

Abstract
We have studied the effect of point mutations of the primary binding residue (P1) at the protein-protein interface in complexes of chymotrypsin and elastase with the third domain of the turkey ovomucoid inhibitor and in trypsin with the bovine pancreatic trypsin inhibitor, using molecular dynamics simulations combined with the linear interaction energy (LIE) approach. A total of 56 mutants have been constructed and docked into their host proteins. The free energy of binding could be reliably calculated for 52 of these mutants that could unambiguously be fitted into the binding sites. We find that the predicted binding free energies are in very good agreement with experimental data with mean unsigned errors between 0.50 and 1.03 kcal/mol. It is also evident that the standard LIE model used to study small drug-like ligand binding to proteins is not suitable for protein-protein interactions. Three different LIE models were therefore tested for each of the series of protein-protein complexes included, and the best models for each system turn out to be very similar. The difference in parameterization between small drug-like compounds and protein point mutations is attributed to the preorganization of the binding surface. Our results clearly demonstrate the potential of free energy calculations for probing the effect of point mutations at protein-protein interfaces and for exploring the principles of specificity of hot spots at the interface.
INTRODUCTION
Protein-protein interactions play essential roles in many biological processes such as enzyme regulation, signal transduction, and immune response. Understanding how protein-protein complexes form and what determines their specificity is not only fundamental to understanding these biological processes but also helpful in inhibitor design. To date, much work has been done to develop computational procedures to predict and evaluate these interactions, and the efforts devoted to protein-protein docking are extensive (for reviews, see, e.g., Halperin et al. (1) or Smith and Sternberg (2)). Typically, these methods utilize surface complementarity, buried surface area, atomic solvation parameters, continuum models, or a combination of the above to find the best docking pose. Once the interaction mode is predicted or known, it is of interest to examine what determines the specificity of the interaction. From alanine-scanning experiments, it has been determined that typically the interface consists of a hot spot of residues that accounts for a major part of the binding free energy (3). Site-directed mutagenesis of these hot spot residues can then give some insight into the factors that determine specificity. Several computational techniques exist that can be used to predict relative binding free energies of protein-protein association, including free energy perturbation (FEP) (see, e.g., Brandsdal et al. (4) or Kollman (5)), linear interaction energy (LIE) (6,7) and MM-PBSA (molecular mechanics-Poisson-Boltzmann surface area) (8) methods as well as those based solely on buried surface area in combination with atomic solvation parameters (9). Although FEP and other similar computer simulation techniques, such as thermodynamic integration, offer a rigorous statistical mechanical way to calculate free energies of binding, they suffer from long computational times and technical difficulties associated with creation and annihilation of atoms. If only the relative binding free energies of a set of mutants is required, and the structural differences between the mutants are small, FEP may nevertheless be a viable method. MM-PBSA approximates free energies using a Poisson-Boltzmann continuum representation of the solvent together with a surface-area-dependent term and molecular mechanics energies using snapshots from MD (molecular dynamics) simulations generated with explicit solvent. This method has been applied to predict, e.g., the relative stabilities of A-DNA and B-DNA (8) and effects of alanine mutations on protein-protein interactions (10). Although the MM-PBSA approach has several appealing features compared to the more rigorous FEP/TI methods, particularly when dealing with binding of diverse sets of ligands that are structurally and chemically different, the free energies of binding are usually computed based on simulations of only the bound state. Hence, the normal implementation of this method assumes that the structure of the receptor and the ligand does not change upon binding. Another problem associated with the MM-PBSA approach is how to determine the contribution from entropy changes involved in binding. If absolute free energies are required the entropic contribution must be determined to yield meaningful results. This is extremely difficult since conformational fluctuations generally change upon binding, and for relative binding free energies the entropic term is often assumed to cancel (11), which is still a severe approximation. Courageous attempts have been made by Gohlke and Case (12) to evaluate all formally relevant MM-PBSA terms for the Ras-Raf complex, which clearly illustrates the magnitude of the convergence problems.
The LIE method was originally developed to predict binding of drug-size ligands to proteins using MD or MC (Monte Carlo) simulations. It relies on the electrostatic linear response approximation to predict the electrostatic contribution to the binding free energy and an empirical scaling of the intermolecular van der Waals (Lennard-Jones) energies to predict the nonpolar contribution to the binding free energy according to
 |
(1) |
where the  's denote MD or MC averages of the ligand-surrounding van der Waals or electrostatic energies, and the Δ's denote the difference between these averages in the bound and free states. The electrostatic energies are scaled by a ligand-dependent parameter, β, which can take on a few different values in the range 0.33–0.50 (13). The van der Waals energies are scaled by an empirically determined coefficient, α. In studies performed previously in our laboratory, an α of 0.18 has reproduced the binding free energies of various ligand-protein systems well, and it has been shown that α is neither system-dependent nor force-field-dependent for the ligand-protein systems and force fields examined to date (14). γ is a constant term reflecting binding-site hydrophobicity that may be required to reproduce absolute binding free energies (14), but is not necessary for relative binding free energies.
's denote MD or MC averages of the ligand-surrounding van der Waals or electrostatic energies, and the Δ's denote the difference between these averages in the bound and free states. The electrostatic energies are scaled by a ligand-dependent parameter, β, which can take on a few different values in the range 0.33–0.50 (13). The van der Waals energies are scaled by an empirically determined coefficient, α. In studies performed previously in our laboratory, an α of 0.18 has reproduced the binding free energies of various ligand-protein systems well, and it has been shown that α is neither system-dependent nor force-field-dependent for the ligand-protein systems and force fields examined to date (14). γ is a constant term reflecting binding-site hydrophobicity that may be required to reproduce absolute binding free energies (14), but is not necessary for relative binding free energies.
The initial idea behind the LIE method was to consider the binding free energy as the change in solvation energy of the ligand when it is transferred from solution to the binding site of the receptor:
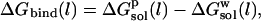 |
(2) |
where l represents a ligand and p and w denote protein and water, respectively. This process can formally be considered to consist of two separate steps: 1), creation of a van der Waals cavity in the given environment; and 2), turning on the electrostatic interactions between the ligand and its surroundings (6). The electrostatic part of the solvation/binding free energies is treated with the linear response approximation, and the linear response result for solvation in a given environment is (see, e.g., Åqvist et al. (6) or Lee and Warshel (15)):
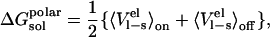 |
(3) |
where the 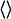 values are MD (or MC) averages sampled with the electrostatic interactions between the ligand and its surroundings (solvated protein or water) turned on and off, respectively. The latter average formally requires an extra simulation of an unphysical “nonpolar ligand” state to be carried out, and a central approximation in the LIE approach is that the term
values are MD (or MC) averages sampled with the electrostatic interactions between the ligand and its surroundings (solvated protein or water) turned on and off, respectively. The latter average formally requires an extra simulation of an unphysical “nonpolar ligand” state to be carried out, and a central approximation in the LIE approach is that the term 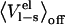 can be neglected, which has been found to hold well in water (13). The
can be neglected, which has been found to hold well in water (13). The 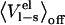 term is often thought of as an electrostatic preorganization term. It is not surprising that this term is zero in water, since the orientations of the water molecules relative to a completely nonpolar solute will be largely random, i.e., the water molecules are not preorganized with respect to the solute electrostatic potential. However, in the binding site of an enzyme, for instance, the backbone and side chains of the amino acids that make up the binding site are most likely not randomly oriented. The extent of this nonrandomness and how much the binding site relaxes upon binding/unbinding of a ligand will greatly affect the size of
term is often thought of as an electrostatic preorganization term. It is not surprising that this term is zero in water, since the orientations of the water molecules relative to a completely nonpolar solute will be largely random, i.e., the water molecules are not preorganized with respect to the solute electrostatic potential. However, in the binding site of an enzyme, for instance, the backbone and side chains of the amino acids that make up the binding site are most likely not randomly oriented. The extent of this nonrandomness and how much the binding site relaxes upon binding/unbinding of a ligand will greatly affect the size of 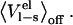
We have previously applied the LIE approach to study the effect of point mutations of the primary binding residue (P1) of bovine pancreatic trypsin inhibitor (BPTI) bound to trypsin (16). The complexes between serine proteinases and their canonical protein inhibitors are excellent systems to test protein-protein recognition models on as there is plenty of experimental binding data on different P1 mutations, and the P1 residue has been found to be responsible for up to 70% of the total association energy (17). By treating the P1 residue as the ligand in the LIE method the effect of mutating it to other amino acids can be estimated. In this study, we report the successful prediction of binding free energies for several mutants of BPTI to trypsin and several mutants of the third domain of the turkey ovomucoid inhibitor (OMTKY3) to elastase and chymotrypsin using the LIE method together with MD simulations. We have further tried to push the computational efficiency by using relatively small simulation systems and short simulations (∼250 ps), while requiring convergence errors in 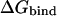 to be <±1.0 kcal/mol. Our previous calculations dealing with P1 mutations of BPTI bound to trypsin used
to be <±1.0 kcal/mol. Our previous calculations dealing with P1 mutations of BPTI bound to trypsin used 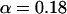 (16). In this study, we have expanded the data set used in our previous study (13 mutations on trypsin-BPTI), which now includes 56 mutants of three different proteinase-protein inhibitor complexes. Our previous calculations also estimated the effect of point mutations relative to P1 Gly, for which the absolute binding energy turns out to have been an outlier, affecting the obtained value for α. Instead of relying on a single reference state we now estimate the absolute binding free energy for each variant. The predicted free energies of binding are in excellent agreement with experiments with average unsigned errors of 0.74, 0.50, and 1.03 kcal/mol for chymotrypsin, elastase, and trypsin, respectively.
(16). In this study, we have expanded the data set used in our previous study (13 mutations on trypsin-BPTI), which now includes 56 mutants of three different proteinase-protein inhibitor complexes. Our previous calculations also estimated the effect of point mutations relative to P1 Gly, for which the absolute binding energy turns out to have been an outlier, affecting the obtained value for α. Instead of relying on a single reference state we now estimate the absolute binding free energy for each variant. The predicted free energies of binding are in excellent agreement with experiments with average unsigned errors of 0.74, 0.50, and 1.03 kcal/mol for chymotrypsin, elastase, and trypsin, respectively.
METHODS
Complexes between bovine β-trypsin and BPTI, and human leukocyte elastase (HLE) and α-chymotrypsin both in complex with OMTKY3, were subjected to MD simulations with the software package Q (18) and the OPLS-AA force field (19). Substitution of the primary binding residue with the remaining natural amino acids gives a total of 20 different complexes for each system. In the case of trypsin-BPTI, 10 different x-ray structures differing only in the nature of the amino acid at the P1 position were available (20), whereas the remaining complexes were constructed by manually changing the P1 residue using the crystallographic software O (21). The series for HLE and chymotrypsin were generated starting from the crystal structures with Protein Data Bank entries 1PPF (22) and 1CHO (23), respectively, and manual adjustments of the P1 side chain, again using O. Complexes in which the P1 residue is proline were omitted in all three series as this residue imposes major changes in the orientation of the backbone. Thus, a total of 19 different complexes were investigated with MD simulations for each enzyme. In the case of OMTKY3 P1 Tyr binding to HLE, the MD simulations were not completed, as it was not possible to obtain a minimized structure without steric clashes at the S1 site of the complex, even though several orientations of the P1 side chain were tested. Hence, 18 MD simulations were carried out for HLE.
The Cα-atom of the P1 residue was defined as the center of a 16-Å sphere for which unrestrained MD simulations were carried out. Atoms in the outermost 3 Å were weakly restrained (harmonic restraint of 5 kcal/mol Å2) to their crystallographic positions, whereas atoms outside the 16-Å sphere were strongly restrained (harmonic restraint of 200 kcal/mol Å2). Nonbonded interactions across the boundary were excluded. The nonbonded potential was truncated at 10 Å while long-range electrostatics were treated using a multipole expansion (24). The interactions involving the P1 residue were not truncated and thus were allowed to interact with the entire simulation sphere. Crystallographic water molecules closer than 12 Å from the reaction center were retained, and additional solvent molecules were added to fill the 16-Å sphere. Water molecules were described using the TIP3P model (25). All systems were heated from 1 to 300 K using a stepwise scheme followed by an equilibration period to stabilize ligand-surrounding energies before the data collection phase. During the heating phase, heavy atoms (protein and crystallographic water) were restrained to the initial positions using a harmonic potential with a force constant of 5 kcal/(mol Å2). The timestep used in the production phase of the simulations was 1.5 fs, and the temperature was set to 300 K using a weak coupling to an external bath. SHAKE (26) was used to constrain bonds and angles on solvent molecules. The total simulation time for the production period was 225 ps for most simulations. Energies were sampled every fifth step in the production part of the simulations. These energies were then used in Eq. 1 to obtain the free energy of binding. The convergence of the simulations is judged by dividing the trajectory in two halves and then calculating the free energy for each of these two halves. If this energy differs by >2.0 kcal/mol, the simulation was further extended (up to 750 ps in a few cases). This also provides an estimate of the convergence error in the computed binding energies, which is on average <1.0 kcal/mol.
To avoid possible Born terms entering into the calculated free energies the net charge, excluding the P1 residue, within the simulation sphere in the complex and the free inhibitors is set to zero. This is done by using the neutral form for ionic residues that are close to the boundary and those outside the simulation sphere as described previously (16,27). Alternatively, counterions can be added to neutralize these ionizable residues. This may, however, slow down the convergence of the free energy calculations as such ions are often highly mobile in the simulations. The contribution from the ionic residues described as neutral is then added to the computed binding energy by using a Coulombic potential with a high dielectric constant (28). This contribution can also be estimated by using continuum electrostatic calculations, but this has been found to yield results virtually identical to those produced using a Coulombic potential with a high dielectric constant (27).
The LIE optimizations are performed by calculating the values of the coefficients that are considered as free, which minimize the root mean square deviation between the calculated and the experimental binding energies. The coefficient of multiple determination, R2, is a measure of the overall fit of the model and is calculated as:  where SSE is the square sum of errors
where SSE is the square sum of errors 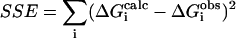 and SST is the total sum of squares
and SST is the total sum of squares  To assess overfitting, the cross-validated statistical figure of merit
To assess overfitting, the cross-validated statistical figure of merit 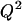 was also calculated.
was also calculated. 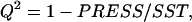 where PRESS is the predictive residual sum of squares:
where PRESS is the predictive residual sum of squares: 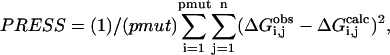 where pmut is the number of permutations of leaving n ligands out of the parameterization.
where pmut is the number of permutations of leaving n ligands out of the parameterization. 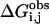 and
and  are the observed and calculated binding free energies of the j'th ligand in the i'th permutation. In this study the number of leave-outs was set to 3. The cross-validated standard deviation,
are the observed and calculated binding free energies of the j'th ligand in the i'th permutation. In this study the number of leave-outs was set to 3. The cross-validated standard deviation,  explicitly takes into account the number of free parameters and is calculated according to:
explicitly takes into account the number of free parameters and is calculated according to: 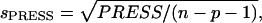 where n is the data set size, and p is the number of optimized parameters in the model.
where n is the data set size, and p is the number of optimized parameters in the model.
RESULTS AND DISCUSSION
X-ray analysis of complexes between serine proteinases and their canonical protein inhibitors (20,29) shows that even though the interactions in the principal interaction zone change in response to the nature of the amino acid at the P1 site, the secondary interactions are virtually identical and independent of the P1 residue. This means that for a given series of complexes differing only in the P1 position of the protein inhibitor, the free energy contribution from the secondary subsites will be similar. Within the LIE framework, the P1 residue is treated as a “ligand”, and its interactions with the surroundings are measured in the bound and unbound states, and the rest of the protein inhibitor is considered as part of the surroundings. The main advantage of using this strategy is that the interaction energies of the P1 residue converge quite rapidly in the MD simulations, which is of course not the case if the entire protein inhibitor is treated as a ligand. Calculation of absolute protein-protein binding energies with the LIE approach would require that one obtains convergent values of the entire interaction energies, and these quantities can be in the order of several thousand kcal/mol. This would require extremely long simulations to get stable averages and result in an inefficient approach with corresponding high uncertainties in the energetics. However, because the secondary interactions are assumed to be identical for all the 20 possible complexes between a given proteinase and a protein inhibitor where only the P1 residue is different, only a constant term (γ) is needed to account for the contribution from the secondary binding sites to the free energy.
Many recent studies of ligand binding (30–36) have determined that a value of 0.18 for α adequately reproduces the free energies of binding for a variety of ligand-protein systems. In those systems the ligands were primarily of drug-like character, particularly their size. That is not the case with this study, where the ligand is a small protein, and hence the standard parameterization with α = 0.18 turns out not to hold. To examine other possible LIE models, two new parameterizations were investigated: model A uses the β-coefficients from Hansson et al. (31) assuming a value of either 0.37, 0.43, or 0.50, depending on the chemical nature of the P1 residue, while α and γ are optimized; in model B, all three parameters are freely optimized. Model C is the standard LIE model, where α = 0.18, β is ligand-dependent (as in model A), and γ reproduces the secondary interactions.
The calculated average ligand-surrounding interaction energies from the MD simulations of the α-chymotrypsin, human leukocyte elastase, and bovine trypsin systems are shown in Tables 1–3. Based on these energies, the three different LIE models were tested (Table 4) and their statistical figures of merit calculated. As is seen by the statistical figures of merit, model C fails for all three systems, whereas models A and B perform very well. Overall, the model that fares best is model A, with only α and γ as free parameters. For chymotrypsin, elastase, and trypsin the values of α necessary to reproduce experimental results are 0.63, 0.56, and 0.54, respectively. Fig. 1 shows the observed versus calculated experimental binding free energies of the three systems using model A. Out of the total 56 complexes, four elastase complexes with large P1 variants that do not fit into the crystallographic S1 binding pocket were excluded in the statistical analysis and these cases are further discussed below.
TABLE 1.
Ligand-surrounding interaction energies from the MD simulations of P1 variants of OMTKY3 free in solution and when bound to chymotrypsin
| Chymotrypsin-OMTKY3
|
OMTKY3
|
|||||
|---|---|---|---|---|---|---|
| P1 residue |  |
 |
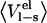 |
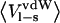 |
 * *
|
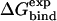 † †
|
| Gly | −69.8 | −4.8 | −70.1 | −3.2 | −8.8 | −9.1 |
| Ala | −67.5 | −8.0 | −69.3 | −4.9 | −9.1 | −10.4 |
| Ser | −80.0 | −11.0 | −84.3 | −3.6 | −11.0 | −10.2 |
| Cys | −78.2 | −13.0 | −77.4 | −5.7 | −12.9 | −12.6 |
| Val | −70.7 | −15.5 | −69.6 | −8.9 | −12.3 | −10.9 |
| Thr | −81.4 | −11.2 | −83.0 | −5.6 | −10.9 | −10.7 |
| Leu | −71.0 | −21.7 | −71.3 | −10.7 | −14.8 | −15.1 |
| Ile | −65.0 | −22.0 | −70.4 | −10.6 | −12.8 | −10.7 |
| Met | −75.6 | −20.9 | −78.0 | −10.2 | −13.7 | −14.8 |
| Asn | −82.9 | −17.4 | −91.8 | −6.5 | −11.0 | −11.8 |
| Asp0‡ | −85.7 | −16.1 | −85.6 | −6.3 | −8.4 | −8.0 |
| Lys0‡ | −80.1 | −21.7 | −83.3 | −9.8 | −10.9 | −10.7 |
| Gln | −84.1 | −21.9 | −92.7 | −8.2 | −12.9 | −12.2 |
| Glu0‡ | −84.5 | −19.3 | −86.3 | −7.9 | −9.4 | −8.6 |
| His | −84.1 | −23.1 | −94.5 | −8.9 | −12.4 | −12.2 |
| Phe | −73.7 | −26.9 | −74.9 | −13.6 | −15.8 | −16.6 |
| Arg0‡ | −107.4 | −18.1 | −104.7 | −7.8 | −9.9 | −11.2 |
| Tyr | −84.0 | −27.8 | −86.1 | −11.9 | −17.2 | −17.3 |
| Trp | −78.7 | −31.6 | −83.6 | −15.8 | −15.8 | −16.8 |
Values are given in kcal/mol.
Energies calculated according to 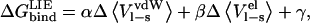 and the error in these values is <1.0 kcal/mol. The coefficients used are from Model A in Table 4.
and the error in these values is <1.0 kcal/mol. The coefficients used are from Model A in Table 4.
Experimental binding data from Lu et al. (38).
0 indicates a neutral side chain.  has been corrected with the free energy required to protonate/deprotonate the ionic side chain according to
has been corrected with the free energy required to protonate/deprotonate the ionic side chain according to 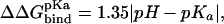 where pH is 8.3 (corresponding to the pH used in the association measurements) and pKa is 12.5, 10.7, 4.5, and 4.0 for Arg(5.7 kcal/mol), Lys(3.2 kcal/mol), Glu(5.1 kcal/mol), and Asp(5.8 kcal/mol), respectively.
where pH is 8.3 (corresponding to the pH used in the association measurements) and pKa is 12.5, 10.7, 4.5, and 4.0 for Arg(5.7 kcal/mol), Lys(3.2 kcal/mol), Glu(5.1 kcal/mol), and Asp(5.8 kcal/mol), respectively.
TABLE 2.
Ligand-surrounding interaction energies from the MD simulations of P1 variants of OMTKY3 free in solution and when bound to elastase
| Elastase-OMTKY3
|
OMTKY3
|
|||||
|---|---|---|---|---|---|---|
| P1 residue |  |
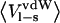 |
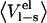 |
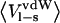 |
 * *
|
 † †
|
| Gly | −69.7 | −5.7 | −70.1 | −3.2 | −10.3 | −9.8 |
| Ala | −70.1 | −9.5 | −69.3 | −4.9 | −12.0 | −12.1 |
| Ser | −82.3 | −9.3 | −84.3 | −3.6 | −11.5 | −10.1 |
| Cys | −78.6 | −11.3 | −77.4 | −5.7 | −12.7 | −13.2 |
| Val | −67.4 | −16.9 | −69.6 | −8.9 | −12.6 | −13.6 |
| Thr | −77.3 | −14.9 | −83.0 | −5.6 | −12.1 | −12.2 |
| Leu | −71.1 | −16.9 | −71.3 | −10.7 | −12.4 | −13.1 |
| Ile | −68.3 | −20.0 | −70.4 | −10.6 | −13.4 | −13.8 |
| Met | −72.2 | −19.0 | −78.0 | −10.2 | −11.5 | −11.9 |
| Asn | −75.7 | −18.0 | −91.8 | −6.5 | −8.6 | −8.0 |
| Asp0‡ | −77.2 | −15.3 | −85.6 | −6.3 | −5.2 | −5.6 |
| Lys0‡ | −74.9 | −19.3 | −83.3 | −9.8 | −7.6 | −7.5 |
| Gln | −82.6 | −18.5 | −92.7 | −8.2 | −10.5 | −9.9 |
| Glu0‡ | −78.3 | −18.3 | −86.3 | −7.9 | −6.8 | −6.2 |
| His | −72.0 | −22.0 | −94.5 | −8.9 | −6.7 | −6.8 |
| Phe | −69.5 | −23.4 | −74.9 | −13.6 | −12.3 | −8.0 |
| Arg0‡ | −85.4 | −20.5 | −104.7 | −7.8 | −2.4 | −6.1 |
| Tyr | ND | ND | ND | ND | ND | −6.7 |
| Trp | −71.2 | −27.2 | −83.6 | −15.8 | −10.2 | −5.7 |
Values are given in kcal/mol.
Energies are calculated according to  and the error in these values is <1.0 kcal/mol. The coefficients used are from Model A in Table 4.
and the error in these values is <1.0 kcal/mol. The coefficients used are from Model A in Table 4.
Experimental binding data from Lu et al. (38).
0 indicates neutral side chain.  has been corrected with the free energy required to protonate/deprotonate the ionic side chain according to
has been corrected with the free energy required to protonate/deprotonate the ionic side chain according to  where pH is 8.3 (corresponding to the pH used in the association measurements) and pKa is 12.5, 10.7, 4.5, and 4.0 for Arg(5.7 kcal/mol), Lys(3.2 kcal/mol), Glu(5.1 kcal/mol), and Asp(5.8 kcal/mol), respectively.
where pH is 8.3 (corresponding to the pH used in the association measurements) and pKa is 12.5, 10.7, 4.5, and 4.0 for Arg(5.7 kcal/mol), Lys(3.2 kcal/mol), Glu(5.1 kcal/mol), and Asp(5.8 kcal/mol), respectively.
TABLE 3.
Ligand-surrounding interaction energies from the MD simulations of P1 variants of BPTI free in solution and when bound to trypsin
| Trypsin-BPTI
|
BPTI
|
|||||
|---|---|---|---|---|---|---|
| P1 residue |  |
 |
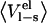 |
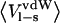 |
 * *
|
 † †
|
| Gly | −71.6 | −5.5 | −70.0 | −3.5 | −5.3 | −5.6 |
| Ala | −71.4 | −10.6 | −69.0 | −5.7 | −7.2 | −7.3 |
| Ser | −90.5 | −10.3 | −82.8 | −4.4 | −9.6 | −10.2 |
| Cys | −81.5 | −12.4 | −78.3 | −6.1 | −8.3 | ND |
| Val | −71.6 | −17.8 | −69.6 | −8.9 | −9.2 | −6.2 |
| Thr | −87.1 | −13.2 | −81.3 | −6.8 | −9.1 | −7.4 |
| Leu | −69.5 | −20.5 | −69.8 | −11.0 | −8.5 | −9.1 |
| Ile | −69.4 | −18.0 | −68.8 | −11.5 | −7.3 | −6.8 |
| Met | −78.6 | −21.1 | −77.0 | −10.9 | −9.7 | −10.3 |
| Asn | −88.5 | −17.8 | −89.4 | −7.1 | −8.9 | −9.9 |
| Asp0‡ | −87.0 | −17.0 | −80.5 | −9.5 | −4.2 | −6.5 |
| Lys§ | −146.7 | −17.9 | −132.7 | −4.1 | −17.8 | −17.9 |
| Gln | −93.5 | −20.4 | −92.2 | −9.0 | −10.2 | −8.6 |
| Glu0‡ | −96.0 | −19.4 | −86.6 | −8.3 | −7.9 | −8.5 |
| His | −84.9 | −22.6 | −93.1 | −9.9 | −6.9 | −9.2 |
| Phe | −70.8 | −28.3 | −73.1 | −14.5 | −10.0 | −10.9 |
| Arg§ | −145.3 | −12.4 | −123.4 | −3.1 | −19.3 | −18.4 |
| Tyr | −88.7 | −26.9 | −88.2 | −12.6 | −11.4 | −11.1 |
| Trp | −76.5 | −33.7 | −83.2 | −15.7 | −10.4 | −9.3 |
Values are given in kcal/mol.
Energies are calculated according to  and the error in these values is <1.0 kcal/mol. The coefficients used are from Model A in Table 4.
and the error in these values is <1.0 kcal/mol. The coefficients used are from Model A in Table 4.
Experimental binding data from Krowarsch et al. (17).
0 indicates a neutral side chain. 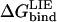 has been corrected with the free energy required to protonate/deprotonate the ionic side chain according to
has been corrected with the free energy required to protonate/deprotonate the ionic side chain according to 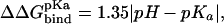 where pH is 8.3 (corresponding to the pH used in the association measurements) and pKa is 4.5 and 4.0 for Glu(5.1 kcal/mol) and Asp(5.8 kcal/mol), respectively.
where pH is 8.3 (corresponding to the pH used in the association measurements) and pKa is 4.5 and 4.0 for Glu(5.1 kcal/mol) and Asp(5.8 kcal/mol), respectively.
To compensate for ionic residues described with neutral topologies +0.2 kcal/mol has been added to the calculated binding free energies of Lys and Arg (see Methods).
TABLE 4.
Optimized LIE models for binding of P1 variants of OMTKY3 to chymotrypsin and elastase, and P1 variants of BPTI to trypsin, along with some statistical figures of merit
| Parameterization | α | β | γ (kcal/mol) | RMS (kcal/mol) | 〈|Err|〉 (kcal/mol) | R2 | Q2 | SPRESS (kcal/mol) | |
|---|---|---|---|---|---|---|---|---|---|
| A | Chymotrypsin | 0.63 | FEP | −8.0 | 0.90 | 0.74 | 0.89 | 0.86 | 1.10 |
| Elastase | 0.56 | FEP | −9.1 | 0.61 | 0.50 | 0.95 | 0.93 | 0.80 | |
| Trypsin | 0.54 | FEP | −3.6 | 1.32 | 1.03 | 0.85 | 0.82 | 1.59 | |
| B | Chymotrypsin | 0.65 | 0.49 | −7.9 | 0.87 | 0.69 | 0.90 | 0.85 | 1.16 |
| Elastase | 0.74 | 0.52 | −8.2 | 0.55 | 0.43 | 0.96 | 0.94 | 0.81 | |
| Trypsin | 0.57 | 0.44 | −3.3 | 1.30 | 1.01 | 0.85 | 0.79 | 1.76 | |
| C
|
Chymotrypsin | 0.18* | FEP | −12.5 | 1.96 | 1.72 | 0.47 | 0.41 | 2.19 |
| Elastase | 0.18* | FEP | −12.2 | 1.20 | 0.92 | 0.81 | 0.78 | 1.38 | |
| Trypsin | 0.18* | FEP | −7.1 | 1.90 | 1.69 | 0.69 | 0.65 | 2.14 | |
The α-parameter is not optimized in Model C.
FIGURE 1.

Scatter diagrams of the calculated versus the experimental binding free energy (kcal/mol) using model A for binding of P1 variants of OMTKY3 to chymotrypsin and elastase and BPTI to trypsin.
The LIE model that provides the best correlation between calculated and experimental binding free energies is virtually identical for the three enzymes studied here. Although the polar contribution is treated in the same fashion as in the previous LIE models (31), the nonpolar contribution needs to be treated with a different weight coefficient (α) than previously (∼0.58 compared to 0.18). In addition, the γ-constant that was originally set to zero is now used to obtain the interaction free energy that arises from secondary subsites through a calibration to reproduce the absolute binding free energy. This constant will of course be system-dependent and it can only be estimated when some experimental data is available. Relative binding free energies can, however, be calculated by leaving out the γ-constant, as this will always cancel out in 
Effect of point mutations
The LIE calculations presented above show that we are able to predict the outcome of point mutations at the protein-protein interface between serine proteinases and their canonical protein inhibitors. These calculations include series of 19, 18, and 19 different amino acids at the P1 position in chymotrypsin-OMTKY3, elastase-OMTKY3, and trypsin-BPTI complexes, respectively, yielding a total of 56 point mutations. These calculations show a remarkable agreement with the experimental binding data, and the amino acid preference at the P1 position is indeed very different among the three serine proteinases. Not only is the protein-protein interface complementary in terms of shape (Fig. 2), but also in terms of polar interactions. This is especially true for binding of BPTI to trypsin with the cognate P1 Lys and Arg variants. The active site of trypsin has been found to possess a negative potential (37), which is matched by the positively charged surface of BPTI. Our calculations also show that mutations of P1 Lys and Arg to Gly has a tremendous effect on the association energy, with a 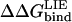 (Lys/Arg → Gly) of ∼12 kcal/mol. Chymotrypsin, on the other hand, is found to prefer large aromatic side chains (Tyr > Phe > Trp) at the S1 site, whereas acidic residues are the most unfavorable. This is also in perfect agreement with the experimental mutagenesis study of Lu et al. (38). HLE prefers Ile at the S1 site according to both experimental and calculated binding data, and again we find that acidic side chains are unfavorable. Fig. 3 shows a structural comparison of the most- and least-favored P1 variants accommodated at the S1 sites of trypsin, chymotrypsin, and elastase. From this figure it is apparent that even though the P1 side chain alters the association energies greatly, the secondary interactions, which are primarily main chain-main chain interactions, are indeed virtually identical in the most- and least-favored P1 variants for all three enzymes.
(Lys/Arg → Gly) of ∼12 kcal/mol. Chymotrypsin, on the other hand, is found to prefer large aromatic side chains (Tyr > Phe > Trp) at the S1 site, whereas acidic residues are the most unfavorable. This is also in perfect agreement with the experimental mutagenesis study of Lu et al. (38). HLE prefers Ile at the S1 site according to both experimental and calculated binding data, and again we find that acidic side chains are unfavorable. Fig. 3 shows a structural comparison of the most- and least-favored P1 variants accommodated at the S1 sites of trypsin, chymotrypsin, and elastase. From this figure it is apparent that even though the P1 side chain alters the association energies greatly, the secondary interactions, which are primarily main chain-main chain interactions, are indeed virtually identical in the most- and least-favored P1 variants for all three enzymes.
FIGURE 2.

Surface representation of the crystal structure of trypsin in complex with the P1 Trp variant of BPTI (Protein Data Bank accession code 3BTW). BPTI is shown in tan with the P1 residue colored red.
FIGURE 3.

Representative snapshot of most and least favored P1 variants bound to chymotrypsin (A and B), elastase (C and D), and trypsin (E and F). Only the binding loop of the inhibitors and the molecular surface of the proteinases are shown for clarity.
The calculations involving large side chains at the interface of OMTKY3 and HLE are, however, problematic, since the S1 site of elastase is much narrower compared to both chymotrypsin and trypsin. This is primarily due to substitution of Gly-216 in chymotrypsin and trypsin for Val-216 in elastase. Docking of large aromatic P1 side chains, such as Phe, Tyr, and Trp, into the S1 site of elastase is therefore not possible without major steric clashes. This situation already suggests that the assumption of constant secondary interactions will not hold for these types of substitutions. Experimentally it is also found that accommodation of the large aromatic side chains to the S1 site of elastase is not favorable and has a deleterious effect on the association energy relative to the P1 Gly (38). When these P1 mutants are forced into the binding site, subsequent LIE calculations indeed give a significantly too negative binding energy with the optimal model (A) for elastase. This strongly suggests that the secondary interactions at the interface are indeed weakened by the required relaxation of the S1 site when Phe, Tyr, and Trp P1 variants bind. This could perhaps also be viewed as a loss of preorganization of the binding site and will be further discussed below. Inspection of the simulated trajectories shows that the S1 site is clearly enlarged when the aromatic side chains are accommodated to elastase. The side chain of Val-216 is also tilted down into the S1 site, making the entrance to the S1 site more accessible (Fig. 4). Fig. 4 also shows the predicted binding mode for P1 Ala, and a comparison with the binding mode for P1 Trp clearly shows the enlargement of the S1 site resulting from introducing Trp into the S1 site. There is unfortunately not any experimental structure available with P1 variants with which to compare our simulation, but it is apparent that the features of the native complex are not reproduced by the simulations of large side chains at the S1 site.
FIGURE 4.

Binding of OMTKY3 P1 Trp (A) results in major structural rearrangements at the S1 site of elastase compared to accommodation of OMTKY3 P1 Ala (B). Only the binding loop of OMTKY3 and the molecular surface of HLE are shown for clarity.
Hot spots of binding tend to be clustered at the center of the interface (3,39), leading to exclusion of bulk solvent from the interacting residues. The scaffold of energetically less important residues surrounding the hot spot regions thus serves to occlude bulk solvent and provide a suitable dielectric environment. Electrostatic interactions and hydrogen bonds will increase in strength as the dielectric constant is reduced at the center of such interfaces. Ionizable side chains (His, Arg, Lys, Asp, and Glu) are generally predicted from our calculations to be accommodated in their neutral form to the S1 sites of the currently investigated proteinases, except for BPTI P1 Lys and Arg to trypsin. This is not very surprising as the binding of ionic P1 variants can be thought of as a transfer of a charge from a medium with a high dielectric constant to a medium with a low dielectric constant, which is, without taking into account specific polar interactions, thermodynamically unfavorable. The P1 residues of both OMTKY3 and BPTI are fully exposed to solvent in the protein when it is in the unbound state, and only minor changes in the pKa of these residues are observed relative to the free amino acid (40). As the free energy calculations require that the protonation state investigated is identical in the bound and unbound states, and the P1 Arg, Lys, Asp, and Glu side chains are expected to be predominantly ionized in the free inhibitor, the calculations with a neutral form of these P1 variants need to be corrected by the free energy required to protonate/deprotonate the side chain of the P1 residue. The free energy of protonation/deprotonation is added to the calculated 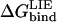 according to
according to  where pH is 8.3 and the pKa of the free amino acid side chain is used. The binding free energy of all the ionizable P1 variants, except for P1 Arg bound to HLE, are in very good agreement with experimental data. Our calculations underestimate accommodation of OMTKY3 P1 Arg to elastase by 3.7 kcal/mol, which is most likely related to the different deprotonation sites on the P1 Arg side chain, as deprotonation of the Arg side chain results in several resonance structures. Still, the calculations show that not only can the effect of point mutations be predicted with a high level of accuracy, but the methodology outlined here also presents an easy and fast procedure to determine the protonation state of residues at protein-protein interfaces.
where pH is 8.3 and the pKa of the free amino acid side chain is used. The binding free energy of all the ionizable P1 variants, except for P1 Arg bound to HLE, are in very good agreement with experimental data. Our calculations underestimate accommodation of OMTKY3 P1 Arg to elastase by 3.7 kcal/mol, which is most likely related to the different deprotonation sites on the P1 Arg side chain, as deprotonation of the Arg side chain results in several resonance structures. Still, the calculations show that not only can the effect of point mutations be predicted with a high level of accuracy, but the methodology outlined here also presents an easy and fast procedure to determine the protonation state of residues at protein-protein interfaces.
Active site preorganization energy
Several factors have been proposed to give rise to different parameterizations of the LIE equation, such as the choice of force field, computational protocols, properties of the active site etc. Possible force field dependencies have been systematically investigated and the method was shown to yield virtually identical coefficients when studying binding of nine ligands to P450cam with three different force fields (14). One of the cornerstones of simplified free energy methods such as LIE (6) and LRA (15) is the electrostatic linear response approximation, which approximates the free energy of charging a solute as in Eq. 3. LIE makes the additional approximation that 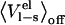 is negligible. The motivation for this is that in the off state the solvent molecules relax, i.e., they orient themselves randomly relative to the solute, and thereby their electrostatic interactions with the solute will average to zero (13). Two major advantages of neglecting
is negligible. The motivation for this is that in the off state the solvent molecules relax, i.e., they orient themselves randomly relative to the solute, and thereby their electrostatic interactions with the solute will average to zero (13). Two major advantages of neglecting 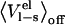 are that only one simulation is needed (of the physical ligand) for bound and unbound states, and the very slow convergence of
are that only one simulation is needed (of the physical ligand) for bound and unbound states, and the very slow convergence of 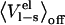 is avoided. The LIE method also utilizes the linear response approximation along with the aforementioned simplification in the complexed state. It is not obvious beforehand that this is a valid simplification in the bound state, since binding sites often have a seemingly preorganized electrostatic environment (in particular, e.g., enzymes for their transition states) which might not relax completely in the off state. For small drug-like solutes it appears that the assumption that
is avoided. The LIE method also utilizes the linear response approximation along with the aforementioned simplification in the complexed state. It is not obvious beforehand that this is a valid simplification in the bound state, since binding sites often have a seemingly preorganized electrostatic environment (in particular, e.g., enzymes for their transition states) which might not relax completely in the off state. For small drug-like solutes it appears that the assumption that  is negligible in the binding site holds. It is not completely clear if this originates from binding site relaxation or randomized orientations of the (unphysical nonpolar) solute relative to its binding site, i.e., ligand relaxation. For protein ligands such as OMTKY3, both ligand and receptor relaxation could be severely reduced since the P1 residue is anchored in place by the secondary interactions between the rest of OMTKY3 and chymotrypsin. In this case, the lack of relaxation would yield a
is negligible in the binding site holds. It is not completely clear if this originates from binding site relaxation or randomized orientations of the (unphysical nonpolar) solute relative to its binding site, i.e., ligand relaxation. For protein ligands such as OMTKY3, both ligand and receptor relaxation could be severely reduced since the P1 residue is anchored in place by the secondary interactions between the rest of OMTKY3 and chymotrypsin. In this case, the lack of relaxation would yield a 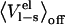 term that is not zero.
term that is not zero.
To elucidate the effects of neglecting the preorganization term we have evaluated 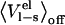 for several ligands in water and bound to their respective proteins. Plotted in Fig. 5 is the free energy contribution of
for several ligands in water and bound to their respective proteins. Plotted in Fig. 5 is the free energy contribution of 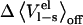 versus
versus 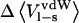 for three different systems: chymotrypsin with P1 mutants of OMTKY3, trypsin with benzamidine analogs, and P450cam with camphor analogs. As can be seen in Fig. 5, neglecting
for three different systems: chymotrypsin with P1 mutants of OMTKY3, trypsin with benzamidine analogs, and P450cam with camphor analogs. As can be seen in Fig. 5, neglecting 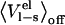 without any compensation will lead to significant errors in the calculated
without any compensation will lead to significant errors in the calculated 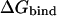 s for the protein-protein system. Also evident in Fig. 5 is a size dependence of
s for the protein-protein system. Also evident in Fig. 5 is a size dependence of  for the protein-protein system. The slow convergence of
for the protein-protein system. The slow convergence of  has most likely imparted large errors in the values of
has most likely imparted large errors in the values of 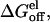 but, nonetheless, the general trend is evident. It is, however, remarkable that the calculations show essentially no preorganization effect for the small ligand data set, which suggests a complete relaxation for these compounds. The protein-protein system has a slope of 0.19, whereas the small ligand data set has a slope close to zero (−0.01). The different slopes of the regressions can be directly related to the different values of α obtained for the protein-protein system and the small-ligand systems. This can bee seen by reparameterizing the LIE model using parameterization A, with the combined values of
but, nonetheless, the general trend is evident. It is, however, remarkable that the calculations show essentially no preorganization effect for the small ligand data set, which suggests a complete relaxation for these compounds. The protein-protein system has a slope of 0.19, whereas the small ligand data set has a slope close to zero (−0.01). The different slopes of the regressions can be directly related to the different values of α obtained for the protein-protein system and the small-ligand systems. This can bee seen by reparameterizing the LIE model using parameterization A, with the combined values of 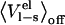 and
and  instead of only
instead of only 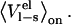 As expected, this reparameterization yields an α of 0.43, but with worse statistics than the original parameterization A (Q2 of 0.72 compared to 0.86), which is probably due to intrinsically bad convergence of
As expected, this reparameterization yields an α of 0.43, but with worse statistics than the original parameterization A (Q2 of 0.72 compared to 0.86), which is probably due to intrinsically bad convergence of 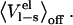 It should be noted here that the convergence problem for this quantity mainly resides in the fact that sampling is done with the unphysical potential (nonpolar ligand), whereas a physical average (polar ligand) is collected.
It should be noted here that the convergence problem for this quantity mainly resides in the fact that sampling is done with the unphysical potential (nonpolar ligand), whereas a physical average (polar ligand) is collected.
FIGURE 5.

(A) The contribution of binding-site preorganization to the binding free energy versus the difference in Lennard-Jones energies between bound and free states. The P1 variants of OMTKY3 complexed with chymotrypsin are shown as diamonds. Small ligands (ligands to p450cam and benzamidine-like ligands) are shown as stars. (B) The size dependence of the electrostatic preorganization energy contribution to the binding free energy for P1 variants of OMTKY3 binding to chymotrypsin (⋄) and small ligands binding to their receptors (▴). On the horizontal axis is the number of heavy atoms in the P1 amino acid or small ligand.
It is thus clear that 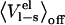 contributes to the binding free energy of OMTKY3 to chymotrypsin and is most likely important for binding of BPTI to trypsin and OMTKY3 to elastase. However, our results suggest that this effect can be incorporated into the empirical α parameter and that this approach yields more accurate results than explicitly evaluating the preorganization term, which requires extra simulations (41) and is associated with intrinsic uncertainties. It appears, however, that the preorganization term does not account for the entire effect observed here on the α parameter, indicating that there may be other size-dependent binding energy contributions that differ between smaller ligands and protein-protein complexes. One such possible factor has to do with translational and rotational entropy losses upon binding. These are clearly also parameterized into Eq. 1 and it may be argued that relative entropy changes between different ligands are more sensitive to ligand size for smaller molecules that are not anchored by secondary interactions (C. Carlsson and J. Åqvist, unpublished). Such an effect would also tend to yield a high α value for protein-protein complexes.
contributes to the binding free energy of OMTKY3 to chymotrypsin and is most likely important for binding of BPTI to trypsin and OMTKY3 to elastase. However, our results suggest that this effect can be incorporated into the empirical α parameter and that this approach yields more accurate results than explicitly evaluating the preorganization term, which requires extra simulations (41) and is associated with intrinsic uncertainties. It appears, however, that the preorganization term does not account for the entire effect observed here on the α parameter, indicating that there may be other size-dependent binding energy contributions that differ between smaller ligands and protein-protein complexes. One such possible factor has to do with translational and rotational entropy losses upon binding. These are clearly also parameterized into Eq. 1 and it may be argued that relative entropy changes between different ligands are more sensitive to ligand size for smaller molecules that are not anchored by secondary interactions (C. Carlsson and J. Åqvist, unpublished). Such an effect would also tend to yield a high α value for protein-protein complexes.
CONCLUDING REMARKS
The experimental association constants for binding of P1 variants of OMTKY3 to chymotrypsin and elastase and of BPTI to trypsin yields free energies of binding ranging from −5.6 to −18.4 kcal/mol depending on the P1 residue accommodated at the S1 site of the enzyme. These complexes thus display some of the highest binding constants measured for noncovalent associations, and it is striking that mutations of a single amino acid can alter the binding energy by as much as 12–13 kcal/mol. To be able to predict the effect of such point mutations before laboratory experiments will therefore have a tremendous impact on the understanding of the specificities of particular interaction sites, and potentially lead to more rationalized research. We have here presented a computational model that accurately predicts the effect of point mutations at protein-protein interfaces. This model is different from the LIE model used to study complexes of smaller ligands with proteins, with the main difference being the treatment of the hydrophobic contribution to the binding free energy. Observations that the nonpolar contribution to solvation free energies of solutes scales linearly with typical size measures, and that 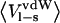 also scales linearly with such size measures, led to the assumption that
also scales linearly with such size measures, led to the assumption that  will scale linearly with nonpolar solvation free energies. It has been shown in many previous studies that this basic idea is sound and the combined proportionality constant is 0.18 for complexes of smaller ligands with proteins. In this work we have found that the binding energetics of mutations at protein-protein interfaces yields a different parameterization of the method and that the reasons for this can be understood. The nonpolar coefficient obtained here is virtually identical for the three systems investigated, with a variation of <0.09. This indicates that the α may be transferable to other proteinase-protein inhibitor systems, but further study is needed to investigate this in detail.
will scale linearly with nonpolar solvation free energies. It has been shown in many previous studies that this basic idea is sound and the combined proportionality constant is 0.18 for complexes of smaller ligands with proteins. In this work we have found that the binding energetics of mutations at protein-protein interfaces yields a different parameterization of the method and that the reasons for this can be understood. The nonpolar coefficient obtained here is virtually identical for the three systems investigated, with a variation of <0.09. This indicates that the α may be transferable to other proteinase-protein inhibitor systems, but further study is needed to investigate this in detail.
One of the central assumptions made in this study is that the contribution from secondary subsites to the interaction free energy does not change when introducing different amino acids at the P1 position. This assumption does not seem to hold when large side chains are inserted into the S1 site of elastase, and the corresponding free energies indicate that the methodology does not work well in such cases. However, these issues cannot be fully resolved until crystal structures of proteinase-protein inhibitor complexes where very large side chains are inserted into narrow active sites become available. This situation also emphasizes the importance of a thorough structural analysis of the simulated trajectories to verify that the assumption holds. Nonetheless, the assumption that the secondary interactions do not change upon complex formation, which is based on crystallographic analysis of several P1 protein-protein complexes displaying virtually identical secondary interactions, seems to hold for 52 out 56 investigated complexes. When experimental binding data is available, the contribution from the secondary interactions can be estimated by fitting the γ-parameter to reproduce the absolute binding free energies. On the contrary, if there are no binding constants available, only relative effects can be computed, as the γ-parameter always cancels out in the relative binding free energies. This constant will also vary depending on which proteins form the complex.
The quantitative agreement with experimental data for our set of 52 mutants is very encouraging and although our data set of mutations is rather large it is, nevertheless, clearly of interest to examine other protein-protein interfaces to determine the generality of the model derived herein. It is also encouraging that 44 of the complexes investigated are homology models, and only one crystal structure was used to generate the full series of complexes for chymotrypsin and one for elastase. This indicates that x-ray structures are not needed to obtain accurate energetic predictions, though more investigations on complexes for which there are no crystal structures available need to be carried out to fully address this. It is highly desirable to have a reliable microscopic computational approach for predicting the energetics of both single and multiple mutations at interfaces and, in this respect, it seems possible to extend our LIE treatment also to multiple mutations. To investigate this in further detail, a study covering binding of the canonical protein inhibitor eglin c to several subtilases with multiple mutations at P1, P4, and P6 has been initiated. Preliminary results show that the LIE model derived herein is indeed capable of dealing with multiple mutations. For two particular proteins, furin and kex2, crystal structures of the complexes with eglin c are not available, but still the energetic effects of point mutations, calculated by the method outlined here, provide relative binding free enegies that are in very good agreement with experimental data (B. O. Brandsdal, S. M. Mekonnen, and A. O. Smalås, unpublished).
Acknowledgments
Support from the Norwegian Research Council to B.O.B. and A.O.S., and from the Swedish Research Council (VR) and the Swedish Foundation for Strategic Research (SSF/Rapid) to J.Å. is gratefully acknowledged. The Norwegian Structural Biology Centre (NorStruct) is supported by the Functional Genomics Program (FUGE) of the Research Council of Norway.
References
- 1.Halperin, I., B. Y. Ma, H. Wolfson, and R. Nussinov. 2002. Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins. 47:409–443. [DOI] [PubMed] [Google Scholar]
- 2.Smith, G. R., and M. J. E. Sternberg. 2002. Prediction of protein-protein interactions by docking methods. Curr. Opin. Struct. Biol. 12:28–35. [DOI] [PubMed] [Google Scholar]
- 3.Bogan, A. A., and K. S. Thorn. 1998. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280:1–9. [DOI] [PubMed] [Google Scholar]
- 4.Brandsdal, B. O., F. Österberg, M. Almlöf, I. Feierberg, V. B. Luzhkov, and J. Åqvist. 2003. Free energy calculations and ligand binding. Adv. Protein Chem. 66:123–158. [DOI] [PubMed] [Google Scholar]
- 5.Kollman, P. 1993. Free energy calculations: applications to chemical and biochemical phenomena. Chem. Rev. 93:2395–2417. [Google Scholar]
- 6.Åqvist, J., C. Medina, and J. E. Samuelsson. 1994. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 7:385–391. [DOI] [PubMed] [Google Scholar]
- 7.Åqvist, J., V. B. Luzhkov, and B. O. Brandsdal. 2002. Ligand binding affinities from MD simulations. Acc. Chem. Res. 35:358–365. [DOI] [PubMed] [Google Scholar]
- 8.Srinivasan, J., T. E. Cheatham, P. Cieplak, P. A. Kollman, and D. A. Case. 1998. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate - DNA helices. J. Am. Chem. Soc. 120:9401–9409. [Google Scholar]
- 9.Horton, N., and M. Lewis. 1992. Calculation of the free-energy of association for protein complexes. Protein Sci. 1:169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massova, I., and P. A. Kollman. 1999. Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J. Am. Chem. Soc. 121:8133–8143. [Google Scholar]
- 11.Kollman, P. A., I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case, and T. E. Cheatham. 2000. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33:889–897. [DOI] [PubMed] [Google Scholar]
- 12.Gohlke, H., and D. A. Case. 2004. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 25:238–250. [DOI] [PubMed] [Google Scholar]
- 13.Åqvist, J., and T. Hansson. 1996. On the validity of electrostatic linear response in polar solvents. J. Phys. Chem. 100:9512–9521. [Google Scholar]
- 14.Almlöf, M., B. O. Brandsdal, and J. Åqvist. 2004. Binding affinity prediction with different force fields: examination of the linear interaction energy method. J. Comput. Chem. 25:1242–1254. [DOI] [PubMed] [Google Scholar]
- 15.Lee, F. S., Z. T. Chu, M. B. Bolger, and A. Warshel. 1992. Calculations of antibody-antigen interactions: microscopic and semi- microscopic evaluation of the free energies of binding of phosphorylcholine analogs to McPC603. Protein Eng. 5:215–228. [DOI] [PubMed] [Google Scholar]
- 16.Brandsdal, B. O., J. Åqvist, and A. O. Smalås. 2001. Computational analysis of binding of P1 variants to trypsin. Protein Sci. 10:1584–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krowarsch, D., M. Dadlez, O. Buczek, I. Krokoszynska, A. O. Smalås, and J. Otlewski. 1999. Interscaffolding additivity: binding of P1 variants of bovine pancreatic trypsin inhibitor to four serine proteases. J. Mol. Biol. 289:175–186. [DOI] [PubMed] [Google Scholar]
- 18.Marelius, J., K. Kolmodin, I. Feierberg, and J. Åqvist. 1998. Q: a molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 16:213–225, 261. [DOI] [PubMed] [Google Scholar]
- 19.Jorgensen, W. L., D. S. Maxwell, and J. Tirado-Rives. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118:11225–11236. [Google Scholar]
- 20.Helland, R., G. I. Berglund, J. Otlewski, W. Apostoluk, O. A. Andersen, N. P. Willassen, and A. O. Smalås. 1999. High-resolution structures of three new trypsin-squash-inhibitor complexes: a detailed comparison with other trypsins and their complexes. Acta Crystallogr. D. 55:139–148. [DOI] [PubMed] [Google Scholar]
- 21.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr. A. 47:110–119. [DOI] [PubMed] [Google Scholar]
- 22.Bode, W., A. Z. Wei, R. Huber, E. Meyer, J. Travis, and S. Neumann. 1986. X-ray crystal-structure of the complex of human-leukocyte elastase (PMN elastase) and the third domain of the turkey ovomucoid inhibitor. EMBO J. 5:2453–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujinaga, M., A. R. Sielecki, R. J. Read, W. Ardelt, M. Laskowski, Jr., and M. N. James. 1987. Crystal and molecular structures of the complex of alpha-chymotrypsin with its inhibitor turkey ovomucoid third domain at 1.8 Å resolution. J. Mol. Biol. 195:397–418. [DOI] [PubMed] [Google Scholar]
- 24.Lee, F. S., and A. Warshel. 1992. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J. Chem. Phys. 97:3100–3107. [Google Scholar]
- 25.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential function for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 26.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 27.Brandsdal, B. O., A. O. Smalås, and J. Åqvist. 2001. Electrostatic effects play a central role in cold adaptation of trypsin. FEBS Lett. 499:171–175. [DOI] [PubMed] [Google Scholar]
- 28.Mehler, E. I., and G. Eichele. 1984. Electrostatic effects in water-accessible regions of proteins. Biochemistry. 23:3887–3891. [Google Scholar]
- 29.Huang, K., W. Lu, S. Anderson, M. Laskowski, Jr., and M. N. James. 1995. Water molecules participate in proteinase-inhibitor interactions: crystal structures of Leu18, Ala18, and Gly18 variants of turkey ovomucoid inhibitor third domain complexed with Streptomyces griseus proteinase B. Protein Sci. 4:1985–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ersmark, K., I. Feierberg, S. Bjelic, J. Hulten, B. Samuelsson, J. Åqvist, and A. Hallberg. 2003. C-2-symmetric inhibitors of Plasmodium falciparum plasmepsin II: Synthesis and theoretical predictions. Bioorg. Med. Chem. 11:3723–3733. [DOI] [PubMed] [Google Scholar]
- 31.Hansson, T., J. Marelius, and J. Åqvist. 1998. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided Mol. Des. 12:27–35. [DOI] [PubMed] [Google Scholar]
- 32.Marelius, J., M. Graffner-Nordberg, T. Hansson, A. Hallberg, and J. Åqvist. 1998. Computation of affinity and selectivity: binding of 2,4-diaminopteridine and 2,4-diaminoquinazoline inhibitors to dihydrofolate reductases. J. Comput. Aided Mol. Des. 12:119–131. [DOI] [PubMed] [Google Scholar]
- 33.Ersmark, K., I. Feierberg, S. Bjelic, E. Hamelink, F. Hackett, M. J. Blackman, J. Hulten, B. Samuelsson, J. Åqvist, and A. Hallberg. 2004. Potent inhibitors of the Plasmodium falciparum enzymes plasmepsin I and II devoid of cathepsin D inhibitory activity. J. Med. Chem. 47:110–122. [DOI] [PubMed] [Google Scholar]
- 34.Graffner-Nordberg, M., K. Kolmodin, J. Åqvist, S. F. Queener, and A. Hallberg. 2001. Design, synthesis, computational prediction, and biological evaluation of ester soft drugs as inhibitors of dihydrofolate reductase from Pneumocystis carinii. J. Med. Chem. 44:2391–2402. [DOI] [PubMed] [Google Scholar]
- 35.Leiros, H. K. S., B. O. Brandsdal, O. A. Andersen, V. Os, I. Leiros, R. Helland, J. Otlewski, N. P. Willassen, and A. O. Smalås. 2004. Trypsin specificity as elucidated by LIE calculations, X-ray structures, and association constant measurements. Protein Sci. 13:1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luzhkov, V. B., and J. Åqvist. 2001. Mechanisms of tetraethylammonium ion block in the KcsA potassium channel. FEBS Lett. 495:191–196. [DOI] [PubMed] [Google Scholar]
- 37.Gorfe, A. A., B. O. Brandsdal, H. K. Leiros, R. Helland, and A. O. Smalås. 2000. Electrostatics of mesophilic and psychrophilic trypsin isoenzymes: qualitative evaluation of electrostatic differences at the substrate binding site. Proteins. 40:207–217. [PubMed] [Google Scholar]
- 38.Lu, W., I. Apostol, M. A. Qasim, N. Warne, R. Wynn, W. L. Zhang, S. Anderson, Y. W. Chiang, E. Ogin, I. Rothberg, K. Ryan, and M. Laskowski, Jr. 1997. Binding of amino acid side-chains to S1 cavities of serine proteinases. J. Mol. Biol. 266:441–461. [DOI] [PubMed] [Google Scholar]
- 39.Clackson, T., and J. A. Wells. 1995. A hot-spot of binding-energy in a hormone-receptor interface. Science. 267:383–386. [DOI] [PubMed] [Google Scholar]
- 40.Qasim, M. A., M. R. Ranjbar, R. Wynn, S. Anderson, and M. J. Laskowski. 1995. Ionizable P1 residues in serine proteinase inhibitors undergo large pK shifts on complex formation. J. Biol. Chem. 270:27419–27422. [DOI] [PubMed] [Google Scholar]
- 41.Sham, Y. Y., Z. T. Chu, H. Tao, and A. Warshel. 2000. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins. 39:393–407. [PubMed] [Google Scholar]