlet states is used: S0 = 1Ag−, S1 = 2Ag−, and S2 = 1Bu+; the recently experimentally detected state with 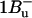 character (7) is assumed to lie above S2 in peridinin (8)). The peridinin-absorption spectrum in polar solvents, as well as in the PCP protein environment, is rather unstructured (Fig. 1). In particular, the presence of a carbonyl group was suggested to result in a broadening of the absorption bands and a pronounced solvent-dependence of the S1-state lifetime (9,10). The latter behavior has been attributed to the formation of an intramolecular charge-transfer state (ICT), which mixes with the S1 state (11,12).
character (7) is assumed to lie above S2 in peridinin (8)). The peridinin-absorption spectrum in polar solvents, as well as in the PCP protein environment, is rather unstructured (Fig. 1). In particular, the presence of a carbonyl group was suggested to result in a broadening of the absorption bands and a pronounced solvent-dependence of the S1-state lifetime (9,10). The latter behavior has been attributed to the formation of an intramolecular charge-transfer state (ICT), which mixes with the S1 state (11,12).
FIGURE 1.

Absorption spectra of the peridinin-chlorophyll a-protein (PCP) and Chl a (in diethyl ether; shaded, red-shifted by 6 nm to match Chl a-absorption in PCP).
Peridinin is generally assumed not to contribute to PCP absorption in the Chl a Qy region (Fig. 1). However, the energetic location as well as the dipole strength of the S0 → S1 transition of peridinin—in particular, in the proteinaceous environment in PCP—is a matter of controversy (e.g., (6,8,10,13–15)). For a recent critical review of the issue of optically dark states of carotenoids and a thorough discussion of the various experimental approaches to detect these states, see Polivka and Sundström (16).
Amino-acid sequences of the C- and N-terminal regions of the PCP apoprotein are only ∼56% identical (1). Hence, the site energies of the attached chromophores (peridinins as well as Chls a) may differ in the C- and N-terminal clusters. The site energies of the four peridinins within one cluster are also expected to vary as a consequence of their different attachment sites in the protein matrix. In the Qy-absorption region of PCP from A. carterae, two subbands (related to the two Chls of one monomer) have been partly resolved at cryogenic temperatures (5,17). Moreover, two different C=O stretching modes (1697 and 1681 cm−1) were observed by fluorescence line-narrowing spectroscopy, suggesting that there are two different emitting Chls a (5). However, Kleima et al. (18) as well as Ilagan et al. (17) state that the two Chls are spectrally indistinguishable at room temperature (RT). A Gaussian deconvolution of the RT fluorescence emission spectrum of PCP from Symbiodinium microadriaticum revealed two well-separated emission bands centered at 675.5 and 679 nm (19,20). An assignment of the spectroscopically different Chls a to the C- or N-terminal clusters has not been achieved, and a comprehensive spectral subband analysis of the PCP Soret/S2 region absorption reflecting all known structural information is not yet possible. An attempt of fitting circular dichroism (CD) and absorption spectra involving all possible inter- and intracluster pigment combinations resulted in only a moderately successful match between experimental and calculated spectra (3). However, four peridinin subbands were identified and excitonic coupling between peridinins was estimated to be of the order of 100–300 cm−1 (3). By combining information from absorption, linear dichroism, CD, and triplet-minus-singlet spectra of PCP with the absorption spectrum of peridinin in solution, four peridinin subbands were also resolved (5) with similar wavelength maxima to those reported in Carbonera et al. (3). So far, none of the above-mentioned peridinin subbands have been assigned to either the N- or C-terminal cluster or to the corresponding Chl a.
In addition, modes and routes of excitation energy transfer (EET) from peridinin to Chl a after excitation via the allowed S0 → S2 transition are not yet firmly established, and the proportion of peridinin → Chl EET that proceeds via the optically dark state(s) is a matter of debate. Several studies (12,13,21) suggest that excitation energy is transferred directly (with an efficiency of 25–50%) from the peridinin S2 state to the Chl a Qx state in addition to a route via the S1 and/or ICT state(s). In contrast, different workers (4,6,22) favor the notion that EET proceeds exclusively via the S1 state after rapid internal conversion from S2.
Fluorescence excitation spectra indicate a high efficiency of EET from peridinin to Chl a in PCP (4,17,23). Wavelength-independence of fluorescence excitation efficiency in the peridinin-absorption region was taken as being consistent with fast EET between the peridinins. This observation would also be in line with strong excitonic coupling among peridinins as suggested by CD spectra (2,24) and structure-based calculations (6). In contrast, Akimoto et al. (22) found a significant wavelength-dependence of the fluorescence excitation efficiency for wavelengths shorter than 520 nm in PCP from Alexandrium cohorticula. Moreover, from unaltered S2 lifetimes in PCP (as compared to peridinin in solution), these authors concluded that EET between peridinin S2 states in PCP is unlikely. However, the fluorescence excitation spectrum alone does not provide information about the EET routes and their relative proportions in PCP. To investigate these issues in more detail, it is necessary to elucidate the photophysical properties of each individual peridinin, i.e., site energy (subband), excited-state lifetime, and the mode of coupling between the pigments. Indeed, spectral substructure models for the peridinin-absorption band have been proposed (3,6,8). Damjanovic et al. (6) have suggested that one peridinin per cluster (per612 or per622, in their notation) does not undergo direct EET to Chl a, but can only transfer energy to a neighboring peridinin at the S2 level. Overall peridinin to Chl EET rates have been derived from femtosecond-transient absorption and fluorescence measurements without being able to differentiate between individual peridinin species (4,12,13,22).
Apparently, the above issues are either not or only partially addressable by conventional spectroscopic approaches. Thus, nonlinear polarization spectroscopy in the frequency domain (NLPF; see Fig. 2) was applied to study pigment-pigment interactions in PCP at RT. The aims were: 1), to determine the mode of coupling between peridinins in the S2 state; 2), to resolve the spectral substructure of the peridinin S2 band and Chl a Qy-absorption regions, as well as to differentiate the routes of EET from peridinin to Chl a; and 3), to elucidate interaction between the peridinin S1 state and Chl a, as well as the energetic location of the former.
FIGURE 2.

Principle of nonlinear polarization spectroscopy in the frequency domain (NLPF). Pump, probe, and signal fields (E) are distinguished by the indices p, t, and s, respectively, as well as the corresponding wave number vectors (k). The NLPF signal is the component of the signal field perpendicular to the incident probe field.
MATERIALS AND METHODS
Sample preparation
PCP was isolated from A. carterae as described (1) and diluted before the experiments in 25 mM Tris-HCl/10 mM KCl, pH 7.5. The sample (OD ≅ 1.1 at 670 nm) was contained in a 5-mm quartz cell (Hellma, Müllheim, Germany), which does not show detectable birefringence.
Experimental setup
NLPF spectra were recorded using a 90° setup as described previously (25). Pump (λp) and probe (λt) beams, characterized by a spectral linewidth of ∼0.05 cm−1, are obtained from dye lasers (DCM in DMSO, or Coumarin 47/307 in methanol, for the Chl Qy and Soret/allowed peridinin-absorption regions, respectively), synchronously pumped by an excimer laser with a pulse duration of 15 ns. The absolute wavelengths, as well as spectral linewidths of the pump and probe laser beams, were assessed with picometer precision using a Wavemeter (Model #450A, Burleigh Instruments, Victor, NY). NLPF spectra were obtained by tuning the pump laser across the spectral region under investigation with an increment of 0.2 nm. The χ3 approach for description of the NLPF spectra is justified for pump-beam intensities of ∼0.7 − 4 × 1022 photons cm−2 s−1. Intensity fluctuations of the pump and probe lasers were on the order of 5%. To obtain a better S/N ratio, 10 repetitive scans were averaged. To compare NLPF signal amplitudes it was essential to keep intensities of the pump and probe fields constant over the entire tuning range. The NLPF signal amplitude was corrected for the dependence on the optical density of the sample at the respective λt. To measure strong-field NLPF spectra, pump-beam intensities were varied by a neutral glass filter set from 1.5 × 1022 to 4.5 × 1024 photons cm−2 s−1. Absorption, CD, and fluorescence spectra were measured before and after the NLPF experiments to monitor sample integrity. All measurements were done at RT.
Information contents of NLPF spectra
The absorption of each individual pigment in a pigment-protein complex is shaped by interaction with its environment. This means that resolution of the spectral substructure of the overall absorption band in connection with the spectroscopic characterization of individual pigments reveals information about pigment-pigment as well as pigment-protein interactions. It has been shown previously that NLPF is a powerful method to elucidate spectral substructure and pigment-pigment interactions (26–28). NLPF is a degenerate four-wave mixing technique. A detailed description of the theoretical background of NLPF spectroscopy, together with a thorough discussion of the information content in NLPF lineshapes, is given in Beenken and Ehlert (29). Briefly, the NLPF-signal results from nonisotropic bleaching of an isotropic sample by a polarized pump laser field (see Fig. 2). The bleaching is probed by a second laser field, whose polarization plane is oriented at 45° with respect to the pump field. The NLPF signal is measured in the propagation direction of the probe beam, but in the polarization plane perpendicular to that of the probe field and has the same wavelength. Conventionally, a NLPF spectrum is measured at a fixed probe wavelength λt = 2πc/ωt by tuning the pump wavelength λp = 2πc/ωp across the absorption band under investigation. Two measuring regimes (with regard to pump intensity) providing different information can be distinguished.
Low pump intensity (χ(3) approach)
In the following, the characteristics of a NLPF spectrum of a homogeneously broadened optical transition (corresponding to an isolated two-level system) will be discussed exemplarily. The theoretical lineshape S(ωp) of the NLPF spectrum measured at λt is given in the limits of a χ(3) approach by the square module of the lineshape function s(ωp) of the nonlinear response to pump and probe frequency (29),
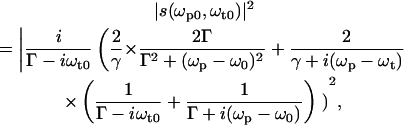 |
(1) |
where Γ = 1/T2 is the dephasing rate and γ = 1/T1 is the energy relaxation rate.
The center frequency of the absorption profile is represented by the transition frequency ω0 = 2πc/λ0 of the underlying two-level system. The spectral displacement between pump and probe beam as well as the center frequency are represented by the terms ωp0 = ωp − ω0 and ωt0 = ωt − ω0. The absolute signal amplitude scales with a factor ∝ In general, the NLPF-spectrum exhibits, besides a narrow T1-peak, a spectrally broad T2-peak. The T1-peak is observed exclusively in the vicinity of ωp ≈ ωt, and its spectral width is determined by the energy relaxation rate, γ. At room temperature, γ is much smaller than the dephasing rate Γ (Γ ≫ γ), which determines the width of the T2-peak. Therefore, the T2-peak for ωp − ωt ≫ γ can be approximated by the first term of the lineshape function and results in a broad spectrum centered at λ0 with a lineshape corresponding to the square module of the absorption profile. The amplitude of the NLPF signal scales with the square module of the pump intensity and excited-state lifetime T1 = 1/γ and, hence, correlates with ground-state depletion. Absorption bands of identical pigments at energetically nonequivalent binding sites are superimposed, resulting in a heterogeneous substructure of the overall absorption band. This situation can be described by an ensemble of (ν) two-level systems, with different transition frequencies ων, dephasing, and energy relaxation rates. Such a heterogeneous substructure results in a lineshape function, which is the sum of the individual lineshape functions weighted by the strength of the transition dipole moment
In general, the NLPF-spectrum exhibits, besides a narrow T1-peak, a spectrally broad T2-peak. The T1-peak is observed exclusively in the vicinity of ωp ≈ ωt, and its spectral width is determined by the energy relaxation rate, γ. At room temperature, γ is much smaller than the dephasing rate Γ (Γ ≫ γ), which determines the width of the T2-peak. Therefore, the T2-peak for ωp − ωt ≫ γ can be approximated by the first term of the lineshape function and results in a broad spectrum centered at λ0 with a lineshape corresponding to the square module of the absorption profile. The amplitude of the NLPF signal scales with the square module of the pump intensity and excited-state lifetime T1 = 1/γ and, hence, correlates with ground-state depletion. Absorption bands of identical pigments at energetically nonequivalent binding sites are superimposed, resulting in a heterogeneous substructure of the overall absorption band. This situation can be described by an ensemble of (ν) two-level systems, with different transition frequencies ων, dephasing, and energy relaxation rates. Such a heterogeneous substructure results in a lineshape function, which is the sum of the individual lineshape functions weighted by the strength of the transition dipole moment 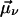 and the concentration nmol,v of individual pigments combined into a weighting factor αν:
and the concentration nmol,v of individual pigments combined into a weighting factor αν:
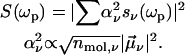 |
(2) |
Thus, the lineshape of the overall NLPF-spectrum does not necessarily conform to the square module of the absorption profile, and may contain a series of more or less distinguishable T2-peaks. By virtue of this, an underlying substructure may become discernible with a resolution exceeding that of conventional spectroscopy (29). Moreover, besides a possible subband analysis, the lineshape of the NLPF spectrum may reveal the mode of pigment-pigment interaction.
Two basic cases of pigment-pigment interaction can be distinguished readily in NLPF spectra (27–29):
Weak dipole-dipole interaction gives rise to incoherent EET. The subbands represent independent species that are connected via Förster-type EET. Since the probe field inspects ground-state depletion, the lineshape of the NLPF spectrum is essentially determined by the excited-state lifetimes (29). In the case of weak coupling, excitation would not be shared instantaneously between pigments; excitation occurs according to the spectral overlap of each individual subband with the pump field. The NLPF signal is enhanced for the subband(s) in which λt is located. Pigments with longer excited-state lifetimes contribute decisively to the overall NLPF signal. Hence, the NLPF lineshape is λt-dependent and structured NLPF spectra will be expected. Probing in a higher-energy subband (relative to the pump field) results in almost the same NLPF lineshape as in the case of a heterogeneous substructure. Probing in a lower-energy subband, the NLPF spectra are distinct; the peak in the spectral region of the higher-energy subband grows with increasing EET rate.
Strong dipole-dipole interaction leads to excitonic coupling which may result in splitting of the transition energies, redistribution of dipole strengths and delocalization of excitation energy. Strongly coupled pigments share a common ground state: a homogeneous substructure emerges. The energy-level scheme is characterized by a common ground state of the coupled pigments. The excitonic states are connected by the exciton downscattering rate. Peaks of nonprobed subbands are considerably more pronounced in NLPF spectra in the case of strong coupling. This may lead—in particular, for multiple coupled pigments—to a uniform/unstructured NLPF lineshape. Additionally, even if there is no spectral overlap between the subbands—the signal corresponding to the nonprobed subband does not disappear. This effect originates from the common ground state of the interacting species.
High pump intensity
The χ(3) approach to describe the NLPF spectrum is not applicable at strong pump intensities (30). Above a certain pump intensity, the amplitude of the NLPF signal no longer follows the square module of the pump intensity, and saturation effects influence the NLPF spectrum. Moreover, the lineshape of the NLPF spectrum broadens, whereas center wavelengths remain unchanged. The onset of these effects is determined by strength of the transition dipole moment 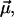 energy relaxation, and dephasing rates. Consequently, the NLPF spectrum differs for subbands representing pigments at nonequivalent binding sites. Thus, a subband with a small transition dipole moment, which would not be detectable in NLPF spectra at low pump intensities, may become obvious at higher pump intensities.
energy relaxation, and dephasing rates. Consequently, the NLPF spectrum differs for subbands representing pigments at nonequivalent binding sites. Thus, a subband with a small transition dipole moment, which would not be detectable in NLPF spectra at low pump intensities, may become obvious at higher pump intensities.
Summarizing, NLPF offers experimental approaches to elucidate spectral substructure and the mode of pigment-pigment coupling in pigment-protein complexes: 1), by measuring NLPF spectra at low intensities (pumping and probing in the same and/or different spectral regions (27,28)); and 2), by measuring NLPF spectra at high pump intensities, NLPF may also reveal subbands with very low transition dipole moment, e.g., the S1 state of peridinin.
RESULTS AND DISCUSSION
Mode of coupling between peridinins in the S2 state
To establish the prevailing mode of coupling in the peridinin S2-absorption band, NLPF spectra were measured by pumping and probing in the respective region.
The RT-absorption spectrum of PCP is shown in Fig. 1. The Chl-absorption regions are indicated (shaded). Peridinin has a broad (S0 → S2) absorption band between ∼350 and 560 nm. The Chl a Soret band absorbs in the same region at wavelengths shorter than 470 nm. The Chl a Qy-absorption band peaks at 670 nm. It is generally assumed that peridinin does not absorb in the vicinity of the Chl a Qy band. The overall absorption of the eight peridinins in the PCP monomer is rather featureless. There is considerable spectral overlap of the constituent subbands as well as no significant difference between the spectral widths of the peridinins (8).
NLPF spectra of PCP pumped exclusively in the peridinin S0 → S2-absorption region (between 480 and 545 nm) were measured by probing either in the peridinin-alone region or in the region where Chl a also absorbs (schematically shown in Fig. 3 A). These spectra are displayed in Fig. 3, B and C. Two main features are immediately obvious:
NLPF spectra for λt in the range 500–530 nm (where exclusively peridinin absorbs) are uniform, and reveal no substructure (Fig. 3 B). The spectra in Fig. 3 B show typical characteristics of excitonically coupled pigments. Strong excitonic coupling creates a common ground state. Excitation is shared instantaneously among the pigments and in the case of spectrally strongly overlapping subbands, the resulting NLPF spectrum should have a smooth, more-or-less uniform lineshape—independent of λt. Apparently, this is the case for all four peridinins within one cluster in PCP.
NLPF spectra for λt in the range 455–470 nm, where both peridinin and Chl a absorb, become nonuniform and show distinct structures characterized by a shoulder at ∼510 nm and rise steeply at wavelengths shorter than 490 nm (Fig. 3 C). These features represent a NLPF signal due to additionally probing Chl. Since peridinins, exclusively, are pumped in this experiment, these signals originate from depletion of the ground states of the two Chls due to EET from peridinin. As mentioned above, the fluorescence-excitation spectrum of PCP shows no wavelength dependence in this pump region, indicating equal EET efficiency from all peridinins to Chl. On the other hand, the additional Chl-derived NLPF signal is substructured, suggesting that the two acceptor Chls must have different excited-state lifetimes.
FIGURE 3.

NLPF experiment to elucidate the mode of coupling between peridinins in the S2 states. (A) Schematic: PCP is pumped in the peridinin S2-absorption region (485–540 nm) and probed in the peridinin S2/Chl Soret region. The shaded arrow symbolizes overall EET from peridinin to Chl a. (B) NLPF spectra obtained for probe wavelengths in the “exclusive peridinin S2-absorption” region. Solid lines represent smoothed data. (C) NLPF spectra obtained for probe wavelengths in the peridinin S2/Chl Soret region.
Apparently, the enhanced Chl-related NLPF signal observed upon pumping between 490 and 520 nm is due to peridinins transferring their energy preferentially to the Chl a with the longer S1 lifetime. These observations show that the peridinins in the two clusters are spectrally distinguishable; and that the two Chls a are not photophysically identical. However, the question of whether peridinin-to-Chl EET proceeds directly from S2 to Qx, or via the dark state(s) of peridinin to Qy, cannot be decided from the above experiment.
Peridinin spectral substructure and EET to Chls
A more straightforward way to assign peridinin subbands to the two Chls in the PCP monomer is to monitor Chl a ground-state depletion in a spectral region with no peridinin absorption after initial peridinin excitation. This was accomplished by tuning λp across the peridinin-absorption region (460–540 nm) and positioning λt in the Qy-absorption region of Chl a (645–680 nm) (Fig. 4 A). In the case of identical peridinin subbands in both clusters, identical NLPF lineshapes, resembling the squared peridinin-absorption profile, independent of λt, would be expected—provided that EET from all peridinins to Chl is uniform. On the other hand, in the case of nonidentical peridinin spectral forms in both clusters and different Chl S1 lifetimes, peridinins transferring energy to the longer-lived Chl are expected to be enhanced in the NLPF spectrum, and vice versa.
FIGURE 4.

NLPF experiment for subband analysis of the peridinin S2-absorption band in PCP. (A) Schematic: PCP was pumped in the peridinin S2-absorption region and probed in the Chl a Qy region. The shaded arrow symbolizes overall EET from peridinin to Chl a. (B) NLPF spectra for probe wavelengths between 645 and 685 nm (and pump wavelengths in the 450–540 nm region). Note that the spectra are composed of two data sets due to tuning-range limitations of the pump lasers. Solid lines represent smoothed data. Individual NLPF spectra are displaced by a constant, offset for better representation.
NLPF spectra obtained for λt from the blue (645 nm) to the red (685 nm) edge of the Chl a Qy band clearly do not reproduce the squared peridinin-absorption profile—but, instead, display a significant substructure (Fig. 4 B). Nevertheless, NLPF signals obtained for λp > 470 nm are exclusively due to peridinin excitation.
Although the NLPF signal is enhanced for λp > 480 nm, it is diminished—but clearly distinct from zero at shorter pump wavelengths (see also Fig. 5 B). Three peaks at ∼490, 500, and 540 nm are apparent in the former region—irrespective of λt (Fig. 4 B). There is also a local minimum at ∼515 nm, being most pronounced for λt being located at the blue edge of the Qy band. This minimum is gradually leveled out for λt > 670 nm. The latter observation indicates the existence of two subbands centered at ∼515 nm being associated with the two different Chls. The 515-nm peridinin feature associated with the longer-lived (red-shifted) Chl is increased with shifting λt away from the blue (short-lived) Chl, gradually filling up the initial gap. The steep rise of the NLPF signal at the short-wavelength edge of the pump region can be assigned to direct pumping of the Chl Bx absorption (being negligible at λ > 470 nm).
FIGURE 5.

NLPF spectrum probed at 645 nm. (A) Model underlying the fit. See text for further details. (B) Fit of the NLPF spectrum. (C) Fit of the PCP-absorption spectrum in the NLPF pump region. Open circles represent the measured data, solid lines represent the best fits; the resolved absorption subbands are additionally shown in panel C.
To extract quantitative spectral subband information, a combined analysis of NLPF spectrum at λt = 645 nm and the absorption spectrum (between 450 and 560 nm) was performed. The analysis is based on the model shown in Fig. 5 A. Four excitonically coupled peridinins are associated with each Chl. The two Chls have different S1 lifetimes (and different site energies, corresponding to ∼5 nm; see below). EET between the Chls is allowed in both directions. Inhomogeneous broadening of the peridinin subbands was not taken into account. We do not differentiate between the two possible EET pathways (via peridinin S2 and Chl Qx or the dark S1 and Qy) finally populating Chl Qy, but instead combine them in one rate (assumed to be equal for all peridinins). The latter assumption is based on the fact that fluorescence excitation efficiency is uniform over the entire peridinin-absorption range.
No lifetime values for the two Chl a subbands can be deduced from these data because these NLPF spectra represent only the broad-band term of the NLPF lineshape formula as shown in Eq. 1. A quantitative determination of the exact ratio would be only amenable by T1 peak (high-resolution) NLPF measurements (29). Nevertheless, lifetime ratios of different components are reflected in broad-band NLPF spectra of multipigment systems; in this particular experiment, subbands transferring energy to the longer-lived Chl-species would appear enhanced in the NLPF spectra (29).
Fits based on the assumption of equal S1 lifetimes for the Chls consistently failed. The model used to evaluate the lineshapes of the NLPF spectra in Figs. 4 and 5 implies that the two Chls a have different S1 lifetimes, in accordance with the other experimental observations (in particular, the subband analyses of the Chl Qy band; see below). Our data indicate a signal ratio of ∼2–3 upon probing in the blue edge of the Chl a Qy band, which is consistent with a lifetime ratio for the Chls of ∼1.5 (as exemplarily used in the fit in Fig. 5, B and C).
The decline of the NLPF signal at λp 470 nm can have two different causes. According to earlier observations of Song et al. (24), the peridinin S2-absorption band of native PCP from Amphidinium rhyncocephaleum splits into two components, a blue- and a red-shifted part. The more blue-shifted peridinins transfer their excitation energy apparently to the shorter-lived Chl whereas the red-shifted peridinins belong to the cluster serving the longer-lived Chl. Higher vibrational bands of the red-shifted peridinins tail into the spectral region <480 nm. Vibrational bands generally give considerably lower contributions to the overall NLPF spectra. Superposition of the contributions from the blue-shifted peridinins and the vibrational bands leads to loss of NLPF substructure in the spectral region <480 nm.
The fit of the NLPF spectrum is shown as a solid line in Fig. 5 B. Three peridinin subbands (centered at ∼535, 501, and 487 nm) can be assigned to the cluster of the longer-lived Chl. A subband centered at ∼515 nm is associated with the shorter-lived Chl a. The λt-dependence of the NLPF spectra also reveals that the cluster associated with the longer-lived Chl must contain a peridinin species peaking at ∼515 nm (see Fig. 4 B).
The peridinin subbands obtained by the fit are indicated with respect to the PCP-absorption spectrum in Fig. 5 C. The subbands partially correspond to those previously reported: Carbonera et al. (3) identified peridinin subbands at 543, 534, 518, and 485 nm and assigned them to per2, per3, per4 and per1, respectively, in the PCP structure. The peridinins were not, however, assigned to the clusters, since the authors assumed that site energies of Chls and peridinins at homologous positions in both clusters are identical. A 543-nm subband was not observed in our study due to measuring-range limitations. A pronounced subband at 501 nm—missing in the analysis in Carbonera et al. (3), as evident in the deviation between their fitted and measured CD spectra in the 500-nm region—was identified in this work. Kleima et al. (5) identified at least four peridinin subbands centered at 520, 529 (or 537), 546, and 555 nm, but found no evidence for a band at 487 nm.
A peridinin response upon Chl Qy excitation
It has been shown repeatedly that peridinin undergoes very efficient (∼90%) EET to Chl a after excitation of the S2 state (e.g., (17)). To date, however, it is still a matter of debate whether EET proceeds directly from S2 of peridinin to Chl Qx, via the dark state(s) of peridinin to Qy of Chl, or to what extent via both channels. Moreover, the mode of interaction (Coulombic versus Dexter mechanism) is not yet established. Furthermore, the possibility of “mixing” between the Chl S1 state and low-lying states of peridinin has been raised (31).
Interactions between Qy of Chls and peridinin dark states should be apparent in a ground-state depletion of the respective peridinin after excitation of the Chls. By pumping in the Chl a Qy region and probing at different wavelengths in the peridinin-alone region (shown schematically in Fig. 6 A), such interaction should generate a NLPF signal corresponding to the S2 energies of interacting peridinins. A comparable approach has been successfully applied by the authors recently to provide evidence for excitonically coupled Chls a and b in the antenna complexes CP29 (27) and LHC II (28).
FIGURE 6.

NLPF experiment to elucidate interactions between Chl a and peridinin S1 states. (A) Schematic: PCP was pumped in the Chl a Qy region and probed in the Chl a-Soret/peridinin S2-absorption region. The shaded arrow symbolizes EET from Chl a to peridinin. (B) Quasi-three-dimensional representation of the (smoothed) NLPF spectra. (C) Intensity courses (along the probe wavelength range) of the NLPF signals pumped at ∼670 nm (solid) and 650 nm (shaded).
Fig. 6 B shows a quasi-three-dimensional plot of the NLPF spectra obtained by pumping between 650 and 685 nm (in the Chl a Qy region) and probing between 450 and 530 nm (Chl a Soret/peridinin S2-absorption region). The NLPF signal for λt in the Chl Soret/peridinin-absorption region would be expected to decrease in parallel to the Chl a Bx absorption, declining to baseline at wavelengths >470 nm. However, an additional NLPF signal is observed for λt around 490 nm and can be attributed to a depletion of the peridinin ground state due to Chl a-peridinin interaction. Under our experimental conditions, intersystem crossing into the Chl triplet-manifold after Chl Qy excitation would be possible, in principle. Triplet EET from Chl a to peridinin was shown to be very effective in PCP with a peridinin triplet-absorption rise-time of 17 ± 7 ns (4,5). Rapid equilibration of triplet excited states among the peridinins of one cluster was demonstrated for PCP by ODMR (2). However, if a peridinin triplet population were the origin of the NLPF signal, it should be observable in the entire λt-range. This is clearly not the case; in particular, no NLPF signal is detectable for λt > 510 nm (see also Fig. 6, B and C).
Lineshapes of the NLPF spectra probed in the immediate vicinity of 490 nm (in contrast to all other spectra) resemble the squared Chl a Qy absorption. We assign these NLPF signals to the 487-nm peridinin species associated with the longer-lived Chl identified by the NLPF measurements described in “Peridinin spectral substructure and EET to Chls”, above. The mechanism of interaction between the excited Chl and peridinin cannot be readily deduced on the basis of the current experiment. However, it has been suggested from structure-based calculations that an electron exchange (Dexter-type) mechanism is far less effective than the Coulomb mechanism in PCP (6,32). In addition, breaking of the C2h symmetry by substitution of the polyene chain in peridinin is expected to result in a significant dipole moment of the S0 → S1 transition (amounting to ∼5% of that of S0 → S2) (33). The latter prediction is supported by the observation of weak fluorescence from S1 of peridinin in solution (11,34). Moreover, distortion of the linear structure of the peridinins upon binding to the apoprotein may further contribute to C2h-symmetry breaking (1,8).
Assuming that a Coulombic mechanism was operative, a NLPF signal is expected in the spectral region where donor fluorescence and acceptor-absorption spectra overlap. Thus, viewing along the λp axis in Fig. 6 B (the lineshape of the observed NLPF signals will indicate the S1 energies of the(se) peridinin(s); see also Fig. 6 C), the maximum of the NLPF signal will be red-shifted or blue-shifted—if peridinin S1 were lower or higher energetic, respectively, than S1 of Chl a. Since the NLPF spectra probed at ∼490 nm have maxima corresponding to the Chl a Qy absorption, we suggest that the dark state is approximately isoenergetic to or slightly above S1,0 of Chl a.
In addition to the features indicating interaction between S1 of Chl a and a dark state of peridinin, another NLPF signal (peaking at ∼500 nm) is observed upon pumping at 650 nm (Fig. 6 C). Fig. 6 C compares the two cross-sections (for λp = 650 and 670 nm) along the λt range. The steep slope at the blue edge of the curve obtained for λp = 670 nm suggests an assignment to a NLPF signal due to pumping and probing Chl. Upon shifting λp to 650 nm, this feature is clearly lacking, indicating that Chl is not pumped. Obviously, a low dipole moment transition of peridinin rather than Chl is directly pumped at ∼650 nm.
Such a peridinin S1/ICT energy would be lower than any value reported so far. In a recent article, Shima et al. (8) locate the S1,0 energy for peridinin dissolved in CS2 at 16,200 cm−1 (∼617 nm) in accordance with previous studies (11,34). Zimmermann et al. (15) deduced from two-photon fluorescence excitation experiments that S1 of peridinin in benzene as well as in PCP is located even more to the blue (peaking at ∼530 nm), mixing with the 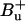 state. Their assignment is apparently in contradiction to one-photon fluorescence studies ((11,34); see also (8)). Shima et al. (8) conclude that the two-photon excitation spectra in Zimmermann et al. (15), as well as their own, are essentially due to the S2 = 1Bu+ state carrying a significant two-photon character in peridinin. Furthermore, it was concluded that the S0 → S2 transitions of peridinins Per612 and Per622 are considerably blue-shifted as compared to the other peridinins and characterized by a 2Ag− state lying above S2 (8). The latter prediction is hard to reconcile with the experimental results presented here. Apparently, at least one of the blue-shifted peridinin species, peaking at ∼495 nm, shows unique interaction with the S1 state of Chl a mediated via the carotenoid's low-lying S1/ICT state.
state. Their assignment is apparently in contradiction to one-photon fluorescence studies ((11,34); see also (8)). Shima et al. (8) conclude that the two-photon excitation spectra in Zimmermann et al. (15), as well as their own, are essentially due to the S2 = 1Bu+ state carrying a significant two-photon character in peridinin. Furthermore, it was concluded that the S0 → S2 transitions of peridinins Per612 and Per622 are considerably blue-shifted as compared to the other peridinins and characterized by a 2Ag− state lying above S2 (8). The latter prediction is hard to reconcile with the experimental results presented here. Apparently, at least one of the blue-shifted peridinin species, peaking at ∼495 nm, shows unique interaction with the S1 state of Chl a mediated via the carotenoid's low-lying S1/ICT state.
Spectral substructure of the Chl a Qy absorption
As mentioned above, the distance between the two Chls a in a PCP monomer is fairly large and interaction between the Chls appears to be weak. Consequently, Chl site energies are determined essentially by pigment-protein interactions. The Chl a Qy-absorption region was investigated by positioning the probe beam at different λt (640–680 nm) and tuning λp at low intensity across the same region (exemplary spectra are shown in Fig. 7). Two main features are observable in these spectra:
The lineshape of the measured NLPF spectra does not correspond to the square module of the Chl a Qy-absorption profile (exemplarily shown for λt = 670 nm, Fig. 7 C).
Lineshapes of the NLPF spectra for λt < 665 nm (Fig. 7 D) deviate from those for λt between 665 and 680 nm. An additional signal component is observed at the blue edge (exemplarily shown in Fig. 7 D).
FIGURE 7.

Spectral substructure analysis of the Chl a Qy-absorption region. Exemplary spectra are shown: (A) NLPF spectrum obtained at λt = 680 nm. (B) NLPF spectrum obtained at λt = 660 nm. (C) The NLPF spectrum obtained at λt = 670 nm is compared to the squared absorption spectrum (dashed line). (D) NLPF spectrum obtained at λt = 650 nm, comparison of two and three subband fits. Solid lines indicate fits assuming two subbands as a result of a global analysis of the spectra obtained for λt > 665 nm, including the absorption spectrum. The shaded line represents a fit with an additional (third) subband.
The observation that the NLPF spectra do not correspond to the squared absorption profile indicates spectral heterogeneity of the Qy band of PCP (meaning that two subspecies exist). However, these subbands are not immediately obvious.
To further characterize the underlying substructure, a global line-shape analysis comprising a fit of the set of NLPF spectra (obtained at different λt) and the absorption spectrum was performed. The procedure is described in detail in Voigt et al. (26).
Fits with only one Chl a Qy band yielded an χ2 = 4.46 and consistently failed. However, a model with two Chl a-subbands results in an χ2 = 2.5 and was sufficient to describe all NLPF spectra for λt in the range from 665 to 680 nm and the Qy-absorption spectrum of PCP simultaneously. An F-test revealed a probability ≫99.9% that the two-subband approach represents an improved model as compared to that of a single band. The best global fit was obtained with center wavelengths of 669 and 675 nm and widths of 320 and 200 cm−1 (full-width at half-maximum) for the two Chl a Qy subbands, respectively. The shorter-wavelength band will consequently have a shorter effective excited-state lifetime. The latter requirement is in line with the results of NLPF experiments to elucidate EET from peridinin to Chl a (obtained by pumping in the allowed peridinin band and probing in the Chl a Qy band; compare to Fig. 4 B).
When attempting to fit the NLPF spectra obtained at λt = 650 and 655 nm, a third subband had to be taken into account. The fit, however, did not identify the peak wavelength of the additional band as precisely as the other two. Nevertheless, the third band must have a center wavelength of ∼660 nm, but cannot be interpreted as a further Chl-Qy-absorption subband, since only two (noninteracting) Chls are found in a PCP monomer.
A distinct NLPF signal originating from peridinin ground-state depletion was observed upon pumping in the Chl Qy region and probing in a spectral region (>470 nm) where exclusively peridinin absorbs (see previous section). On the basis of the above results, it cannot be decided whether this peridinin ground-state depletion is a result of a direct excitation of a low dipole moment S0 → S1/ICT transition of certain peridinins and/or due to EET from a neighboring Chl.
In any case, the(se) peridinin(s) must have a non-negligible S0 → S1/ICT transition dipole moment and show considerable spectral overlap with the Chl a Qy band. An S1 transition dipole moment of up to five Debye (depending on parameterization) has been inferred previously from theoretical considerations of breaking of the C2h symmetry for peridinins in PCP (6). Moreover, Krueger et al. (13) have deduced a peridinin S1 transition dipole moment of approximately three Debye in PCP from transient absorption measurements. However, a dipole moment of that size should be detectable in absorption—which is in contradiction to experimental results with peridinin in solution and the interpretation of the PCP Qy-region absorption spectrum by previous workers (e.g., Kleima et al. (5)). Nevertheless, a low-dipole moment peridinin transition may become visible in NLPF spectra, measured at strong pump intensities exceeding the range of validity of the χ(3) approach for the dominant Chls (see also Beenken and May (30)).
Additional evidence for spectral heterogeneity of the Chl Qy region
Another approach to reveal spectral heterogeneity and (hidden) low dipole-moment species in complex spectra is provided by the pump-intensity dependence of the NLPF spectral profiles. In the case of Chl spectral heterogeneity (including different S1 lifetimes of the subbands), contributions of the (two) spectral subbands to the overall NLPF spectrum are expected to vary with pump intensity (30). With increasing pump intensity, the subband with the longest S1 lifetime (usually the lowest energetic band) becomes progressively saturated, and contributions from other, shorter-lived and/or low dipole-moment bands may become apparent (see also Schubert et al. (35)). Hence, a blue-shift of the overall NLPF spectrum, in addition to an increase in amplitude, would be expected. Both phenomena are observed with PCP; Fig. 8 A shows NLPF spectra of PCP obtained at pump intensities between 1.5 × 1022 and 4.5 × 1024 photons cm−2 s−1 (probed at λt = 660 nm). Note that the spectra obtained at lower pump intensities (traces a–c in Fig. 8 A) are magnified 250 times. The NLPF spectra display a clear shift toward shorter wavelengths with increasing pump intensity. The dependence of the NLPF signal amplitude on pump intensity is shown for λt = 660 and 680 nm (i.e., on the blue and red edges of the PCP Qy-absorption region, respectively) in Fig. 8 B. The NLPF signal obtained at λt = 660 nm displays later commencement of saturation effects (leading to deviation from the almost quadratic pump intensity dependence) than the signal at λt = 680 nm, indicating heterogeneity of the Qy-absorption region.
FIGURE 8.

Pump intensity dependence of NLPF spectra of PCP. (A) NLPF spectra probed at λt = 660 nm (smoothed). Actual data points are shown exemplarily in trace g (open circles, with typical SD). Spectra obtained at the three lowest pump intensities (a–c) are magnified 250-fold. Intensity variation was between 1.5 × 1022 (a) to 4.5 × 1024 (g) photons cm−2 s−1. (B) Intensity dependence of the NLPF signal amplitude pumped and probed at 680 nm (open symbols) and at 660 nm (solid symbols). Dotted line indicates strict quadratic intensity dependence. (C) Normalized NLPF spectra from A.
This blue-shift is even more obvious when the NLPF spectra are normalized (Fig. 8 C). The moderate shift (observed in the NLPF spectrum at intermediate intensity in trace d) is consistent with the existence of two subbands with different excited-state lifetimes and different (by a few nanometers) center-wavelengths. Traces f and g in Fig. 8 C display a more pronounced blue-shift (toward ∼660 nm). A further blue-shift at even higher pump intensities is not expected since saturation of the NLPF signal is observed in this pump intensity range (Fig. 8 B). The shift, however, cannot be explained on the basis of the two Chl a subbands at 669 and 675 nm but requires a third, low dipole moment band centered at ∼660 nm. The blue-shift is also observed upon probing at 680 nm, but at this λt it is, however, less pronounced (data not shown).
The latter observation would be in line with the different slopes and saturation behavior of the NLPF signals observed in the blue and red edges of the Qy band, and is consistent with the requirement of a third subband to fit the low pump-intensity NLPF spectra obtained at λt = 650 and 655 nm. The ∼660-nm band is tentatively assigned to the S1/ICT-state of peridinin(s) and most likely corresponds to the peridinin molecule with a blue-shifted S2 state (at 495 nm). Assignment of this subband to the previously resolved S1 → Sn transient absorption (10) can be excluded by a comparison of the respective maximum positions (∼590 vs. 660 nm).
Acknowledgments
We thank Frank Sharples for assistance in growing the algae and preparing PCP.
Financial support by the Deutsche Forschungsgemeinschaft (grant No. SFB 429, TP A2) and the Australian Research Council (grant No. A00000264 to R.G.H.) is gratefully acknowledged. M.K. was supported by postgraduate scholarships of the Deutscher Akademischer Austausch Dienst (DAAD) and the Stifterverband für die Deutsche Wissenschaft (Dr. Egon und Hildegard Diener-Stiftung).
Maria Krikunova's present address is Institut für Experimentalphysik, Universität Hamburg, Luruper Chaussee 149, D-22761 Hamburg, Germany.
Heiko Lokstein's present address is Institut für Biochemie und Biologie/Pflanzenphysiologie, Universität Potsdam, Karl-Liebknecht-Str. 24-25, Haus 20, D-14476 Golm, Germany.
Dieter Leupold's and Bernd Voigt's present address is Institut für Physik/Photonik, Universität Potsdam, Postfach 601553, 1-14415 Potsdam, Germany.
References
- 1.Hofmann, E., P. M. Wrench, F. P. Sharples, R. G. Hiller, W. Welte, and K. Diederichs. 1996. Structural basis of light harvesting by carotenoids: peridinin-chlorophyll-protein from Amphidinium carterae. Science. 272:1788–1791. [DOI] [PubMed] [Google Scholar]
- 2.Carbonera, D., G. Giacometti, and U. Segre. 1996. Carotenoid interactions in peridinin chlorophyll a proteins from dinoflagellates. Evidence for optical excitons and triplet migration. J. Chem. Soc. Faraday Trans. 92:989–993. [Google Scholar]
- 3.Carbonera, D., G. Giacometti, U. Segre, E. Hofmann, and R. G. Hiller. 1999. Structure-based calculations of the optical spectra of the light-harvesting peridinin-chlorophyll-protein complexes from Amphidinium carterae and Heterocapsa pygmaea. J. Phys. Chem. B. 103:6349–6356. [Google Scholar]
- 4.Bautista, J. A., R. G. Hiller, F. P. Sharples, D. Gosztola, M. Wasielewski, and H. A. Frank. 1999. Singlet and triplet energy transfer in the peridinin-chlorophyll a-protein from Amphidinium carterae. J. Phys. Chem. A. 103:2267–2273. [Google Scholar]
- 5.Kleima, F. J., M. Wendling, E. Hofmann, E. J. G. Peterman, R. van Grondelle, and H. van Amerongen. 2000. Peridinin chlorophyll a protein: relating structure and steady-state spectroscopy. Biochemistry. 39:5184–5195. [DOI] [PubMed] [Google Scholar]
- 6.Damjanovic, A., T. Ritz, and K. Schulten. 2000. Excitation transfer in the peridinin-chlorophyll-protein of Amphidinium carterae. Biophys. J. 79:1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
7.Sashima, T., Y. Koyama, T. Yamada, and H. Hashimoto. 1999. The
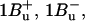 and
and 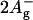 energies of crystalline lycopene, β-carotene, and mini-9-β-carotene as determined by resonance-Raman excitation profiles: dependence of
energies of crystalline lycopene, β-carotene, and mini-9-β-carotene as determined by resonance-Raman excitation profiles: dependence of 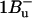 -state energy on the conjugation length. J. Phys. Chem. B. 104:5011–5019. [Google Scholar]
-state energy on the conjugation length. J. Phys. Chem. B. 104:5011–5019. [Google Scholar] - 8.Shima, S., R. P. Ilagan, N. Gillespie, B. J. Sommer, R. G. Hiller, F. P. Sharples, H. A. Frank, and R. R. Birge. 2003. Two-photon and fluorescence spectroscopy and the effect of environment on the photochemical properties of peridinin in solution and in the peridinin-chlorophyll-protein from Amphidinium carterae. J. Phys. Chem. A. 107:8052–8066. [Google Scholar]
- 9.Frank, H. A., J. A. Bautista, J. Josue, Z. Pendon, R. G. Hiller, F. P. Sharples, D. Gosztola, and M. R. Wasielewski. 2000. Effect of the solvent environment on the spectroscopic properties and dynamics of the lowest excited states of carotenoids. J. Phys. Chem. B. 104:4569–4577. [Google Scholar]
- 10.Zigmantas, D., R. G. Hiller, F. P. Sharples, H. A. Frank, V. Sundström, and T. Polivka. 2004. Effect of a conjugated carbonyl group on the photophysical properties of carotenoids. Phys. Chem. Chem. Phys. 6:3009–3016. [Google Scholar]
- 11.Bautista, J. A., R. E. Connors, B. B. Raju, R. G. Hiller, F. P. Sharples, D. Gosztola, M. R. Wasielewski, and H. Frank. 1999. Excited-state properties of peridinin: observation of solvent dependence of the lowest excited singlet-state lifetime and spectral behavior unique among carotenoids. J. Phys. Chem. B. 103:8751–8758. [Google Scholar]
- 12.Zigmantas, D., R. G. Hiller, V. Sundström, and T. Polivka. 2002. Carotenoid to chlorophyll energy transfer in the peridinin-chlorophyll-a-protein complex involves an intramolecular charge transfer state. Proc. Natl. Acad. Sci. USA. 99:16760–16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krueger, B. P., S. S. Lampoura, I. H. M. van Stokkum, E. Papagiannakis, J. M. Salverda, C. C. Gradinaru, D. Rutkauskas, R. G. Hiller, and R. van Grondelle. 2001. Energy transfer in the peridinin chlorophyll-a protein of Amphidinium carterae studied by polarized transient absorption and target analysis. Biophys. J. 80:2843–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zigmantas, D., T. Polivka, R. G. Hiller, A. Yartsev, and V. Sundström. 2001. Spectroscopic and dynamic properties of the peridinin lowest singlet excited states. J. Phys. Chem. A. 105:10296–10306. [Google Scholar]
- 15.Zimmermann, J., P. A. Linden, H. M. Vaswani, R. G. Hiller, and G. R. Fleming. 2002. Two-photon excitation study of peridinin in benzene and in the peridinin chlorophyll a-protein (PCP). J. Phys. Chem. B. 106:9418–9423. [Google Scholar]
- 16.Polivka, T., and V. Sundström. 2004. Ultrafast dynamics of carotenoid excited states—from solution to natural and artificial systems. Chem. Rev. 104:2021–2071. [DOI] [PubMed] [Google Scholar]
- 17.Ilagan, R. P., S. Shima, A. Melkozernov, S. Lin, R. E. Blankenship, F. P. Sharples, R. G. Hiller, R. R. Birge, and H. A. Frank. 2004. Spectroscopic properties of the main-form and high-salt peridinin-chlorophyll a proteins from Amphidinium carterae. Biochemistry. 43:1478–1487. [DOI] [PubMed] [Google Scholar]
- 18.Kleima, F. J., E. Hofmann, B. Gobets, I. H. M. van Stokkum, R. van Grondelle, K. Diederichs, and H. van Amerongen. 2000. Förster excitation energy transfer in peridinin-chlorophyll-a-protein. Biophys. J. 78:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iglesias-Prieto, R., N. S. Govind, and R. K. Trench. 1991. Apoprotein composition and spectroscopic characterization of the water-soluble peridinin-chlorophyll a-proteins from three symbiotic dinoflagellates. Proc. R. Soc. Lond. B Biol. Sci. 246:275–283. [Google Scholar]
- 20.Iglesias-Prieto, R., and R. K. Trench. 1996. Spectroscopic properties of chlorophyll a in the water-soluble peridinin-chlorophyll a-protein complexes (PCP) from the symbiotic dinoflagellate Symbiodinium microadriaticum. J. Plant Physiol. 149:510–516. [Google Scholar]
- 21.Linden, P. A., J. Zimmermann, T. Brixner, N. E. Holt, H. M. Vaswani, R. G. Hiller, and G. R. Fleming. 2004. Transient absorption study of peridinin and peridinin chlorophyll a-protein after two-photon excitation. J. Phys. Chem. B. 108:10340–10345. [Google Scholar]
- 22.Akimoto, S., S. Takaichi, T. Ogata, Y. Nishimura, I. Yamazaki, and M. Mimuro. 1996. Excitation energy transfer in carotenoid-chlorophyll protein complexes probed by femtosecond fluorescence decays. Chem. Phys. Lett. 260:147–152. [Google Scholar]
- 23.Koka, P., and P. Song. 1977. The chromophore topography and binding environment of peridinin-chlorophyll a-protein complexes from marine dinoflagellate algae. Biochim. Biophys. Acta. 495:220–231. [DOI] [PubMed] [Google Scholar]
- 24.Song, P., P. Koka, B. B. Prezelin, and F. T. Haxo. 1976. Molecular topology of the photosynthetic light-harvesting pigment complex, peridinin-chlorophyll a-protein, from marine dinoflagellates. Biochemistry. 15:4422–4427. [DOI] [PubMed] [Google Scholar]
- 25.Voigt, B., F. R. Nowak, and W. Beenken. 1999. A new set-up for nonlinear polarization spectroscopy in the frequency domain: experimental examples and theoretical background. Meas. Sci. Technol. 10:N7–N11. [Google Scholar]
- 26.Voigt, B., F. R. Nowak, J. Ehlert, W. Beenken, D. Leupold, and W. Sandner. 1997. Substructures and different energy relaxation time within the first electronic transition of pinacyanol. Chem. Phys. Lett. 278:380–390. [Google Scholar]
- 27.Voigt, B., K.-D. Irrgang, J. Ehlert, W. Beenken, G. Renger, D. Leupold, and H. Lokstein. 2002. Spectral substructure and excitonic interactions in the minor photosystem II antenna complex CP29 as revealed by nonlinear polarization spectroscopy in the frequency domain. Biochemistry. 41:3049–3056. [DOI] [PubMed] [Google Scholar]
- 28.Krikunova, M., B. Voigt, and H. Lokstein. 2002. Direct evidence for excitonically coupled chlorophylls a and b in LHC II of higher plants by non-linear polarization spectroscopy in the frequency domain. Biochim. Biophys. Acta. 1556:1–5. [DOI] [PubMed] [Google Scholar]
- 29.Beenken, W., and J. Ehlert. 1998. Subband analysis of molecular electronic transitions by nonlinear polarization spectroscopy in the frequency domain. J. Chem. Phys. 109:10126–10137. [Google Scholar]
- 30.Beenken, W., and V. May. 1997. Strong field theory of nonlinear polarization spectroscopy. Fundamentals and the two-level system. J. Opt. Soc. Am. B. 14:2804–2810. [Google Scholar]
- 31.Salverda, J. M. 2003. Interacting pigments in light-harvesting complexes studied with nonlinear spectroscopy. PhD thesis. Free University. Amsterdam, The Netherlands.
- 32.Nagae, H., T. Kakitani, A. Murakami, M. Watanabe, M. Erata, M. M. Watanabe, M. Tokutomi, and I. Yamazaki. 1993. Calculation of the excitation transfer matrix elements between the S2 or S1 state of carotenoid and the S2 or S1 state of bacteriochlorophyll. J. Chem. Phys. 98:8012–8023. [Google Scholar]
- 33.Ritz, T., A. Damjanovic, K. Schulten, J.-P. Zang, and Y. Koyama. 2000. Efficient light harvesting through carotenoids. Photosynth. Res. 66:125–144. [DOI] [PubMed] [Google Scholar]
- 34.Mimuro, M., U. Nagashima, S. Takaichi, Y. Nishimura, I. Yamazaki, and T. Katoh. 1992. Molecular structure and optical properties of carotenoid for the in vivo energy transfer function in the algal photosynthesis pigment system. Biochim. Biophys. Acta. 1098:271–274. [Google Scholar]
- 35.Schubert, A., W. Beenken, H. Stiel, B. Voigt, D. Leupold, and H. Lokstein. 2002. Excitonic coupling of chlorophylls in the plant light-harvesting complex LHC II. Biophys. J. 82:1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]