

Abstract
It is now well established that characteristic properties of excitation-contraction (EC) coupling in cardiac myocytes, such as high gain and graded Ca2+ release, arise from the interactions that occur between L-type Ca2+ channels (LCCs) and nearby ryanodine-sensitive Ca2+ release channels (RyRs) in localized microdomains. Descriptions of Ca2+-induced Ca2+ release (CICR) that account for these local mechanisms are lacking from many previous models of the cardiac action potential, and those that do include local control of CICR are able to reconstruct properties of EC coupling, but require computationally demanding stochastic simulations of ∼105 individual ion channels. In this study, we generalize a recently developed analytical approach for deriving simplified mechanistic models of CICR to formulate an integrative model of the canine cardiac myocyte which is computationally efficient. The resulting model faithfully reproduces experimentally measured properties of EC coupling and whole cell phenomena. The model is used to study the role of local redundancy in L-type Ca2+ channel gating and the role of dyad configuration on EC coupling. Simulations suggest that the characteristic steep rise in EC coupling gain observed at hyperpolarized potentials is a result of increased functional coupling between LCCs and RyRs. We also demonstrate mechanisms by which alterations in the early repolarization phase of the action potential, resulting from reduction of the transient outward potassium current, alters properties of EC coupling.
INTRODUCTION
Myocyte contraction occurs in response to a series of events that are initiated and coordinated by the cardiac action potential (AP). Initial membrane depolarization during the upstroke of the AP activates voltage-gated L-type Ca2+ channels (LCCs), which provide for Ca2+ influx across the sarcolemma. The influx of Ca2+ ions triggers the release of Ca2+ from the junctional sarcoplasmic reticulum (SR) in the process known as Ca2+-induced Ca2+ release (CICR). CICR leads to a 10-fold increase in cytosolic Ca2+ concentration, and hence, provides the necessary signal to initiate mechanical shortening of the cell's contractile machinery. The elucidation of the mechanisms that underlie the process of CICR has become possible in recent years with the development of experimental techniques for simultaneous measurement of LCC currents and Ca2+ transients (1–3), and detection of local Ca2+ release events (4,5). These experimental observations gave rise to and later verified the local control theory of excitation-contraction (EC) coupling (1,6–8). The mechanism of local control predicts that within discrete regions of the dyadic cleft, the opening of an individual LCC located in the t-tubule membrane will trigger Ca2+ release from a small cluster of SR Ca2+ release channels (known as ryanodine receptors, i.e., RyRs) that reside in close proximity directly across the dyad in the junctional SR membrane. Tight regulation of CICR is achieved because LCCs and RyRs are regulated by the local dyadic Ca2+ signal, rather than by bulk cytosolic Ca2+ levels. The amplitude and profile of the whole-cell intracellular Ca2+ transient results from the recruitment and temporal summation of elementary Ca2+ release events, known as Ca2+ sparks, in these discrete regions (4,9). The recruitment of Ca2+ sparks via this mechanism allows for graded control of SR Ca2+ release, in which amplitude of the Ca2+ release flux is a smooth, continuous function of LCC Ca2+ influx (6,9).
Until recently, computational models of the cardiac myocyte have been unable to capture the fundamental mechanism of local control of CICR. Many recent models (10–14) can be classified as common pool models (6), in which all LCC Ca2+ influx and RyR release flux is directed into a common Ca2+ compartment, and it is this Ca2+ pool that controls RyR activity. Stern (6) demonstrated that in any such model exhibiting gain at or near the high level observed physiologically, the initiation of SR Ca2+ release will stimulate regenerative all-or-none, rather than graded, Ca2+ release. That is, graded release and high gain are incompatible. This limitation of common pool models has important implications for Ca2+-mediated LCC dynamics. An increase in local Ca2+ concentration will promote Ca2+-dependent inactivation of nearby LCCs (15). Recent experiments have shown that inactivation of LCCs is mediated primarily by a Ca2+-dependent process (15,16), whereas voltage-dependent inactivation is relatively slow and incomplete during the time course of the AP (15,17). We have shown that incorporation of an LCC model with this experimentally validated balance between voltage- and Ca2+-dependent inactivation into a common pool myocyte model destabilizes the plateau of the AP (18). In fact, recent common pool models have incorporated strong voltage-dependent inactivation of the L-type Ca2+ current (ICaL) to stabilize the AP (19). For this reason, common pool models are not good candidates for the study of CICR. As an alternative to the common pool formulation, models in which Ca2+ release flux is represented as an explicit function of sarcolemmal Ca2+ influx (20–22) are able to achieve both high gain and graded Ca2+ release. However, because these phenomenological formulations lack mechanistic descriptions of the processes that are the underlying basis of CICR, their predictive ability is limited.
We have recently formulated a computational model of the cardiac ventricular myocyte that includes a mechanistic implementation of local control of CICR (18). The model consists of a population of Ca2+ release units (CaRUs), each of which represents a discrete dyadic Ca2+ release site at the junction of t-tubule and SR membranes. The gating of individual LCCs and RyRs within each CaRU are simulated stochastically, allowing for each CaRU to be independently modulated by its own local Ca2+ dynamics. Integration of this CICR model into a myocyte model describing membrane ionic and pump/exchanger currents, SR Ca2+ uptake, and time-varying cytosolic ion concentrations (10) yielded a model that accurately reconstructs stable APs and describes important features of CICR including graded release and voltage-dependent EC coupling gain. In addition, the stochastic model implementation has allowed for further study of the role of LCC gating noise in the generation of early after depolarizations (23). A limitation of this model, however, is that it is far more computationally demanding than its predecessors, and therefore requires significantly more computational resources. To overcome this issue, we recently introduced an approach to simplifying the local control model of CICR by applying a carefully chosen set of approximations that allow for the ensemble behavior of CaRUs to be represented by a low dimensional system of ordinary differential equations (ODEs), eliminating the need for computationally expensive stochastic simulations. This model, termed the coupled LCC-RyR model (24), retains the biophysically based description of local control of CICR, and captures its key properties including graded Ca2+ release and voltage-dependent EC coupling gain.
In this study, we formulate an integrative model of the canine ventricular myocyte based on the theory of local control of SR Ca2+ release. This is achieved by applying the method described by Hinch et al. (24) to the model of Greenstein and Winslow (18). The approach is demonstrated using a simplified CaRU model containing only a single LCC and a single RyR. We demonstrate that the resulting model faithfully reproduces features of LCC voltage- and Ca2+-dependent gating (8,17,25), microscopic EC coupling (1,26–29), and macroscopic whole-cell AP and Ca2+ cycling properties (30,31). This formulation can be considered a multiscale model because of its ability to capture details on the scale of local EC coupling events as well as on the scale of whole-cell phenomena.
Recent experiments have indicated that the quantitative features of EC coupling gain may be dependent upon local redundancy of LCC channel gating (32). Local redundancy in LCC open probability refers to the probability that multiple LCCs open simultaneously within a single release site. Manipulation of redundancy in LCC open probability (which depends upon channel-gating properties as well as the number of channels in a local cluster) is difficult to achieve experimentally. To study this phenomenon using this approach to modeling CICR, we generalize the method of Hinch et al. (24) to accommodate LCC and RyR models of arbitrary complexity, and to accommodate a CaRU containing multiple LCCs and RyRs. This is accomplished by designing a software algorithm that builds a coupled LCC-RyR model corresponding to given definitions of individual LCC and RyR models, and the numbers of each channel that resides in the CaRU. This allows for comparison of models of CICR containing different numbers of LCCs and RyRs, and therefore acts as a powerful tool for elucidating the relationship between physical RyR/LCC coupling ratio (i.e., RyR/LCC channel stoichiometry) and functional coupling ratio (i.e., the number of RyRs triggered per activated LCC, on average), and hence the underlying determinants of EC coupling gain. Simulations suggest that the steep rise in EC coupling gain at hyperpolarized potentials, as observed experimentally (1,29), is a result of increased functional coupling between LCCs and RyRs and reflects the maximal number of RyRs that can be triggered by a single LCC.
Finally, Sah et al. (33) have recently demonstrated that slowing early repolarization, in a manner consistent to that which occurs with inhibition of the transient outward K+ current (Ito1) in larger mammals, leads to blunted Ca2+ transients with slower rise times, decreased efficiency of EC coupling, and impaired contractility. Although a general mechanism by which Ito1 regulates properties of CICR has been described (18,33), we use the model presented here to demonstrate that a slowing of early repolarization of the AP via alteration in Ito1 leads to a reduction in SR Ca2+ release by not only a reduction of ICaL trigger influx (18,33), but also by a loss of EC coupling gain as a result of an increase in local redundancy of LCC openings. Thus this model provides insight into the mechanisms by which alteration of a membrane current that is seemingly unrelated to EC coupling can have a significant impact on CICR and hence, contractility.
METHODS
Our recent model formulation incorporating stochastic, local control of SR Ca2+ release (18) was designed to capture the fundamental properties of CICR (e.g., graded Ca2+ release) emerging from the local interaction of LCCs and RyRs in the dyad. The basic unit of that model is the calcium release unit (CaRU), consisting of LCCs, RyRs, and the dyadic volume within which these channels communicate via the local Ca2+ concentration. The CaRU mimics the properties of Ca2+ sparks in the t-tubule/SR junction. The CaRU was formulated based on the following experimental constraints:
RyR/LCC stoichiometry is set to five RyRs to one LCC, based on estimates indicating that a single LCC typically triggers the opening of 4–6 RyRs (34).
CaRUs are simulated independently in accordance with the observation that RyR clusters (i.e., dyads) are physically separated at the ends of sarcomeres (35).
LCC and RyR channel models are based on previously developed and validated models (11,36,37).
The current model formulation aims to retain all of the fundamental features of our previous model (18), but to eliminate the need for computationally intensive stochastic simulation of individual ion channels. This is accomplished using a method that we have described recently (24). Briefly, LCC and RyR models composed of the minimum number of states necessary to capture fundamental dynamics of channel gating are designed and constrained based on experimental data sets. Application of a steady-state approximation for Ca2+ diffusion in the dyad allows the resulting CaRU to be simplified into a single Markov state model in which each state represents a unique configuration of channel states for the combined set of LCCs and RyRs in the CaRU. All parameters of this compound Markov state model are derived analytically from the parameter sets describing individual LCCs, RyRs, and CaRU Ca2+ fluxes. In the resulting formulation, the dynamics of a population of CaRUs can be described by a set of coupled ODEs. The computationally expensive Monte Carlo methods used for single-channel simulations in our previous model (18) are therefore not required to simulate the model presented here.
LCC and RyR channel models
The L-type Ca2+ channel model (Fig. 1 A) is a simplified version of the 12-state Markov model presented and validated previously (see Fig. 12 A of Greenstein and Winslow (18)), which is based on the mode-switching model developed by Jafri et al. (11). The simplified LCC model retains all of the essential features by which it has been characterized, such as:
Forward and reverse activation rates (α and β) based on ICaL measured in canine midmyocardial cells (38);
Experimentally based balance between voltage- and Ca2+-dependent inactivation processes (15,17,42); and
Channel permeability consistent with measured values of single-channel slope conductance and unitary current (39,43).
Whereas the original model contained five states in the activation pathway before the open state (18), the current model reduces the activation pathway to two states. The normal gating mode (i.e., Mode Normal) is therefore represented by states 1–3, where state 3 is the open state. Ca2+-dependent inactivation (CDI) occurs upon downward transitions from Mode Normal states into states 4–5 (i.e., Mode CDI). The Mode CDI open state from the original 12-state model has been excluded from this model version because its occupancy probability is nearly zero. States 6–10 represent voltage-dependent inactivation (i.e., Mode VDI) and are accessible from states 1–5, respectively. This process was previously modeled using an independent channel gate (see Fig. 12 B of Greenstein and Winslow (18)). The methods implemented here require VDI to be incorporated within the LCC Markov-state model; however, steady-state and kinetic properties of this process are identical to those in our previous LCC model description (18).
FIGURE 1.

State diagrams for the LCC and RyR Markov models and schematic diagrams of the CaRU. (A) State model of the LCC which represents a simplification of the model presented previously (see Fig. 12, A and B, of Greenstein and Winslow (18)). Upper states (green outline, states 1–3, and 6–8) encompass Mode Normal, lower states (red outline, states 4, 5, 9, and 10) encompass Mode CDI, and the backplane states (gray shading, states 6–10) encompass Mode VDI. The open state (state 3) is denoted by a circle, whereas closed states are denoted by squares. Depolarization rapidly promotes transitions from left to right, and more slowly promotes transitions into Mode VDI states, whereas downward transitions into Mode CDI states are promoted by elevation in local Ca2+ level. Pairs of transitions with the same arrow color share identical transition rates. (B) State model of the RyR based on a simplification described previously (see Fig. 13 C of Greenstein and Winslow (18)), where states 1 and 2 are the resting states, state 3 is the open state, and state 4 is the refractory state. Rate constants are Ca2+-dependent based on the assumption that four Ca2+ ions must bind the RyR before opening. Rate definitions for both the LCC and RyR models are provided in the Supplementary Material. (C) Schematic representation of the CaRU, which is the basis of the baseline-coupled LCC-RyR model. In the baseline model, a CaRU consists of a single LCC on the t-tubule membrane and a single RyR on the SR membrane, both facing into the dyadic space. (D) Visualization of the state diagram for the baseline-coupled LCC-RyR model. The “outer” model represents the four-state RyR model of B, and for each RyR state, there are 10 possible LCC states, shown within each RyR state as the LCC model of A. The model therefore consists of 40 states, where each state represents a unique pairing of LCC and RyR states. Each state transition of the coupled model represents a single state transition of either the LCC or the RyR. The full set of equations describing the 40-state coupled LCC-RyR model is provided in the Supplementary Material.
The free parameters of the model were determined using a Nelder-Mead simplex search algorithm (44) implemented in MatLab (The MathWorks, Cambridge, MA). The optimization algorithm was implemented using a cost function that quantified the difference in behavior of the simplified LCC model to the previous 12-state LCC model (18) based on measures of steady-state open probability and Mode Normal occupancy, peak and time-to-peak of open probability and Mode Normal occupancy in response to a depolarizing voltage-clamp, and rate of recovery from inactivation. All protocols were performed over a large range of voltages and Ca2+ concentrations. The number of active LCCs was chosen such that the amplitude of whole-cell ICaL corresponds to measurements in canine myocytes (38), yielding a value of 50,000, which agrees with experimental estimates of LCC density (39,45).
The RyR is represented by a model developed by Keizer and Smith (36) and later modified by Rice et al. (37). The RyR model implemented in this study (Fig. 1 B) is identical to the reduced four-state model used in our previous local control myocyte model (see Fig. 13 C of Greenstein and Winslow (18)). It is a highly simplified model that is intended to describe Ca2+ release fluxes under physiologically relevant stimuli, but does not explicitly incorporate additional features such as regulation by Mg2+ or luminal Ca2+ (see Discussion). Upon influx of trigger Ca2+ into the dyad, RyRs will transition rapidly from state 1 through state 2 to state 3, the open state. Open channel flux is driven by the concentration gradient between SR luminal Ca2+ and the local dyadic Ca2+. Open channels will close via an inactivating transition into state 4. After local Ca2+ levels subside, RyRs will recover by transitioning back to state 1. RyR model parameters were constrained based on experimentally obtained properties of EC coupling (1,29), and are in agreement with the findings of Sham et al. (26) suggesting that RyR inactivation into an absolute refractory state occurs in vivo and is the primary mechanism of SR Ca2+ release termination. The Ca2+ dependence of the model state transition rates have been adjusted based on the assumption that four Ca2+ ions must bind to the channel before it can enter the open state (46). In most cases, it will be possible to model a small cluster of RyRs using a single model RyR. In these instances, the open channel Ca2+ flux rate and activation rates (transition rates between states 1 and 2) are adjusted accordingly based on the number of channels the single RyR model represents (see below).
The coupled LCC-RyR model
We have recently described a method for formulating a simplified model of the CaRU (24). Briefly, a minimal model of the CaRU (Fig. 1 C) is devised consisting of a single LCC in the sarcolemmal membrane, a single RyR in the SR membrane, and the dyadic volume within which these channels reside. The single-model RyR represents an approximation to a cluster of five RyRs (see Fig. 4 C) in agreement with estimates of RyR/LCC stoichiometry (34). This representation of the CaRU requires that the SR is considered as a single volume compartment, and no distinction is given to junctional versus network SR. This simplification can be justified by recent experimental observations that demonstrate that junctional SR Ca2+ is in quasiequilibrium with network SR Ca2+ during a Ca2+ release event, that is, the Ca2+ refilling rate from network to junctional SR is rapid relative to the release flux (47). Since the diffusion rate of Ca2+ between the dyad and cytosol is relatively fast compared to the gating rates of LCC and RyR channels (48,49), and because dyad volume is very small, it can be assumed that dyadic space Ca2+ concentration ([Ca2+]ds) equilibrates very rapidly. In other words, it can be assumed that Jxfer = JRyR + JLCC, where JRyR and JLCC are the Ca2+ fluxes into the dyad via the RyRs and LCCs, respectively, and Jxfer is the Ca2+ flux from the dyad to the bulk cytosol (Fig. 1 C). Note that this assumption does not imply that [Ca2+]ds = [Ca2+]cyto. Rather, this assumption allows [Ca2+]ds to change instantaneously upon a change in dyadic Ca2+ flux due to the opening or closing of an LCC or RyR. The above expression can be expanded and rearranged to yield an expression for [Ca2+]ds (in terms of cytosolic and SR [Ca2+], and membrane potential) which can be used to evaluate [Ca2+]ds in the transition rate expressions. This step transforms [Ca2+]ds into a dependent variable, rather than an independent variable, in each CaRU. The LCC and RyR models can then be combined into a single coupled LCC-RyR model, in which each state represents the joint configuration of the LCC-RyR pair, and whose parameters are derived analytically from the individual LCC and RyR models (see (24) for a detailed example). The coupled LCC-RyR model of the CaRU described here consists of a 40-state Markov process (Fig. 1 D), and will be able to capture nearly all of the biophysical details of CICR associated with local control of SR Ca2+ release. This 40-state model will be referred to the baseline-coupled LCC-RyR model below. All equations for the baseline-coupled LCC-RyR model are provided in the Supplementary Material. The model is available for download at the Center for Cardiovascular Bioinformatics and Modeling website (www.ccbm.jhu.edu).
FIGURE 4.

Voltage dependence of LCC Ca2+ influx, RyR Ca2+ release flux, and EC coupling gain. (A) Peak LCC Ca2+ flux (solid circles) and peak RyR release flux (open circles) as a function of membrane voltage. (B) Peak Ca2+ fluxes (data in A) normalized by their respective maxima. (C) EC coupling gain as a function of membrane potential for the baseline-coupled LCC-RyR model (solid circles), in which a single model RyR is used to represent a cluster of five RyRs is compared to a model in which the five RyRs are each modeled individually (open circles).
The coupled LCC-RyR model lacks Ca2+ buffers in the dyadic space. In general, the binding/unbinding of Ca2+ to buffers alters the rate at which free Ca2+ concentration changes in any given compartment. Ca2+ buffers, however, do not affect the steady-state concentration of free Ca2+ in a compartment (i.e., at steady-state, Ca2+ influx equals Ca2+ efflux and the fraction of buffer that has Ca2+ bound to it remains constant). Since this model assumes that Ca2+ in the dyad is always at its steady-state value (i.e., rapid equilibrium with neighboring compartments), implicit in this assumption is that any Ca2+ buffers that may be present have fast kinetics and are not of sufficient capacity to significantly alter the rate of equilibration between the dyad and its neighboring compartments. Therefore, no dyadic Ca2+ buffers are explicitly included in this model.
The method described above for formulating a coupled LCC-RyR model is not limited to a dyad containing only a single LCC and single RyR. Investigation of the quantitative role of microdomain LCC-RyR signaling on CICR requires the ability to construct more complex coupled LCC-RyR gating models based on a variety of individual LCC and RyR models and/or the number and ratio of RyRs and LCCs in a CaRU. To do this, we have devised an automated method for generating a set of ODEs describing the joint behavior of all channels in a CaRU. This method is sufficiently general that it can be applied to construct a CaRU model using individual LCC and RyR models of arbitrary complexity. The generalized method is given in the Supplementary Material and is described briefly as follows. If each CaRU is composed of NLCC LCCs, each represented by an identical MLCC-state model, and NRyR RyRs, each represented by an identical MRyR-state model, then it can be shown that MCaRU, the number of unique states in the coupled LCC-RyR model, is given by
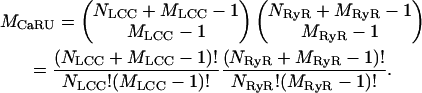 |
A transition between any two of these states occurs as the result of a single-state transition of a single channel (either LCC or RyR) within the CaRU. Each pair of connected states in the group of MCaRU states is identified and assigned transition rate expressions that are derived from the transition rates of the underlying channel models. Some of these rate expressions will depend upon [Ca2+]ds. The value of [Ca2+]ds is determined analytically based on the number of open LCCs and RyRs corresponding to each CaRU state and the assumption that Ca2+ flux-balance is in rapid equilibrium (as described above). A data structure is formed that stores the relevant parameters needed to correctly evaluate the rate expressions for each connected pair of states in the coupled LCC-RyR model. The velocity field of the ODEs that define the CaRU can then be evaluated within any Solver routine on a term-by-term basis where the contribution of each state pair is incrementally added to the right-hand side of the appropriate state equation. This entire process of model derivation and code implementation has been automated in software to eliminate errors in model formulation. This is a critical step, since manual derivation and implementation of models of this complexity would be tedious and error-prone. For example, using the 10-state LCC and four-state RyR models described above, evaluation of the above equation for MCaRU shows that a CaRU containing five RyRs and one LCC will require a 560-state coupled model. Such a model can be rapidly formulated and simulated using our generalized methods. In addition, multiple model variations that differ in LCC and/or RyR number can be rapidly generated and compared, whereas a manually derived coupled LCC-RyR model must be completely reformulated upon a change in channel number.
Whole-cell simulations are performed by incorporating the coupled LCC-RyR model into the canine ventricular myocyte model described previously (18) and based on the model of Winslow et al. (10). Some membrane currents and ionic fluxes have been scaled to adjust cytosolic ionic concentrations, AP shape, and rate dependence of AP duration. All equations and parameters describing the myocyte model are provided in the Supplementary Material. The set of ODEs defining the myocyte model are solved using a trapezoidal rule algorithm (MatLab function ode23t, The MathWorks).
RESULTS
L-type Ca2+ current
To correctly capture the strong negative feedback of SR Ca2+ release on the dynamics of ICaL, the simplified LCC model presented here (Fig. 1 A) has been configured to match the kinetic properties of our previous detailed LCC model and the experimental data against which it was validated (18). Simulated whole-cell currents elicited by a family of depolarizing voltage steps from −30 mV to +40 mV in 10-mV increments are shown in Fig. 2 A. Currents activate rapidly (<8 ms) and decay over ∼100 ms. Fig. 2 B shows the LCC peak I-V relationship for ICaL, where peak currents were determined in response to voltage steps from a holding potential of −100 mV. Maximum inward Ca2+ current is produced at a test potential of +10 mV. The bell-shaped peak current profile is in agreement with peak currents measured in canine (38,50,51), Guinea pig (39), and human (52) ventricular myocytes. Fig. 2 C demonstrates the underlying processes that govern the time course of ICaL during a voltage-clamp to 0 mV. The quantities shown are LCC open probability (po, solid line), probability that CDI has not occurred (pnot_CDI, shaded line), and the probability that VDI has not occurred (pnot_VDI, dashed line). LCC po reaches a peak value of ∼0.09, consistent with studies that indicate that peak po with Ca2+ as the charge carrier is in the range of 0.05–0.15 (39,40). A comparison of the time progression of pnot_CDI and pnot_VDI clearly demonstrates that at 0 mV, Ca2+-mediated inactivation of ICaL develops more rapidly and progresses more completely than voltage-dependent inactivation, consistent with recent experiments (17,53,25,15,42) and our previous model (see Fig. 3 C of Greenstein and Winslow (18)). Steady-state inactivation properties of ICaL are measured using a standard double-pulse protocol. Normalized peak currents are shown as a function of the prepulse potential in Fig. 2 D. Simulations were performed under normal conditions (solid circles) and with subspace Ca2+ clamped to diastolic levels to mimic an alternative charge carrier such as a monovalent cation that would not significantly promote CDI (open circles). Under normal conditions, the ICaL steady-state inactivation curve is U-shaped, whereas upon removal of CDI, the inactivation curve decreases monotonically with depolarization. Note that LCC inactivation is incomplete, and asymptotically approaches ∼48% at the most depolarized potentials. These features agree well with inactivation curves obtained experimentally in native myocytes (17,50,53) and demonstrate the role of graded CICR on CDI, particularly in the range of potentials that correspond to the AP plateau (–10 mV to +30 mV). The degree of inactivation attained is more than would be expected theoretically due to VDI alone (maximum of 40%, see Eq. 5 of the Supplementary Material) because there is a small degree of CDI that remains present under these conditions. Single-channel properties of the LCC model under normal physiological conditions (i.e., with EC coupling intact and 2 mM extracellular Ca2+) display a voltage-independent mean open time of ∼0.5 ms, an open-channel slope conductance of 8.2 pS, and a unitary current of ∼0.12 pA at 0 mV (data not shown), in agreement with experiments (39–41,54,55) and our previous model (see Fig. 2 of Greenstein and Winslow (18)).
FIGURE 2.

Properties of ICaL. (A) Simulated whole-cell currents as a function of time in response to a family of depolarizing voltage steps from −30 mV to 40 mV in 10-mV increments. (B) Peak I-V relation for ICaL. (C) LCC open probability (po, solid line), probability that Ca2+-dependent inactivation has not occurred (pnot_CDI, shaded line), and probability that voltage-dependent inactivation has not occurred (pnot_VDI, dashed line), in response to a voltage-clamp to 0 mV. (D) Steady-state inactivation curve obtained using a double-pulse protocol with (black line) and without (shaded line) Ca2+ as the charge carrier.
FIGURE 3.

Ca2+ dependence of peak (solid line) and steady-state (dashed line) RyR open probability (po) for simulated maintained changes in free dyadic Ca2+ concentration. The response at each [Ca2+] level was initiated from a resting [Ca2+] of 0.1 μM.
Fig. 3 shows the model RyR peak and steady-state open probabilities in response to a range of Ca2+ concentration stimuli. The peak open probability for this model is most sensitive to Ca2+ stimuli in the range of 1–20 μM and approaches a maximal value of ∼50% for higher Ca2+ concentrations. This behavior is consistent with recent experimental measurements of RyR po under simulated physiological conditions in the presence of 0.6 mM Mg2+ (56). The small overshoot in peak po that occurs in the range of 15–30 μM Ca2+ is result of model simplification. For Ca2+ concentrations >∼1 μM, the steady-state po is <1%, indicating that the process of inactivation/adaptation is nearly complete for a wide range of trigger Ca2+ concentrations.
Excitation-contraction coupling
Previous experiments (1,27,28,57) and mathematical models (6,18,58) have demonstrated the relationship between LCC Ca2+ influx and SR Ca2+ release flux, in which SR release is graded and there is a characteristic shift in the voltage-dependent release flux with respect to LCC trigger influx. As a result, EC coupling gain, defined as the ratio of peak SR Ca2+ release flux to peak LCC Ca2+ influx in response to a voltage-clamp step (1), decreases monotonically with increasing voltage. Fig. 4 demonstrates that the coupled LCC-RyR model captures these fundamental quantitative properties of CICR. Peak LCC trigger influx and SR release flux (solid circles and open circles, respectively, Fig. 4 A), display similar voltage dependence; however, normalization of these curves (Fig. 4 B) reveals that maximal SR release flux occurs at ∼5 mV, whereas maximal trigger influx occurs at ∼10 mV. The EC coupling-gain curve for this coupled LCC-RyR model (containing only a single LCC and a single RyR per CaRU, Fig. 4 C, solid circles) agrees with experimentally observed measurements of whole-cell Ca2+ measurements (1,27–29,57). As stated in Methods, this coupled LCC-RyR model contains a single LCC and a single RyR, where the RyR model is altered to approximate the behavior of a cluster of five RyRs. The validity of this approximation is demonstrated by the similarity in EC coupling gain predicted by a model in which this approximation is not applied, i.e., a model that contains one LCC and five RyRs (Fig. 4 C, open circles). Such a model requires 560 states to describe the CaRU (see Methods), and is therefore far less computationally efficient than the baseline 40-state model. The approximation of a cluster of RyRs by a single model RyR is effectively the implementation of the assumption that neighboring RyRs are functionally coupled and gate synchronously as has been observed experimentally (59). Due to the fact that Ca2+-release is locally regenerative in the model, the EC coupling gain curve does not depend strongly on whether RyRs are allowed to gate independently or are constrained to gate synchronously. Regardless of the number of modeled RyRs, as membrane potential decreases, EC coupling gain in the model increases approximately linearly and does not exhibit a steep rise at potentials <−10 mV as has been observed in experiments (1,29). The mechanism underlying the steep slope of the gain curve at negative potentials as observed in experimentally obtained measurements of EC coupling is addressed below. The results of Figs. 2–4 demonstrate that the baseline-coupled LCC-RyR model (i.e., the 40-state model) adequately reproduces average properties of local Ca2+-dynamics and can therefore reliably simulate CICR at the level of the whole myocyte.
The coupled LCC-RyR model provides a unique opportunity to understand the mechanistic determinants of the shape of the EC coupling-gain curve. The effect of CaRU (i.e., dyad) size on EC coupling gain is demonstrated in Fig. 5 A. A CaRU that is n-fold larger in size than the baseline model contains an n-fold larger dyadic volume, and n-fold more LCCs and RyRs. However, regardless of CaRU size, the RyR/LCC stoichiometry of 5:1 remains unchanged. The total number of CaRUs in the cell is reduced n-fold, such that the total number of LCCs and RyRs, as well as the total aggregate dyadic volume in the whole myocyte, remains unchanged. Coupled LCC-RyR models for larger CaRUs were generated using the generalized algorithm for coupled LCC-RyR models as described in Methods. These manipulations allow for the study of the role of dyad size on EC coupling independently from the role of RyR/LCC stoichiometry. Gain for the baseline-coupled LCC-RyR model (Fig. 5 A, solid line) is compared to that for a population of CaRUs, which are twofold (shaded line) or threefold (dashed line) larger than the baseline model. The simulation results demonstrate that EC coupling gain at potentials >−10 mV increases by only a small to moderate amount with and increase in CaRU size. However, at potentials more negative than −10 mV, the increase in CaRU size yields a substantial increase in gain and, hence, the EC coupling-gain curve acquires the steep slope similar to those observed experimentally in this range of potentials (1,29). This demonstrated relationship between CaRU size and EC coupling gain arises due to the local redundancy of LCC po. Local redundancy of LCC po refers to the probability that there are multiple LCCs open simultaneously within a single CaRU. In the coupled LCC-RyR model, local redundancy of po is defined as the fraction of open LCCs that have at least one open neighboring LCC within the same CaRU. As shown in Fig. 5 B, in response to a voltage-clamp step to 0 mV, redundancy of po is a time-dependent quantity that has a peak that approximately coincides with peak trigger LCC Ca2+ influx (see Fig. 2). Peak redundancy of po as a function of voltage-clamp potential is shown in Fig. 5 C using the CaRU that is increased in size by twofold (i.e., 2 LCCs, 10 RyRs). At negative potentials, local redundancy of po is nearly zero due to relatively low LCC po. This increases the functional RyR/LCC ratio because it is likely that a single LCC opening will trigger regenerative release from all RyRs in a CaRU. The functional RyR/LCC ratio will therefore increase with an increase in CaRU size (i.e., an increase in the number of dyadic LCCs and RyRs) leading to a substantial increase in gain at negative potentials (Fig. 5 A). At more positive potentials, the local redundancy of po is increased significantly, and approaches 30% (note that redundancy never approaches 100% because peak LCC po never approaches unity). Under these conditions, the functional RyR/LCC ratio approaches the stoichiometric ratio of 5:1 due to increased likelihood of simultaneous opening of neighboring LCCs. As a result, EC coupling gain displays little dependence on CaRU size at positive potentials (Fig. 5 A). Recent experiments have demonstrated that a reduction in LCC unitary current (iLCC), achieved via reduction of extracellular Ca2+ concentration ([Ca2+]o), increases EC coupling gain, suggesting that the high gain at negative potentials is due to less redundancy in po rather than larger iLCC (32). Fig. 5 D demonstrates model simulations in which EC coupling gain is measured as a function of [Ca2+]o using the model with twofold-increased CaRU size. At negative potentials, successive decreases in [Ca2+]o from 2.0 mM (solid line) to 1.4 mM (shaded dashed line) lead to successive increases in EC coupling gain, in agreement with experiments (32) suggesting that EC coupling gain relies on the redundancy of po rather than the magnitude of iLCC. At higher potentials (>0 mV), however, the model predicts that the dependence of EC coupling gain on [Ca2+]o reverses direction and gain declines as [Ca2+]o is reduced. This suggests that the local coupling efficiency between LCCs and RyRs within a dyad becomes progressively weaker as iLCC falls to relatively small amplitudes.
FIGURE 5.

Properties of EC coupling gain. (A) EC coupling gain as a function of membrane potential for the baseline-coupled LCC-RyR model (solid line) is compared to gain for models in which dyad size and number of channels is increased twofold (shaded line), and threefold (dashed line). The number of CaRUs in the cell is scaled such that the number of LCCs and RyRs (and total dyad volume) in the whole cell remains identical for all models. (B) Redundancy of LCC po as a function of time in the model with twofold increase in dyad size in response to a voltage-clamp to 0 mV. (C) Peak redundancy of LCC po as a function of voltage in the model with a twofold increase in dyad size. (D) EC coupling gain in the model with a twofold increase in dyad size for [Ca2+]o of 2.0 mM (solid line), 1.8 mM (shaded line), 1.6 mM (black dashed line), and 1.4 mM (shaded dashed line).
Action potentials and whole-cell Ca2+ transients
The baseline-coupled LCC-RyR model of CICR has been incorporated into a global model of the myocyte (18) and Fig. 6 demonstrates the ability of this model to reconstruct action potentials and Ca2+ transients of normal canine midmyocardial ventricular myocytes. Steady-state APs are shown (Fig. 6 A) for pacing cycle lengths (CL) of 500 ms, 1000 ms, 2000 ms, and 4000 ms, with the corresponding cytosolic Ca2+ transients (Fig. 6 B). Model AP shape and rate dependence of AP duration (APD) agree with experimental recordings obtained from isolated canine midmyocardial myocytes (31,60), and demonstrate an APD of ∼300 ms at CL of 1000 ms (30). The rate-dependence of model Ca2+ transient amplitude is similar to that measured in experiments (50), exhibiting peak amplitudes that increase with increasing pacing rate. Fig. 6 C compares the shape of the cytosolic Ca2+ transient (solid line) with the mean subspace Ca2+ transient (dashed line) at CL of 1000 ms. Although the cytosolic Ca2+ transient peaks at ∼0.73 μM, and lasts longer than the duration of the AP, Ca2+ in the subspace reaches a level of ∼15.5 μM on average, and after SR Ca2+ release, rapidly returns to near-cytosolic levels. It is important to emphasize that the [Ca2+]ds signal shown in Fig. 6 C is an average over all CaRUs (including units that were not activated). Maximal [Ca2+]ds in a single dyad during a release event is ∼100 μM under control conditions in this model (data not shown). Fig. 6 D shows ICaL (solid line) corresponding to the AP at CL of 1000 ms. ICaL peaks at ∼5.2 pA pF−1 and has a sustained component of ∼0.5–1.0 pA pF−1, which lasts for nearly the entire duration of the AP. The underlying LCC inactivation process is shown as pnot_CDI (shaded line) and pnot_VDI (shaded dashed line). The quantities demonstrate that, during the plateau, ∼80% of LCCs become unavailable due to Ca2+-dependent inactivation, whereas only ∼35% become unavailable due to voltage-dependent inactivation. This balance between voltage- and Ca2+-dependent inactivation is consistent with experimental data, indicating that LCC voltage-dependent inactivation is slow and incomplete, whereas Ca2+-mediated inactivation is strong and dominates the inactivation process (15,17,25,42), and is a key mechanism in determining how graded SR Ca2+ release influences AP properties and whole-cell Ca2+ dynamics (18). The results of Fig. 6 demonstrate that the baseline-coupled LCC-RyR model can successfully simulate CICR within a model of an AP, and retains features associated with local control of Ca2+ release.
FIGURE 6.

Action potentials and Ca2+ transients. (A) Steady-state model APs for CLs of 500 ms (shaded line), 1000 ms (solid line), 2000 ms (shaded dashed line), and 4000 ms (black dashed line). (B) Cytosolic Ca2+ transients corresponding to the APs of A. (C) Mean subspace (dyadic) Ca2+ transient (dashed line) demonstrates that microdomain Ca2+ levels reach significantly higher levels than that in the cytosol (solid line) during the AP at 1000 ms CL. (D) Properties of ICaL (solid line) during the AP at 1000 ms CL, demonstrating relatively strong Ca2+-dependent inactivation (pnot_CDI, shaded line) but weak voltage-dependent inactivation (pnot_VDI, dashed line).
Effect of AP shape on efficiency of EC coupling
The canine myocyte model presented here provides the ability to study the mechanistic details underlying the tight coupling between AP morphology and CICR. In a previous study, we examined the role of the transient outward potassium current Ito1 in determining AP shape and duration in a canine myocyte model and showed that reductions in Ito1 lead to progressive slowing of phase-1 repolarization and elimination of the notch (61). We subsequently showed that in a model which correctly reproduces graded SR Ca2+ release, the loss of the notch leads to a reduction in ICaL peak amplitude, and therefore a reduction in SR Ca2+ release flux (18). In agreement with these model predictions, Sah et al. (33) demonstrated that slowing early repolarization, in a manner consistent to that which occurs with Ito1 inhibition in larger mammals, leads to blunted Ca2+ transients with slower rise times, decreased efficiency of EC coupling, and impaired contractility. These findings have led to a number of hypotheses regarding the quantitative dependence of SR Ca2+ release on AP profile and ICaL. Specifically, it has been suggested that the reduction and slowing of ICaL in response to slowed phase-1 repolarization is associated with a reduction in local coupling efficiency at Ca2+ release sites and reduced synchronization of Ca2+ release events, indicating that in normal myocytes of large mammals (e.g., human and canine), Ito1, and the associated AP notch may be required to transiently increase ICaL to synchronize and optimize SR Ca2+ release (62). Fig. 7 demonstrates the role of Ito1 amplitude on EC coupling properties in a canine myocyte model. This model incorporates the twofold larger CaRUs (i.e., containing two LCCs and 10 RyRs per dyad). Model simulations are compared in Fig. 7 under control conditions (left panels) and with a threefold reduction in Ito1 density (right panels). This degree of reduction in Ito1 is based on that observed in failing canine myocytes (51). Both simulations were run using the same set of steady-state (CL of 1000 ms) control initial conditions. Under normal conditions, the AP (Fig. 7 A) has a duration of ∼300 ms and Ito1 reduction leads to a modest shortening of APD to ∼270 ms and a less pronounced phase-1 notch, consistent with our previous findings (61). The change in AP shape due to the altered rate of early repolarization results in a 40% reduction in peak ICaL from ∼5.5 pA pF−1 to ∼3.3 pA pF−1 (Fig. 7 B). The redundancy of LCC po displays a similar peak value in both cases (Fig. 7 C). However, in the control case, po redundancy is considerably reduced (by as much as fourfold) during the AP notch. The loss of the AP notch associated with reduced Ito1 results in an increased LCC po redundancy over this time window, suggesting an increased tendency for simultaneous openings of LCCs in the same CaRU. This effect would be expected to degrade efficiency of EC coupling and hence, reduce EC coupling gain. LCC Ca2+ influx (shaded line) and SR Ca2+ release flux (solid line, Fig. 7 D) indicate that the reduction of Ito1 leads to a 50% reduction in peak SR Ca2+ flux. The reduction in SR Ca2+ flux in Fig. 7 D occurs not only as a result of the diminished trigger influx Ca2+ signal, but also as a result of a reduction of EC coupling gain. EC coupling gain during the AP (i.e., the ratio of peak SR release flux to trigger influx) is reduced from 7.0 in control to 5.6 as a result of Ito1 reduction. The net effect of these changes yields a cytosolic Ca2+ transient (Fig. 7 E) with a dramatically slowed rise-time and an 18% reduction in peak amplitude. Such alterations would be expected to reduce contractility. The simulations of Fig. 7 demonstrate a reduction in EC coupling efficiency in the presence of identical SR Ca2+ load. Upon pacing the model with threefold reduction in Ito1 to steady state, the drop in ICaL amplitude leads to a net reduction in SR Ca2+ load and hence, an even more pronounced reduction in SR Ca2+ release flux and cytosolic Ca2+ transient amplitude (data not shown). These data indicate that the repolarization rate during phase 1 of the AP has a marked influence on the process of CICR, and agree with the hypothesis that Ito1 plays an important role in optimizing SR Ca2+ release (62). The scope of these results, however, is limited to species and cell types that exhibit longer, notched AP morphologies. The role of Ito1 on CICR may in fact be reversed (i.e., reduction in Ito1 amplitude enhances SR Ca2+ release) in species with short triangular APs such as mouse and rat (62). In addition, some myocytes lack Ito1 completely (e.g., Guinea pig and most endocardial myocytes), which indicates that, although Ito1 plays an important role in modulating CICR, its presence is not always crucial for normal myocyte function.
FIGURE 7.

Role of Ito1 on EC coupling during the AP. Control simulations (left panels) are compared to simulations in which Ito1 density is reduced threefold (right panels). (A) AP; (B) ICaL; (C) redundancy of LCC po; (D) LCC Ca2+ influx (shaded line) and RyR Ca2+ release flux (solid line); and (E) cytosolic Ca2+ transient.
DISCUSSION
In this study, we present a biophysically detailed model of the normal canine ventricular myocyte, which incorporates local control of EC coupling. This myocyte model is formulated by replacing a population of independent stochastically simulated CaRUs (18) with a formulation of CICR, which we refer to as a generalized coupled LCC-RyR gating model (24). The procedure for deriving the coupled LCC-RyR model has been described in detail previously (24). In this study, these methods have been generalized to allow for formulation (via automated software generation) of a coupled LCC-RyR model using any predefined LCC and RyR models and with CaRUs consisting of an arbitrary number of each of these channels. The resulting model of CICR consists of a set of ODEs that describe the state-occupancy probabilities of a population of CaRUs and retains essential properties of CICR (e.g., voltage-dependent EC coupling gain) that arise due to local control of SR Ca2+ release, as described in our previous model (18). This approach yields a number of advantages over previous model formulations. Ease of use is substantially increased due to the dramatically decreased computational cost and hence decreased runtime for this formulation. APs stimulated at 1-Hz can be generated two-to-threefold faster than real-time on a desktop computer. It is important to note that the runtime for this model is similar (within 20%) to that for our previous canine myocyte model, which was based on a common-pool formulation for CICR (10). An essential feature of the coupled LCC-RyR model is that it is based only on the known biological features of CICR, and does not require the introduction of any new or complex assumptions. The recent model of Bondarenko et al. (63) achieved graded Ca2+ release by assuming a distribution of CaRUs that contain different numbers of LCCs and RyRs. Such an assumption should not be necessary as we have demonstrated recently (24). The only assumptions used in the coupled LCC-RyR model are that [Ca2+]ds is in rapid equilibrium with Ca2+ in its neighboring compartments (48,49), and that junctional SR Ca2+ is in quasiequilibrium with network SR Ca2+ (47), both of which are justified by experimental and theoretical considerations. Finally, parameters of a coupled LCC-RyR gating model are analytically determined from those of the underlying LCC and RyR models. Thus, there is no need for additional parameter fitting once the individual LCC and RyR model parameters have been independently constrained. The coupled LCC-RyR model is, therefore, a novel approach to simulation of EC coupling within a model of the cardiac AP, which captures the biophysical detail of CICR without requiring computationally intensive stochastic simulation of independent dyads.
The baseline-coupled LCC-RyR model of CICR presented here is a 40-state model built from the 10-state LCC model and four-state RyR model shown in Fig. 1. Each of these models are minimal-model approximations to more detailed channel models (11,36,37) where the degree of reduction in model complexity was limited to retain fundamental dynamics of channel gating that were present in the original models. In the case of the LCC, the reduced model was required to reproduce experimentally measured activation kinetics, the peak I-V relationship, and the appropriate balance between voltage- and Ca2+-dependent inactivation. Further simplification of this model, as was performed by Hinch et al. (24), would have sacrificed some of these critical features and therefore would not be appropriate for use in the canine myocyte model. In the case of the RyR, the model reduction has been described previously (see Fig. 13 of Greenstein and Winslow (18)), and involved reduction of a six-state model to a four-state model (Fig. 1 B) based on the difference in timescales of transition rates at higher [Ca2+]ds levels. It should be noted that the simplified RyR model used in this study does not specifically account for all experimentally characterized features of RyR gating such as regulation by Mg2+ (56), low and high activity gating modes (64), and regulation by luminal SR Ca level (65,66). However, the RyR model is designed to operate as would be expected under physiological conditions. Fig. 3 demonstrates that the RyR model implemented here is consistent with recent measurements of open probability under simulated physiological conditions (56). The results therefore indicate that a simple RyR model that accounts for activation, inactivation and/or adaptation, and recovery is adequate to describe many features of EC coupling under both voltage-clamp (e.g., shape of the gain curve) and during an AP. Incorporation of additional mechanistic details of RyR gating would yield more complex channel models with a larger number of states, and hence would greatly amplify the complexity of the coupled LCC-RyR model (i.e., increase greatly the number of required states for the coupled model). We therefore chose to use a simplified model that has worked well for us in past studies, and left the study of the role of additional RyR gating features to future work. In the CaRU, the RyR/LCC stoichiometry was assumed to be 5:1 (34); however, the five RyRs were simulated with a single RyR model altered to approximate the behavior of a cluster of five RyRs (fivefold increase in open channel Ca2+ flux rate and increased Ca2+ sensitivity). The results of Figs. 2–4 demonstrate that a CaRU composed of a coupled LCC-RyR model based on these simplified channel models can reliably reproduce the salient features of local control of CICR which previously required a far more complex model (18).
A number of reports indicate that the Na+-Ca2+ exchanger (NCX) is primarily located within the t-tubule membrane (67,68). However, it remains unclear whether NCX actually senses the very high Ca2+ concentrations in the junctional space of the dyad (69). One recent study that looked specifically at the role of NCX in shaping the AP in canine myocytes (70) found from both experiments and model simulations that NCX is unlikely to sense Ca2+ in the dyadic cleft. This agrees with the findings of Lopez-Lopez et al. (71) that Ca2+ influx via reverse mode NCX current cannot alone trigger Ca2+ sparks. The recent experiments of Weber et al. (72) do suggest the existence of a local submembrane compartment whose Ca2+ is sensed by NCX; however, the experimental results of Armoundas et al. (70) are not in full agreement with these findings. Clearly, the issue regarding which pool of Ca2+ is sensed by NCX is not yet well understood, and may be species-dependent. We therefore chose to locate NCX outside of the dyad in this study consistent with measurements obtained in canine myocytes (70) and previous canine myocyte models (10,18).
The EC coupling gain curve for the baseline model (Fig. 4 C) does not show the steep increase in slope at hyperpolarized potentials as seen in experiments (1,29). To understand how the higher gain may arise at these potentials, we investigated the role of CaRU size on gain. Expanding the size of the CaRU (while maintaining a constant RyR/LCC stoichiometry) yields an increase in gain predominantly at hyperpolarized potentials, although it has only a small effect on gain at positive potentials (Fig. 5 A). This corresponds to an increase in slope at negative potentials by a factor similar to the increase in CaRU size. These results suggest that the slope of the EC coupling gain curve may be proportional to the functional coupling ratio between RyRs and LCCs (i.e., the number of RyRs activated per activated LCC). At depolarized membrane potentials, there is a high redundancy of LCC po (Fig. 5 C), indicating that many LCCs will open in a synchronous manner. Under these conditions, the functional RyR/LCC coupling ratio would be expected to approach the physical RyR/LCC stoichiometry (e.g., 5:1 in this model), and will not be affected by the size of the release unit. However, at hyperpolarized potentials, where redundancy of LCC po approaches zero (Fig. 5 C), only a small fraction of LCCs will be activated. The functional RyR/LCC coupling ratio will therefore be greatly enhanced, and will approach a value that represents the maximal number of RyRs that can be triggered by a single LCC (e.g., 15:1 in the model with threefold increase in CaRU size). This number can be thought of as the maximal-reach of a single trigger LCC. In the model, an increase in CaRU size translates to an increase in the maximal reach of an LCC, and results in a similar increase in the slope of the EC coupling gain curve. These results suggest that the change in slope of the EC coupling gain curve from the positive potential range to the negative potential range yields information about the relative change in the number of RyRs activated per active LCC. For example, for the CaRU increased in size by threefold (Fig. 5 A, dashed line), the slope of the gain curve in the range from −40 mV to −10 mV is −0.81 mV−1, whereas the slope in the range from +10 mV to +40 mV is −0.30 mV−1—and these differ by a factor of 2.7, indicating an approximately threefold change in the number of RyRs activated per LCC at negative potentials as compared to positive potentials. Unfortunately, the actual RyR/LCC stoichiometry cannot generally be derived from measures of EC coupling gain alone because the relationship between gain and stoichiometry depends upon additional unknown quantities such as single-channel Ca2+ flux rates and open probabilities for both LCCs and RyRs.
The above results indicate that the larger CaRU models better reproduce experimentally measured EC coupling gain curves, with steeply increasing gain at negative potentials (1,29). It is important to note that the 10 RyR to 2 LCC CaRU requires a 550-state model and that the 15 RyR to 3 LCC CaRU requires a 4400-state model. Clearly, these models will require significantly more computational resources than the 40-state baseline model, and can no longer be considered as reduced models of CICR. For this reason, the baseline model of CICR was chosen for implementation within the canine myocyte model, and should be sufficient for nearly all types of AP simulations. In cases where redundancy of LCC po is of interest, the CICR model can be upgraded to one containing a larger CaRU (e.g., see Fig. 7).
As in our previous canine myocyte model (18), graded SR Ca2+ release in this model is an emergent property based on the consideration of mechanistic details of the interaction of LCCs and RyRs in their local environment. Some other recent models have achieved graded SR Ca2+ release (73,74); however, this was accomplished in a phenomenological manner by formulating the RyR Ca2+ flux and gating kinetics as a complex function of ICaL, rather than solely as a function of the Ca2+ concentrations that are in the direct vicinity of the RyRs. Although such formulations can reproduce features of CICR, they are not formulated based on the known mechanisms underlying CICR and therefore would not be appropriate for the study of the detailed interaction of EC coupling processes with whole-cell Ca2+ cycling and the AP. The study of heart failure serves as one example where it is critical to accurately capture the mechanisms of EC coupling because heart-failure-related defects have been identified in a number of EC coupling proteins. Single LCCs from failing myocytes exhibit increased availability and open probability compared to nonfailing myocytes (55), consistent with the observation that macroscopic ICaL is unchanged in the face of reduced channel density (75). RyRs have been found to be hyperphosphorylated in failing hearts, resulting in increased Ca2+ sensitivity and open probability (76). In fact, alterations of any target protein of the β-adrenergic signaling pathway must be considered, given its heightened state of stimulation in heart failure. Finally, heart-failure-related alterations in proteins/currents not directly involved in CICR, such as Ito1, can have a significant impact on EC coupling properties as demonstrated in Fig. 7 and in experiments (62). These observations serve as a strong motivation to build mechanistic models of CICR to maximize their predictive ability in both normal and diseased states.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Antti Tanskanen for helpful discussions.
This work is supported by grants from the National Institutes of Health (No. RO1 HL-61711, No. RO1 HL-60133, No. RO1 HL-72488, No. P50 HL-52307, and No. N01 HV-28180); The Falk Medical Trust; The Whitaker Foundation; and IBM Corporation.
References
- 1.Wier, W. G., T. M. Egan, J. R. Lopez-Lopez, and C. W. Balke. 1994. Local control of excitation-contraction coupling in rat heart cells. J. Physiol. 474:463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cannell, M., J. Berlin, and W. Lederer. 1987. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 283:1419–1423. [DOI] [PubMed] [Google Scholar]
- 3.Nabauer, M., G. Gallewaert, L. Cleeman, and M. Morad. 1989. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. 244:800–803. [DOI] [PubMed] [Google Scholar]
- 4.Cheng, H., W. J. Lederer, and M. B. Cannell. 1993. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 262:740–744. [DOI] [PubMed] [Google Scholar]
- 5.Lopez-Lopez, J. R., P. S. Shacklock, C. W. Balke, and W. G. Wier. 1994. Local stochastic release of Ca2+ in voltage-clamped rat heart cells: visualization with confocal microscopy. J. Physiol. (Lond.). 480:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stern, M. D. 1992. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 63:497–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bers, D. M. 1993. Excitation Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Press, Boston, MA.
- 8.Sham, J. S. K. 1997. Ca2+ release-induced inactivation of Ca2+ current in rat ventricular myocytes: evidence for local Ca2+ signalling. J. Physiol. 500:285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wier, W. G., and C. W. Balke. 1999. Ca2+ release mechanisms, Ca2+ sparks, and local control of excitation-contraction coupling in normal heart muscle. Circ. Res. 85:770–776. [DOI] [PubMed] [Google Scholar]
- 10.Winslow, R. L., J. J. Rice, M. S. Jafri, E. Marban, and B. O'Rourke. 1999. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. II. Model studies. Circ. Res. 84:571–586. [DOI] [PubMed] [Google Scholar]
- 11.Jafri, M. S., J. J. Rice, and R. L. Winslow. 1998. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 74:1149–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandit, S. V., R. B. Clark, W. R. Giles, and S. S. Demir. 2001. A mathematical model of action potential heterogeneity in adult rat left ventricular myocytes. Biophys. J. 81:3029–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noble, D., A. Varghese, P. Kohl, and P. Noble. 1998. Improved Guinea-pig ventricular cell model incorporating a dyadic space, IKr and IKs, and length- and tension-dependent processes. Can. J. Cardiol. 14:123–134. [PubMed] [Google Scholar]
- 14.Iyer, V., R. Mazhari, and R. L. Winslow. 2004. A computational model of the human left-ventricular epicardial myocyte. Biophys. J. 87:1507–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson, B., C. DeMaria, J. Adelman, and D. Yue. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 22:549–558. [DOI] [PubMed] [Google Scholar]
- 16.Alseikhan, B. A., C. D. DeMaria, H. M. Colecraft, and D. T. Yue. 2002. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc. Natl. Acad. Sci. USA. 99:17185–17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linz, K. W., and R. Meyer. 1998. Control of L-type calcium current during the action potential of Guinea-pig ventricular myocytes. J. Physiol. (Lond.). 513:425–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenstein, J. L., and R. L. Winslow. 2002. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys. J. 83:2918–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winslow, R. L., J. L. Greenstein, G. F. Tomaselli, and B. O'Rourke. 2001. Computational models of the failing myocyte: relating altered gene expression to cellular function. Philos. Trans. R. Soc. Lond. A. 359:1187–1200. [Google Scholar]
- 20.Luo, C. H., and Y. Rudy. 1994. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74:1071–1096. [DOI] [PubMed] [Google Scholar]
- 21.Priebe, L., and D. J. Beuckelmann. 1998. Simulation study of cellular electric properties in heart failure. Circ. Res. 82:1206–1223. [DOI] [PubMed] [Google Scholar]
- 22.Faber, G. M., and Y. Rudy. 2000. Action potential and contractility changes in [Na+]i overloaded cardiac myocytes: a simulation study. Biophys. J. 78:2392–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanskanen, A. J., J. L. Greenstein, B. O'Rourke, and R. L. Winslow. 2005. The role of stochastic and modal gating of cardiac l-type ca2+ channels on early after-depolarizations. Biophys. J. 88:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinch, R., J. L. Greenstein, A. J. Tanskanen, L. Xu, and R. L. Winslow. 2004. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys. J. 87:3723–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sipido, K., G. Callewaert, and E. Carmeliet. 1995. Inhibition and rapid recovery of Ca2+ during Ca2+ release from sarcoplasmic reticulum in Guinea pig ventricular myocytes. Circ. Res. 76:102–109. [DOI] [PubMed] [Google Scholar]
- 26.Sham, J., L.-S. Song, Y. Chien, L.-H. Deng, M. D. Stern, E. G. Lakatta, and H. Cheng. 1998. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc. Natl. Acad. Sci. USA. 95:15096–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cannell, M. B., H. Cheng, and W. J. Lederer. 1995. The control of calcium release in heart muscle. Science. 268:1045–1049. [DOI] [PubMed] [Google Scholar]
- 28.Janczewski, A. M., H. A. Spurgeon, M. D. Stern, and E. G. Lakatta. 1995. Effects of sarcoplasmic reticulum Ca2+ load on the gain function of Ca2+ release by Ca2+ current in cardiac cells. Am. J. Physiol. 268:H916–H920. [DOI] [PubMed] [Google Scholar]
- 29.Song, L.-S., S.-Q. Wang, R.-P. Xiao, H. Spurgeon, E. G. Lakatta, and H. Cheng. 2001. β-Adrenergic stimulation synchronizes intracellular Ca2+ release during excitation-contraction coupling in cardiac myocytes. Circ. Res. 88:794–801. [DOI] [PubMed] [Google Scholar]
- 30.O'Rourke, B., D. A. Kass, G. F. Tomaselli, S. Kaab, R. Tunin, and E. Marban. 1999. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. I. Experimental studies. Circ. Res. 84:562–570. [DOI] [PubMed] [Google Scholar]
- 31.Liu, D.-W., G. A. Gintant, and C. Antzelevitch. 1993. Ionic basis for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circulation. 72:671–687. [DOI] [PubMed] [Google Scholar]
- 32.Altamirano, J., and D. M. Bers. 2004. Cardiac E-C coupling gain: roles of unitary ICa and open probability. Biophys. J. 86:224a (Abstract).14695264 [Google Scholar]
- 33.Sah, R., R. J. Ramirez, and P. H. Backx. 2002. Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate. Role of phase-1 action potential repolarization in excitation-contraction coupling. Circ. Res. n press. [DOI] [PubMed]
- 34.Wang, S. Q., L. S. Song, E. G. Lakkata, and H. Cheng. 2001. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 410:592–596. [DOI] [PubMed] [Google Scholar]
- 35.Franzini-Armstrong, C., F. Protasi, and V. Ramesh. 1999. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 77:1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keizer, J., and G. D. Smith. 1998. Spark-to-wave transition: saltatory transmission of calcium waves in cardiac myocytes. Biophys. Chem. 72:87–100. [DOI] [PubMed] [Google Scholar]
- 37.Rice, J. J., M. S. Jafri, and R. L. Winslow. 1999. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac dyadic space. Biophys. J. 77:1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hobai, I. A., and B. O'Rourke. 2001. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 103:1577–1584. [DOI] [PubMed] [Google Scholar]
- 39.Rose, W. C., C. W. Balke, W. G. Wier, and E. Marban. 1992. Macroscopic and unitary properties of physiological ion flux through L- type Ca2+ channels in Guinea-pig heart cells. J. Physiol. (Lond.). 456:267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herzig, S., P. Patil, J. Neumann, C.-M. Staschen, and D. Yue. 1993. Mechanisms of β-adrenergic stimulation of cardiac Ca2+ channels revealed by discrete-time Markov analysis of slow gating. Biophys. J. 65:1599–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Handrock, R., F. Schroder, S. Hirt, A. Haverich, C. Mittmann, and S. Herzig. 1998. Single-channel properties of L-type calcium channels from failing human ventricle. Cardiovasc. Res. 37:445–455. [DOI] [PubMed] [Google Scholar]
- 42.Peterson, B. Z., J. S. Lee, J. G. Mulle, Y. Wang, M. d. Leon, and D. T. Yue. 2000. Critical determinants of Ca2+-dependent inactivation within an EF-hand motif of L-type Ca2+ channels. Biophys. J. 78:1906–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yue, D. T., and E. Marban. 1990. Permeation in the dihydropyridine-sensitive calcium channel. Multi-ion occupancy but no anomalous mole-fraction effect between Ba2+ and Ca2+. J. Gen. Physiol. 95:911–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelder, J., and R. Mead. 1965. A simplex method for function minimization. Comput. J. 7:308–315. [Google Scholar]
- 45.McDonald, T. F., A. Cavalie, W. Trautwein, and D. Pelzer. 1986. Voltage-dependent properties of macroscopic and elementary calcium channel currents in Guinea pig ventricular myocytes. Pflugers Arch. 406:437–448. [DOI] [PubMed] [Google Scholar]
- 46.Zahradnikova, A., I. Zahradnik, I. Gyorke, and S. Gyorke. 1999. Rapid activation of the cardiac ryanodine receptor by submillisecond calcium stimuli. J. Gen. Physiol. 114:787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shannon, T. R., T. Guo, and D. M. Bers. 2003. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 93:40–45. [DOI] [PubMed] [Google Scholar]
- 48.Sobie, E. A., K. W. Dilly, J. dos Santos Cruz, W. J. Lederer, and M. S. Jafri. 2002. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys. J. 83:59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hinch, R. 2004. A mathematical analysis of the generation and termination of calcium sparks. Biophys. J. 86:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sipido, K. R., P. G. A. Volders, S. H. M. de Groot, F. Verdonck, F. Van de Werf, H. J. J. Wellens, and M. A. Vos. 2000. Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation. 102:2137–2144. [DOI] [PubMed] [Google Scholar]
- 51.Kääb, S., H. B. Nuss, N. Chiamvimonvat, B. O'Rourke, P. H. Pak, D. A. Kass, E. Marban, and G. F. Tomaselli. 1996. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ. Res. 78:262–273. [DOI] [PubMed] [Google Scholar]
- 52.He, J.-Q., M. Conklin, J. Foell, M. Wolff, R. Haworth, R. Coronado, and T. Kamp. 2001. Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc. Res. 49:298–307. [DOI] [PubMed] [Google Scholar]
- 53.Hadley, R. W., and J. R. Hume. 1987. An intrinsic potential-dependent inactivation mechanism associated with calcium channels in Guinea-pig myocytes. J. Physiol. 389:205–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yue, D. T., S. Herzig, and E. Marban. 1990. β-Adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proc. Natl. Acad. Sci. USA. 87:753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schroder, F., R. Handrock, D. J. Beuckelmann, S. Hirt, R. Hullin, L. Priebe, R. H. Schwinger, J. Weil, and S. Herzig. 1998. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 98:969–976. [DOI] [PubMed] [Google Scholar]
- 56.Zahradnikova, A., M. Dura, I. Gyorke, A. L. Escobar, I. Zahradnik, and S. Gyorke. 2003. Regulation of dynamic behavior of cardiac ryanodine receptor by Mg2+ under simulated physiological conditions. Am. J. Physiol. Cell Physiol. 285:C1059–C1070. [DOI] [PubMed] [Google Scholar]
- 57.Santana, L. F., H. Cheng, A. M. Gomez, M. B. Cannell, and W. J. Lederer. 1996. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ. Res. 78:166–171. [DOI] [PubMed] [Google Scholar]
- 58.Stern, M. D., L. S. Song, H. Cheng, J. S. Sham, H. T. Yang, K. R. Boheler, and E. Rios. 1999. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 113:469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marx, S. O., J. Gaburjakova, M. Gaburjakova, C. Henrikson, K. Ondrias, and A. R. Marks. 2001. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ. Res. 88:1151–1158. [DOI] [PubMed] [Google Scholar]
- 60.Liu, D. W., and C. Antzelevitch. 1995. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial cells: a weaker IKs contributes to the longer action potential of the M cell. Circ. Res. 76:351–365. [DOI] [PubMed] [Google Scholar]
- 61.Greenstein, J. L., R. Wu, S. Po, G. F. Tomaselli, and R. L. Winslow. 2000. Role of the calcium-independent transient outward current Ito1 in shaping action potential morphology and duration. Circ. Res. 87:1026–1033. [DOI] [PubMed] [Google Scholar]
- 62.Sah, R., R. J. Ramirez, G. Y. Oudit, D. Gidrewicz, M. G. Trivieri, C. Zobel, and P. H. Backx. 2003. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (Ito). J. Physiol. (Lond.). 546:5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bondarenko, V. E., G. C. L. Bett, and R. L. Rasmusson. 2004. A model of graded calcium release and L-type Ca2+ channel inactivation in cardiac muscle. Am. J. Physiol. Heart Circ. Physiol. 286:H1154–H1169. [DOI] [PubMed] [Google Scholar]
- 64.Saftenku, E., A. J. Williams, and R. Sitsapesan. 2001. Markovian models of low and high activity levels of cardiac ryanodine receptors. Biophys. J. 80:2727–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shannon, T. R., F. Wang, J. Puglisi, C. Weber, and D. M. Bers. 2004. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys. J. 87:3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gyorke, I., and S. Gyorke. 1998. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys. J. 75:2801–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Despa, S., F. Brette, C. H. Orchard, and D. M. Bers. 2003. Na/Ca exchange and Na/K-ATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys. J. 85:3388–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kawai, M., M. Hussain, and C. H. Orchard. 1999. Excitation-contraction coupling in rat ventricular myocytes after formamide-induced detubulation. Am. J. Physiol. Heart Circ. Physiol. 277:H603–H609. [DOI] [PubMed] [Google Scholar]
- 69.Scriven, D. R. L., P. Dan, and E. D. W. Moore. 2000. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys. J. 79:2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Armoundas, A. A., I. A. Hobai, G. F. Tomaselli, R. L. Winslow, and B. O'Rourke. 2003. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ. Res. 93:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lopez-Lopez, J. R., P. S. Shacklock, C. W. Balke, and W. G. Wier. 1995. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 268:1042–1045. [DOI] [PubMed] [Google Scholar]
- 72.Weber, C. R., V. Piacentino III, K. S. Ginsburg, S. R. Houser, and D. M. Bers. 2002. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ. Res. 90:182–189. [DOI] [PubMed] [Google Scholar]
- 73.Fox, J. J., J. L. McHarg, and R. F. Gilmour, Jr. 2002. Ionic mechanism of electrical alternans. Am. J. Physiol. Heart Circ. Physiol. 282:H516–H530. [DOI] [PubMed] [Google Scholar]
- 74.Hund, T. J., and Y. Rudy. 2004. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 110:3168–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen, X., V. Piacentino III, S. Furukawa, B. Goldman, K. B. Margulies, and S. R. Houser. 2002. L-Type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 91:517–524. [DOI] [PubMed] [Google Scholar]
- 76.Marx, S. O., S. Reiken, Y. Hisamatsu, T. Jayaraman, D. Burkhoff, N. Rosemblit, and A. R. Marks. 2000. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 101:365–376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.