

Abstract
The energy transfer and charge separation kinetics in several core Photosystem I particles of Chlamydomonas reinhardtii with point mutations around the PA and PB reaction center chlorophylls (Chls) have been studied using ultrafast transient absorption spectroscopy in the femtosecond to nanosecond time range to characterize the influence on the early electron transfer processes. The data have been analyzed in terms of kinetic compartment models. The adequate description of the transient absorption kinetics requires three different radical pairs in the time range up to ∼100 ps. Also a charge recombination process from the first radical pair back to the excited state is present in all the mutants, as already shown previously for the wild-type (Müller, M. G., J. Niklas, W. Lubitz, and A. R. Holzwarth. 2003. Biophys. J. 85:3899–3922; and Holzwarth, A. R., M. G. Müller, J. Niklas, and W. Lubitz. 2005. J. Phys. Chem. B. 109:5903–59115). In all mutants, the primary charge separation occurs with the same effective rate constant within the error limits as in the wild-type (»350 ns−1), which implies an intrinsic rate constant of charge separation of <1 ps−1. The rate constant of the secondary electron transfer process is slowed down by a factor of ∼2 in the mutant B-H656C, which lacks the ligand to the central metal of Chl PB. For the mutant A-T739V, which breaks the hydrogen bond to the keto carbonyl of Chl PA, only a slight slowing down of the secondary electron transfer is observed. Finally for mutant A-W679A, which has the Trp near the PA Chl replaced, either no pronounced effect or, at best, a slight increase on the secondary electron transfer rate constants is observed. The effective charge recombination rate constant is modified in all mutants to some extent, with the strongest effect observed in mutant B-H656C. Our data strongly suggest that the Chls of the PA and PB pair, constituting what is traditionally called the “primary electron donor P700”, are not oxidized in the first electron transfer process, but rather only in the secondary electron transfer step. We thus propose a new electron transfer mechanism for Photosystem I where the accessory Chl(s) function as the primary electron donor(s) and the A0 Chl(s) are the primary electron acceptor(s). This new mechanism also resolves in a straightforward manner the difficulty with the previous mechanism, where an electron would have to overcome a distance of ∼14 Å in <1 ps in a single step. If interpreted within a scheme of single-sided electron transfer, our data suggest that the B-branch is the active branch, although parallel A-branch activity cannot be excluded. All the mutations do affect to a varying extent the energy difference between the reaction center excited state RC* and the first radical pair and thus affect the rate constant of charge recombination. It is interesting to note that the new mechanism proposed is in fact analogous to the electron transfer mechanism in Photosystem II, where the accessory Chl also plays the role of the primary electron donor, rather than the special Chl pair P680 (Prokhorenko, V. and A. R. Holzwarth. 2000. J. Phys. Chem. B. 104:11563–11578).
INTRODUCTION
Photosystem I (PS I) is one of the two photosystems of oxygenic photosynthetic organisms which are connected by the sequential Z-scheme mechanism for electron transfer (1–3). PS I receives electrons from the plastoquinone pool on the acceptor side, and its physiological function is to catalyze light-driven transfer of electrons from reduced plastocyanin or cytochrome c6 to ferredoxin (see Brettel and Leibl (4) for a review). There now exist high resolution x-ray structures for both cyanobacterial PS I (5,6) as well as for higher plant PS I (7). The higher plant PS I is monomeric and contains 93 Chls in the core, including the six reaction center (RC) Chls, with the remainder of the total 167 Chls located in four peripheral light-harvesting complexes (LHC I) (7). The core of cyanobacterial PS I is organized as a trimer, comprising 96 Chls per monomer, and lacks any peripheral chlorophyll (Chl) antennae (5). The core structure is highly conserved between the two types of organisms. No x-ray structure is available for Chlamydomonas reinhardtii PS I, but it is expected that the core antenna organization is quite similar to that of higher plant PS I.
The mechanism and kinetics of the ultrafast events of energy transfer from the antenna to the RC and for the electron transfer within the RC are still a matter of intensive debate and controversy. Since the antenna Chls and the RC Chls are both bound to the same polypeptide units (5), an intact well-defined PS I RC devoid of antenna Chls cannot be isolated. In view of this situation, the electron transfer processes cannot be studied separately from the energy transfer processes. Thus the energy transfer and the electron transfer dynamics are tightly linked and intertwined, which presents the key difficulty in the analysis and interpretation of ultrafast optical data for PS I. The problem is aggravated further by the fact that energy and electron transfer processes occur on comparable picosecond timescales, which makes the assignment of the different lifetime components even more difficult.
Based on femtosecond transient absorption data on PS I cores of the green algae C. reinhardtii, we recently showed that both the apparent energy equilibration between the core antenna and the RC, and the apparent trapping by charge separation, occur with lifetimes which are by a factor of 10 and 4, respectively, faster than previously assumed (8). We thus proposed a new kinetic scheme for the energy transfer and early electron transfer processes where the apparent energy equilibration lifetime between the PS I core antenna and the RC is 1–2 ps and the apparent primary charge separation lifetime is 7–10 ps (8) (see that article also for an extensive discussion of the problems with previous kinetic and mechanistic interpretations of early PS I kinetics. A detailed updated definition of kinetic terms, e.g., effective charge separation rate constant, apparent charge separation lifetime, etc. is provided in our recent fluorescence kinetics work (9)). Furthermore, we demonstrated the necessity to include an additional radical pair before the so far assumed first radical pair 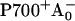 in the kinetic scheme, and we also demonstrated that the first electron transfer step is reversible, giving rise to charge recombination fluorescence (8,9). The kinetic scheme for the energy transfer and electron transfer processes that we developed based on these findings is shown in Fig. 1. After clarifying the kinetics of the antenna/RC energy transfer and the electron transfer steps in the wild-type (wt), a new avenue is opened to study in more detail the mechanism of the early electron transfer processes in PS I, which is an area of considerable controversy.
in the kinetic scheme, and we also demonstrated that the first electron transfer step is reversible, giving rise to charge recombination fluorescence (8,9). The kinetic scheme for the energy transfer and electron transfer processes that we developed based on these findings is shown in Fig. 1. After clarifying the kinetics of the antenna/RC energy transfer and the electron transfer steps in the wild-type (wt), a new avenue is opened to study in more detail the mechanism of the early electron transfer processes in PS I, which is an area of considerable controversy.
FIGURE 1.

Kinetic scheme of the energy transfer and electron transfer processes, including two antenna compartments, the equilibrated excited RC state (RC*, comprising 6 Chls), three radical pairs (RP1 …. RP3), and the charge recombination process (scheme reproduced from Müller et al. (8)). The rate constants are given in units of ns−1. The corresponding lifetimes for this scheme are shown in parentheses below. Note that the lifetimes are the inverse eigenvalues of the kinetic matrix which contains the rate constants. They are not per se the lifetimes of a particular species in the scheme. Thus the eigenvalues and lifetimes are in principle functions of all the rate constants in the scheme (8,52).
The RC of PS I consists of six Chls organized in two almost symmetric branches (5,7). The two central Chls, PA and PB, believed to form a dimer traditionally called “P700”, upon oxidation give rise to the typical 700 nm maximum in the absorption difference spectrum of what is believed to be the oxidized primary electron donor P700 (10,11) (see Brettel and Leibl (4) and Webber and Lubitz (12) for reviews). The primary acceptor is believed to be another Chl called “A0”. It is one or both of the two Chls that are located next to the phylloquinones, which in this mechanism function as the secondary acceptor(s) A1 (4). This interpretation involves basically two radical pairs which differ in the oxidation state of the RC Chls (i.e., 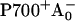 and
and 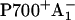 ).
).
Until recently, no details of the early electron transfer steps in the PS I RC had been resolved directly by ultrafast transient absorption measurements on intact PS I particles. Rather, only the rise kinetics of the final radical pair (on the timescale up to ∼100 ps), considered to reflect the 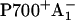 state, had been resolved directly (see Melkozernov (13) and Brettel and Leibl (4) for reviews), whereas the purported first radical pair had only been assigned spectroscopically from difference measurements between open and closed RCs. This often employed difference technique (14,15) for assigning the nature and kinetics of the first radical pair is, however, untenable. The reason is the underlying assumption of identical energy transfer kinetics for open and closed RCs, which should thus be removed from the overall kinetics by the subtraction method, is incorrect in view of the long range antenna quenching by P700+ (16,17). A new picture arose when we showed in direct measurements on open RCs that the first radical pair (RP1) in fact rises with a lifetime of only ∼7–9 ps in wt C. reinhardtii PS I cores and decays with a lifetime of ∼20–25 ps, forming another radical pair (RP2). Our RP2 species is similar both spectroscopically and kinetically to the previously resolved first radical pair
state, had been resolved directly (see Melkozernov (13) and Brettel and Leibl (4) for reviews), whereas the purported first radical pair had only been assigned spectroscopically from difference measurements between open and closed RCs. This often employed difference technique (14,15) for assigning the nature and kinetics of the first radical pair is, however, untenable. The reason is the underlying assumption of identical energy transfer kinetics for open and closed RCs, which should thus be removed from the overall kinetics by the subtraction method, is incorrect in view of the long range antenna quenching by P700+ (16,17). A new picture arose when we showed in direct measurements on open RCs that the first radical pair (RP1) in fact rises with a lifetime of only ∼7–9 ps in wt C. reinhardtii PS I cores and decays with a lifetime of ∼20–25 ps, forming another radical pair (RP2). Our RP2 species is similar both spectroscopically and kinetically to the previously resolved first radical pair 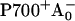 (15,18–21) (see also Brettel and Leibl (4) for a review). A relatively large edge-to-edge distance of ∼14 Å exists between the PA and PB chls and the A0 chls. This large distance makes it highly unlikely that it can be overcome in a single ultrafast electron transfer step whose intrinsic rate constant is believed to be larger than 1 ps−1 (22–25). This situation calls for the involvement of an intermediary state, possibly comprising one or both of the accessory Chls (Chlacc) located between the P and the A0 Chls. It was thus interesting that our recent work demonstrated for the first time that a third RP, which also differs in the redox state of the Chls from the other two RPs, was indeed required for the adequate description of the kinetics (8). We were not able, though, to give a definite assignment of the nature of these radical pairs based on the wt data alone. Rather we proposed several possibilities which could not be distinguished at that time, as is shown in Fig. 2. This left the mechanism of the early electron transfer processes within PS I unsolved and demanded an explanation within a more complex electron transfer scheme than assumed so far.
(15,18–21) (see also Brettel and Leibl (4) for a review). A relatively large edge-to-edge distance of ∼14 Å exists between the PA and PB chls and the A0 chls. This large distance makes it highly unlikely that it can be overcome in a single ultrafast electron transfer step whose intrinsic rate constant is believed to be larger than 1 ps−1 (22–25). This situation calls for the involvement of an intermediary state, possibly comprising one or both of the accessory Chls (Chlacc) located between the P and the A0 Chls. It was thus interesting that our recent work demonstrated for the first time that a third RP, which also differs in the redox state of the Chls from the other two RPs, was indeed required for the adequate description of the kinetics (8). We were not able, though, to give a definite assignment of the nature of these radical pairs based on the wt data alone. Rather we proposed several possibilities which could not be distinguished at that time, as is shown in Fig. 2. This left the mechanism of the early electron transfer processes within PS I unsolved and demanded an explanation within a more complex electron transfer scheme than assumed so far.
FIGURE 2.

Three possible sequences of redox intermediates corresponding to the three radical pairs found in our previous work (8) (top). An alternative relaxed radical pair scheme with only two different redox intermediates is shown at the bottom. The latter scheme is not very likely, given the significant differences in the spectra of radical pairs RP2 and RP3. (Note that in Fig. 2 D, a relaxed radical pair scheme is shown containing two formally identical but energetically different RPs.) (Reproduced from Müller et al. (8).)
The situation described above poses anew the question as to the nature of the primary electron donor and the redox state of the first radical pair. It is generally assumed that the redox state termed “P700+” is formed already in the first electron transfer step. In fact all previous interpretations of ultrafast data on PS I assume a model requiring the oxidation of one of the central Chls of the RC, i.e., PA or PB, in the first electron transfer step. We note, however, that this is an unproven hypothesis since there exists no solid experimental data so far to support such an assignment. The spectral characteristics of P700+ have typically been measured and tested only at times substantially longer than the formation time of the first radical pair RP1 (see Müller et al. (8) for an extensive discussion). Note that in strict terms, P700+ is only defined in the difference spectrum (P700+ – RCground state). It was only in our recent work that the kinetics of what we believe to be the first radical pair and its difference spectrum have been resolved directly on open RCs (8). Recent EPR/ENDOR studies and a molecular orbital calculation showed that in the oxidized state the electron hole is preferentially located on the Chl PB of the RC, with a partial delocalization on PA (26,27). However from Fourier transform infrared (FTIR) spectroscopy it was concluded that the charge should be more equally distributed among the two P700 Chls (28). We note that the final charge distribution of the electron hole on the P700 Chls may be different from the initially formed one. Thus other initial electron transfer steps and sequences than assumed so far are possible and are not excluded by the available data. It appears possible inter alia that P700+ forms only in the secondary electron transfer step. An analogous scheme was, for example, proposed recently for isolated Photosystem II (PS II) RCs (29). In contrast to general assumptions that the primary electron donor should be the pseudodimer PD1/PD2 (30), we demonstrated that the primary electron donor is the accessory Chl and that the P680+ cation is only produced in the second electron transfer step. Additional results in support of that mechanism in PS II have in the meantime emerged also from other groups (31,32). It should also be noted that a similar electron transfer process was observed earlier in bacterial RCs (33,34), although it plays only a minor role in wt RCs due to the energetically high lying and very short-lived excited state of the accessory BChl. Clearly similar electron transfer schemes cannot be excluded at present for PS I either. Thus one should consider the nature of the first two radical pairs in PS I as an open question.
So far we have entirely ignored another problem that has been discussed in the literature quite controversially for PS I, i.e., the question whether electron transfer occurs on one side only, as in bacterial and PS II RCs (35,36), or whether both of the quite symmetric branches in PS I are involved in electron transfer. The traditional view, based mainly on the analogy to bacteria and PS II, was that electron transfer is one-sided only (see Brettel and Leibl (4) for a review). However recent data suggested that actually both sides may be active (37–39), although these results have been questioned by other authors (40). A careful interpretation so far would be that different results have been obtained for PS I from different species. Although a species dependence cannot be ruled out, it would be surprising if the mechanism was drastically different in PS I RCs from different species, given the high similarity in optical spectra of the intermediates.
In view of these open questions in our understanding of the early electron transfer processes in PS I, the purpose of this work is twofold: First, we aim to reexamine critically the current interpretations regarding the mechanism of charge separation in the light of the new transient absorption data. Second, we intend to assign the nature of the first radical pair(s). In particular we need to ask the question whether any of the PA or PB Chls are indeed oxidized in the first electron transfer step and what is likely the nature of the first electron acceptor. This cannot be done without the use of mutants designed to introduce specific changes in certain electron transfer steps. We thus performed ultrafast transient absorption spectroscopy on several PS I mutants with open PS I RC from C. reinhardtii, and we analyzed these kinetic data using the kinetic compartment modeling demonstrated earlier (8,9). The mutants that we have used here all involve modifications close to the P700 Chl pair. The location of the modified amino acids is shown in Fig. 3. Specifically, these are the PsaB-H656C (B-H656C) mutant, which replaces the His central ligand on the PB Chl with cysteine; the PsaA-T739V (A-T739V) mutant, which removes the hydrogen bonding ligand to the keto group of the PA Chl; and a mutation PsaA-W679A (A-W679A), which replaces the aromatic amino acid near the PA and PB Chls by alanine and may thus perhaps modify the charge delocalization on P700+. All mutants are still photosynthetically competent. Most importantly, however, these mutations modify the redox potential of P700+ to various extents (41–44). The change in redox potential of P700+ is expected to change the effective rate constant of electron transfer from the PA or PB Chls in the transfer step where these Chls(s) get oxidized. These mutations are furthermore expected to change the rate constants of charge recombination due to a change in the energy level of either the radical pair or the average RC excited state energy or both. The other important aspect which is helpful for a specific assignment is that the RC spectra (absorption difference, circular dichroism (CD), P700+ difference spectra, etc.) are quite different for these mutants which, together with detailed exciton level calculations, should be another important tool to assign the nature of the intermediates.
FIGURE 3.

Structural arrangement of the RC cofactors and amino acids relevant for the mutations studied in this work. The structure is based on the Synechococcus PS I structure (5) but using the modified numbering scheme required for the adaptation to C. reinhardtii (43). The three mutated amino acids are shown in red along with their numbering. The P Chls are displayed in green, the accessory (Chlacc) Chls in blue, and the A0 Chls in black. The phylloquinones are shown in yellow.
Similar mutants as studied here have been applied earlier for other studies using fluorescence and transient absorption methods. Gibasiewicz et al., using femtosecond spectroscopy at low temperature, characterized the exciton coupling in the RC (45). FTIR and EPR have been used to characterize the influence of the mutations on the charge distribution in P700+ (43,44,46,47). Optical and ADMR measurements have been used to characterize the differences in the optical spectra of the RCs, including triplet-singlet spectra (41–44). Thus there is already a variety of data available which can be referred to when interpreting the transient absorption data.
MATERIALS AND METHODS
Mutant strains, site directed mutagenesis, and cell growth
The strain C. reinhardtii CC2696 was used as recipient of donor plasmids. In contrast to wt it carries the nuclear mutation DS-521, causing a deficiency in the Cab proteins, and a deletion in psbA, leading to a loss of PS II (48). Therefore, this strain is well suited for the preparation and analysis of PS I core complexes. Site-directed mutagenesis was performed according to Witt et al. (43,44). The B-H656C mutant has been constructed by Webber and co-workers (43).
Biochemical preparation and measurement conditions
Cells of C. reinhardtii CC2696 and the various mutant strains were grown and the PS I complexes prepared as described previously (44). The cells were harvested and thylakoids were prepared (41). Isolation of the PS I complex was performed according to the method of Hippler et al. (49) with slight modifications as described (8). Chl concentrations were determined according to the method of Porra et al. (50). For the measurements, the PS I complexes were diluted to an OD676 = 0.73–0.89/mm in 5 mM Tricine-NaOH (pH 7.5), 0.02% dodecyl-β-D-maltoside (DM), 100 mM NaCl, 40 mM Na ascorbate, and 50 μM phenoxy-methosulfate (PMS) as redox mediator to ensure faster reopening of the RCs. The presence of PMS does not perturb, however, the observed electron transfer kinetics. Integrity of the sample was checked by absorption spectroscopy before and after the measurements. No changes in the spectrum were observed. All measurements were performed at room temperature (22°C).
Femtosecond measurements and analysis
Transient spectra in the femtosecond to nanosecond time range have been measured with open (reduced) P700 on these PS I particles at room temperature as described (8). The primary data analysis has been performed by lifetime distribution analysis (using an exponential basis set with 500 exponentials) as described previously in detail (8,51). Kinetic compartment modeling, which results in a set of discrete exponential lifetimes, was performed with a home-written program directly on the kinetics obtained from the lifetime density maps (lifetime distributions) and not on a limited set of precalculated decay-associated difference spectra (DADS) and lifetimes, which a priori would restrict the number of possible models. The advantages of this approach have been described in detail (8,52).
RESULTS AND DISCUSSION
The ground state absorption spectra of the wt and the mutant PS I cores are shown in Fig. 4. In the Qy band the differences are only minor, indicating that the same antenna composition is present in the mutants and the wt Small differences are present primarily ∼450–460 nm, which reflects slightly different amounts of the energetically uncoupled LHC I antenna complex in the samples (8) and possibly also some changes in the RC absorption. The differences in RC absorption, which makes only a small contribution to the overall spectrum in the Qy region, have been characterized previously by various kinds of spectroscopy (41–43). There are large differences in the light-minus-dark absorption, CD, T-S, and P700+ difference spectra between these mutants and with respect to the wt RC (41–43). In general, the mutations cause a complex blue shift in the overall RC absorption and the P700+ difference spectrum relative to the wt and introduce new short-wave bands. The largest change in redox potential occurs for the B-H656C mutant, followed by the A-T739V mutant, whereas for the A-W679A mutant the change of redox potential is small.
FIGURE 4.

Ground state absorption spectra at room temperature of the wt and the three mutant PS I core complex preparations as used in this work.
Experimental kinetics
Transient absorption spectra have been measured for 700 nm (primarily RC excitation) excitation wavelength for open (P700 reduced) PS I particles from C. reinhardtii and mutants on the timescale from a few tens of femtoseconds to hundreds of picoseconds at room temperature. The excitation intensity was low enough to primarily cause single excitations per PS I particle and pulse. Detailed calculations show that our excitation conditions on average provide <0.4 excitations per PS I particle. We chose the 700 nm excitation wavelength for several reasons: First it excites preferentially the RCs and thus reduces contributions from the energy transfer processes in the antenna (see below for details). This is convenient since in this work we are mainly interested in characterizing the details of the electron transfer processes in the RC and not in the energy transfer. At the same time, a 700 nm wavelength avoids excitation of the small amount of energetically uncoupled LHC I antenna present in these samples and thus avoids kinetic complications due to these additional kinetic contributions (8). Note that the “RC” in our notation used throughout this work comprises all six Chls of the electron transfer chain.
Fig. 5 shows the original femtosecond data. It can be seen that formation of the long-lived charge separated state (>50 ns on our timescale) is occurring efficiently in all the samples, including the mutants, in agreement with earlier observations (41–43). The difference spectra of the long-lived RPs are substantially different for the mutants and the wt (compare also with Müller et al. (8) for the wt data) as demonstrated in Fig. 6, which shows the time-dependent difference spectra at various delay times. Mutant B-H656C has the main bleaching for the difference spectrum of the long-lived radical pair at 695 nm and shows two additional bands at shorter wavelength, in good agreement with steady-state data (41,43). Mutant A-W679A shows two about equally intense bleaching bands for the long-lived radical pair located at ∼697 and 682 nm. The A-T739V mutant shows the main bleaching in the short-wavelength band around 682 nm, whereas the long-wavelength bleaching band is smaller. The early bleaching bands (∼1 ps range) are quite similar for all the samples, showing only small shifts in the maxima due to the modified RC absorption properties.
FIGURE 5.

Three-dimensional plots of the femtosecond transient absorption kinetics for an excitation wavelength of 700 nm (see Materials and Methods for details) for PS I core particles from C. reinhardtii mutants at room temperature. (A) B-H656C, (B) A-W679A, and (C) A-T739V. Note that the ΔA scale is reversed (negative is up) for better visibility of the surfaces. The figures show clearly the large differences in the long-lived radical pair at a time of 300 ps.
FIGURE 6.

Transient absorption difference spectra at various delay times for PS I particles using excitation at 700 nm after correction for chirp (time dispersion) of the detection pulse and deconvolution with the excitation pulse (cf. the original data in Fig. 5). (A) B-H656C, (B) A-W679A, and (C) A-T739V.
The lifetime density maps shown in Fig. 7 reveal all the relevant lifetime components: Processes on the ultrafast timescale up to 300 fs are caused mainly by energy transfer among the initially excited RC pigments and to a smaller extent by energy transfer processes in the partially excited PS I core antenna. We have ignored these processes in this analysis, since they are not relevant on the timescales of the electron transfer processes discussed here. On the longer timescales, the dominant processes are seen in the 7–70 ps range. These reflect only the electron transfer processes. A smaller amplitude component is seen in the 7–10 ps range (dominant in the range 710–740 nm), which has been assigned previously to the apparent lifetime of the primary charge separation and trapping process (8). Relatively weak contributions due to energy transfer are seen in the time range around 1 ps. As expected, the spectral signature of this component shows that there is energy transfer uphill, i.e., from the RC to the antenna, as indicated by the negative amplitude of the components around 695–700 nm and the positive component around 675 nm (Fig. 7, A–D). As already shown in our previous work, the relative energy transfer contribution to the difference spectra and kinetics is much less for 700 nm excitation than for preferential antenna excitation. This has two reasons: First, less excitation is delivered back to the antenna due to the fact that charge separation occurs quite rapidly from the directly excited RC. Second, the signal in the difference spectra for the energy transfer components is even more reduced due to the fact that the—still present—directly excited antenna contribution produces a negative/positive signal (blue/red bands), whereas the predominant RC excitation and the subsequent energy transfer back to the antenna produces the opposite kinetic pattern (positive/negative). Thus compensation of signals occurs (note that the data show that ∼20% of the excitation is absorbed by the antenna for 700 nm excitation, with slight differences for the different samples). This situation is actually quite desirable for our purpose, since we do not have to deal with strong energy transfer components interfering with the electron transfer components.
FIGURE 7.

Lifetime density maps (see Materials and Methods for details) of the transient absorption decays of PS I particles as obtained from the data shown in Fig. 5. (A) wt, (B) B-H656C, (C) A-W679A, and (D) A-T739V. Blue-black color indicates negative amplitudes; yellow color indicates positive amplitude. The zero-level is colored in orange.
With respect to the 7–70 ps range, at first glance all the lifetime density maps for the different PS I samples look surprisingly similar, showing only minor differences in a few details. Upon closer inspection, one notices that the main difference between the samples occurs in the width of the lifetime distribution of the strong component in the 50 ps range around 680–690 nm (blue, i.e., negative band, in Fig. 7, A–D). For all the mutants and also for the wt, this distribution extends from 20 ps to ∼50 ps. However, for the B-H656C mutant this distribution range is shifted upward in lifetime, ranging from ∼30 ps up to 70 ps. This reflects a substantial slowing of at least one of the electron transfer rate constants, most likely the effective rate constant of the secondary step. The exact details of the differences can only be revealed by the kinetic target analysis which is presented below. Qualitatively, the 7–10 ps component—assigned in our previous measurements on the wt to the apparent primary charge separation lifetime from the equilibrated RC—is present in all the samples in the same wavelength range and with very similar intensity. This indicates qualitatively that the mutations did not substantially change the rates of the primary charge separation process.
Kinetic modeling
We now proceed with kinetic modeling of the transient absorption data presented above. The modeling will take into account kinetic components in the lifetime range from ∼800 fs to nanoseconds. It has been shown previously also for some of these mutants that ultrafast equilibration in the range of 150 fs occurs among the exciton states in the PS I RC for red excitation (8,19,53). This view is well supported by these data. Thus the first electron transfer step would occur from a completely equilibrated RC* excited state. (Note that the RC* compartment in our modeling comprises all six Chls of the RC, and should not be confused with the two P700 Chls.) The suppression of the energy transfer contributions in the difference spectra for 700 nm excitation, as discussed above, allows for a slight simplification of the full kinetic scheme that has been developed in our previous work (cf. Fig. 1) (8). To not complicate the kinetic model unnecessarily, we have thus ignored here the details of the antenna energy transfer processes and used a description with only one antenna pool (Ant*). This model (although not yet including the charge recombination process) has actually been tested in our previous work for the wt and was shown to be a good model for describing processes in the time range from a few picoseconds up to nanoseconds. Some small deviations from the full model only occur for spectral components ∼1 ps due to the simplification of the description of the energy transfer processes. This simplification is quite tolerable in view of our focus on the electron transfer processes and the fact that all energy transfer processes occur on a substantially faster timescale. Indeed the analysis shows that a model with one antenna compartment comprising 93 Chls, the RC* compartment comprising six Chls, and three radical pairs describes the data for all samples very well, as is shown in Fig. 8, A–D for the kinetic analysis within such a scheme. All the fittings are excellent except for slight deviations in the ultrafast time range from ∼800 fs to ∼2 ps. In contrast, models involving only two radical pairs did not result in very good kinetic fits (not shown).
FIGURE 8.


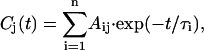 |
The compartment modeling reveals that the kinetics for all the PS I particles can be described within the same formal kinetic scheme, although with different sets of effective rate constants (Fig. 8, A–D). All data, including the effective rate constants, the fitted lifetime sets, the species-associated difference spectra (SADS), and the time-dependent populations of the various intermediates, and the weighted eigenvectors for the kinetic schemes are given in these figures. The effective rate constants of the electron transfer processes are summarized in Table 1 together with the P700 redox potential changes.
TABLE 1.
Summary of the rate constants ki [ns−1] of the electron transfer processes for the various PS I core preparations and changes in redox potential 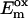 [mV] relative to the wt
[mV] relative to the wt
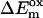
The qualitative assignment of the apparent charge separation step to a lifetime in the range 7–10 ps (note that the apparent charge separation lifetime is essentially equal to the apparent energy trapping lifetime (9)) is fully supported by the results of the modeling. The first kinetic change observed is for the effective energy transfer rate constant from the RC* to the antenna, which is increased for the B-H656C mutant (700 vs. 500 ns−1), possibly due to the blue-shifted RC absorption as compared to wt (the overall RC spectral change is rather complex, however (41,43)). The RC spectral change on average both decreases the energy difference to the antenna excited state and increases the spectral overlap RC*/antenna, which in turn may increase the effective uphill energy transfer rate constant to the antenna. The effective forward rate constants of antenna ⇒ RC transfer are about the same in all the samples within the 15% error limits. We point out again that for a full kinetic characterization of the energy transfer processes, the use of shorter wavelength excitation data and at minimum a two-antenna compartment model are required, as shown previously (8). We intentionally ignore these details of the energy transfer dynamics, which appear on an earlier timescale in this work, for the sake of focusing on the electron transfer processes. Nevertheless, all the conclusions put forward here with respect to the electron transfer processes also hold within the more complex two-antenna compartment model.
The most interesting and striking result of our analysis is the fact that the effective rate constant of primary charge separation is identical within the error limits for all the PS I particles studied here, i.e., no mutation-induced change is observed within the error limits. However, the rate constant of charge recombination differs substantially for the different samples. This rate constant is highest for the wt (40 ns−1), by a factor of 4 smaller for the B-H656C mutant (10 ns−1), and between these extremes for the other two mutants. We note, however, that there is a larger uncertainty in the rate constants of charge recombination as determined from the modeling of the transient absorption data than for the other rate constants. This problem has been discussed extensively in our previous work for the wt, and this discussion also applies here. For this reason, additional time-resolved fluorescence measurements as performed for the wt (9) should also be carried out for a more accurate determination of the effective charge recombination rate constants of the mutants. We estimate a maximal error of 30% for this rate constant (see Table 1) in this analysis. In view of this situation, we will not engage in a very detailed discussion regarding the exact origin of the differences in charge recombination rates. We only note here that in view of the equal forward electron transfer rate constants for the primary step, the changes must be caused by a change in the energy difference ΔG(RP1 – RC*), and changes in the energies of either level can contribute to the change in the rate constant. Likely, the average energy level of the equilibrated RC* state in the B-H656C mutant is the highest for all the different PS I particles, thus explaining the most extreme recombination rate constant in this mutant. To which extent these average energy levels may be affected by the redox potential of P700 is an open question. Interestingly Tang et al. (54) found that the recombination rate in bacterial RCs was not influenced by the P/P+ redox potential.
Apart from this difference in the charge recombination rate constants, the largest changes in electron transfer rate constants between the mutants and the wt occur in the secondary electron transfer step from RP1 to RP2. This rate constant is 70 ns−1 for the wt, 40 ns−1 for the B-H656C mutant, 80 ns−1 for the A-W679A mutant, and 62 ns−1 for the A-T739V mutant. Clearly the upshift in the redox potential for the B-H656C mutant causes a substantial reduction of this rate constant, whereas the slight downshift in redox potential decreases this rate constant only to a minor extent. In the A-W679A mutant, the rate constant is at best only slightly larger than in the w.t. Since we observe a large change in this rate constant for the B-H656C mutant which also shows the largest effect on the redox potential, it appears reasonable to assume that the PB Chl gets oxidized in the secondary electron transfer step only and not in the first one. The rate constant of the latter step is not affected at all by any of the mutations. Interestingly the PB Chl is the site where most of the positive charge on the P700 Chls is located (26,43,44,46,55,56) although substantial delocalization is present (28,57). To a first approximation, it seems reasonable to assume that the electron transfer occurs preferentially in the B-branch of the RC (see below for a more detailed discussion). Despite the smaller changes in the secondary rate constant for the other mutants, the results point in the same direction, i.e., that the PA and PB Chls are not oxidized in the first electron transfer step. Clearly it is not only the redox potential of P700 that controls the electron transfer rate constant, but other factors also play a significant role, like change in electronic overlap, the nature of the excitonic coupling, etc. Thus a quantitative explanation for the changes in this rate constant for the different mutations is not possible at present. Finally the tertiary electron transfer rate constants are not significantly affected by the mutations, and they are all in the ∼22 ns−1 range.
The difference spectra for the various radical pairs also differ substantially between the samples, as was already expected from the differences in the RC spectra. RP1 for the wt has a two-banded spectrum (see also our earlier work (8)) with a maximum bleaching at 693 nm. The radical pairs RP1 for the mutants are essentially single-banded, with a shoulder on the red side at 693 nm. These spectral changes predominantly reflect the shift in the energy levels of the excited PA and PB (P700) Chls which are blue shifted on average in the mutants. However, the details of these spectral changes are complex and cannot be understood without taking the detailed exciton coupling among the pigments into account. Radical pairs RP2 and RP3 differ also for all the samples studied. Again any detailed discussion of these spectral differences must await a detailed exciton calculation. It is important to note, however, that the SADS for the different systems fulfill the central requirements set out in our previous work (8). These limiting requirements are that the excited state SADS of the antenna and the RC* must show a bleaching in the wavelength range 720–750 nm due to stimulated emission. In contrast the radical pairs must show an absorption increase above 720 nm as is expected for Chl cations and anions. This further supports the assignment of the intermediates and confirms that the principal mechanism for energy and electron transfer is the same in all the samples. The energy equilibration antenna/RC* occurs with 0.9–1.5 ps lifetime in agreement with our previous data (8,9).
The apparent lifetime for primary charge separation ranges from 6.3 ps to 10 ps. This lifetime is influenced and determined by all effective rate constants of all processes up to the formation of RP2. In all systems, the maximal antenna excited state population reaches ∼40%, except for the B-H656C mutant where it attains ∼50% due to the faster back-transfer of energy to the antenna (vide supra). The RP1 population reaches its maximum in ∼7–8 ps in all PS I cores except for the B-H656C mutant, where it takes ∼13 ps for the RP1 to reach its maximal population. This is due to the much slower electron back-transfer in this mutant, which slows down the equilibration process. The appearance kinetics of RP1 is biphasic as can be seen from inspection of the weighted eigenvectors in Fig. 8, A–D. The largest contribution to the apparent rise time is by the component of 7–10 ps lifetime, and a smaller (15–20% of the amplitude) part rises with the ∼900 fs component. The decay of RP1 and the rise of RP2 occurs with the lifetime of 17–29 ps, depending on the mutation. The population maximum in RP2 is reached at ∼30–35 ps for all samples, again with the exception of the B-H656C mutant where it takes more than 50 ps for the RP2 to reach its maximum. This slowing down of the appearance kinetics of the state which we assign to RP2 was detected earlier by Melkozernov et al. in a similar mutant (15,21) but was assigned in their work to a slowing of the primary electron transfer rate within the traditional electron transfer scheme. However, our data show clearly that the slowing occurs in the effective rate constant of the secondary electron transfer process.
Implications of the mutation-induced rate constant changes
Our results are clearly in contrast with this model of the electron transfer mechanism and the sequence of radical pairs in PS I considered so far. Our results thus require a new mechanism as already indicated in our previous work (8,9) (cf. also Fig. 2, to which we refer in the following discussion). This new mechanism must involve three radical pairs which differ in the redox state of one or more of the six RC Chls. From the above discussion, we can exclude that the primary electron donor is any of the P700 Chls. Rather these Chls are oxidized only in the secondary electron transfer step, whose rate constants are highly sensitive to the mutations introduced here. Thus we exclude scheme 2a (Fig. 2), which leaves only schemes 2b and 2c as possible electron transfer mechanisms. In both of the latter schemes, the primary electron donor is one of the accessory Chls. The difference between them is in the sequence of the second and third electron transfer step. In scheme 2b the secondary process involves oxidation of the P700 Chl(s). This mechanism is supported by the mutation-induced changes in the secondary electron transfer rate constants whereas the tertiary rates were not affected. Thus we assign the third electron transfer process (rate constant of ∼22 ns−1) to reduction of A1 from the initially reduced A0 Chl as shown in scheme 2b (Fig. 2). The same conclusion can also be derived from an entirely different argument. The close distances among the RC Chls and the larger distance of the A0 Chls to the A1 phylloquinone acceptors would make it unlikely that A0 transfers an electron to A1 before the P700 Chl(s) are oxidized, as implied in scheme 2c. Furthermore, the rate constant of the third electron transfer process in our preferred scheme 2b also agrees with the rate constant of A1 reduction measured directly by Savikhin et al. (58). Thus scheme 2b is left as the only sequence of electron transfer processes and radical pairs compatible with our data, provided that we consider only asymmetric electron transfer. In this case, it also seems more likely that the B-branch is the active branch rather than the A-branch, since the mutation at PB showed the largest effect on the effective electron transfer rate constant. This conclusion would be in contrast to the recent interpretation of EPR data by Cohen et al. (40) stating that the A-branch is the active branch, at least in cyanobacterial PS I. However, we need to point out here that our data do not really allow us to exclude a two-branched electron transfer mechanism as proposed earlier (38,46). To test such models, more mutant studies are required. In fact, we are quite open to such an electron transfer mechanism for PS I RCs. However, even if a two-branched mechanism were in fact present or—alternatively—even if the A-branch should turn out to be the exclusive route for electron transfer (40), this would hardly invalidate our notion that the P700 Chls are not the primary electron donor(s) and that this role is played in fact by one or both of the accessory Chls.
Our new electron transfer mechanism solves a severe problem inherent in the hitherto accepted electron transfer scheme. There has been general agreement that the intrinsic primary step of electron transfer from the excited donor to the first acceptor should take ∼1 ps or less (18,22,25,58–60). Assigning this electron transfer step to a transfer from one of the P700 Chls to the nearest A0 acceptor would imply an edge-to-edge electron transfer distance between these cofactors of ∼14 Å, according to the structural data (5). It is highly unlikely, if not impossible, that electron transfer could occur over such a large distance in <1 ps. The new scheme proposed here obviates the necessity for such an ultrafast long range electron transfer step. In this scheme, electron transfer occurs only among neighboring cofactors and—barring any detailed calculations —the observed effective rate constant (∼350 ns−1), and in particular the implied intrinsic rate constants of the primary electron transfer step of >1 ps−1 do appear to be quite feasible within the RC structure.
In our previous work, we discussed the involvement of a protein relaxation step after initial charge separation as an alternative—although unlikely—explanation for the three radical pairs occurring in our new ET mechanism (8). In view of the data obtained on the mutants, we now exclude that possibility as the dominant explanation for the observation of the three early RPs. Protein relaxation after charge separation has been discussed both for PS I and for PS II earlier (see scheme Fig. 2 d). For isolated PS II RCs (D1-D2-cyt-b559 RC), this process is well documented (61–63) and it plays an important role for the stabilization of the radical pairs and for enhancing the forward electron transfer processes. Similar processes are known from isolated bacterial RCs (54,64–66). Radical pair relaxation has also been invoked to explain some kinetic features in intact PS II cores (67). It is possible that similar mechanisms also play a role in PS I RCs, but the timescale of such relaxations is unclear at present. A protein relaxation process might actually be necessary to explain, at least in part, the differences seen in the effective charge recombination rate constant for the different mutants, which may be hard to explain on the basis of redox potential changes alone in the P700 Chls. However, radical pair relaxation as an exclusive or even dominant mechanism to explain the mutant induced changes in the effective rate constants of electron transfer, in particular of the secondary electron transfer rate constant, can be safely excluded. In all cases of protein relaxation steps studied so far, the spectra of the radical pairs did not change significantly, which is in fact expected, since such relaxation steps are due to subtle global and local changes of the protein conformation. However, we observed substantial differences of the cofactor spectra between radical pairs 2 and 3 which is not compatible with an interpretation invoking primarily protein relaxation. We want to point out that recent data reporting an electrochromic shift do in fact suggest a protein relaxation process in PS I radical pairs, although mainly on the longer time range after the reduction of A1 (68).
CONCLUSIONS
Our data strongly suggest that the P700 Chls are not the primary electron donor(s) and the best possible interpretation at present assigns this function to the accessory Chl(s). However, it remains unclear whether electron transfer is indeed highly asymmetric—as was assumed in our minimal analysis here—or whether it is more symmetric thus involving both branches, although not necessarily with a 1:1 activity ratio. The available data are conflicting. Evidence for B-branch activity comes from C. reinhardtii PS I samples (38). The data in this work support substantial B-branch activity but are inconclusive about a possible two-branch mechanism. If there should be a more symmetric or balanced two-branch electron transfer mechanism in action, the effective rate constants determined in this work for the electron transfer processes should be interpreted as average effective rate constants for the two branches.
Evidence for exclusive A-branch activity and asymmetric electron transfer comes from studies on cyanobacterial PS I (40). We would like to discuss here briefly whether the available data in the literature would support opposite preferences for the two branches in different species. On the one hand the absorption spectra of the RCs, the absorption difference spectra of the P700+ state, and the T-S spectra are substantially different for PS I from different species (43). On the other hand species differences are not more, but generally less, pronounced than between the wt and the mutant PS I RCs studied in this work. In view of the fact that the electron transfer processes for all the particles studied here can be modeled quite well within the same general scheme, in the absence of clear indications for significant changes in reaction mechanism between the different mutants, it would be surprising, in our view, if there existed drastic species differences in the electron transfer mechanism in different PS I particles. Perhaps all the existing data from different species, mutants, and different measuring techniques could be best explained within a unified model taking into account two-sided electron transfer. Any species-dependent and/or mutation-induced differences could probably be explained easily with modest changes in the relative contributions of the two branches. At present the existing data are inconclusive, but it will be a challenge for the coming years to put that hypothesis to the test.
Acknowledgments
We acknowledge the excellent work by Claudia Schulz (Max-Volmer-Institut, Technische Universität, Berlin) in the preparation of the PS I particles, and we thank Michael Reus (Max-Planck-Institut für Bioanorganische Chemie, Mülheim an der Ruhr) for help with the measurements and characterization of the particles. Dr. Heike Witt, Berlin, is gratefully acknowledged for providing the mutants and for valuable comments.
References
- 1.Cramer, W. A., and A. R. Crofts. 1982. Electron and proton transport. In Photosynthesis. 1: Energy Conversion by Plants and Bacteria. Govindjee, editor. Academic Press, New York. 387–467.
- 2.Avron, M. 1981. Photosynthetic electron transport and photophosphorylation. In The Biochemistry of Plants. Photosynthesis. 8. M. D. Hatch, N. K. Boardman, P. K. Stumpf, and E. E. Conn, editors. Academic Press, New York. 163–191.
- 3.Haehnel, W. 1984. Photosynthetic electron transport in higher plants. Ann. Rev. Plant Physiol. 35:659–693. [Google Scholar]
- 4.Brettel, K., and W. Leibl. 2001. Electron transfer in Photosystem I. Review. Biochim. Biophys. Acta. 1507:100–114. [DOI] [PubMed] [Google Scholar]
- 5.Jordan, P., P. Fromme, H. T. Witt, O. Klukas, W. Saenger, and N. Krauβ. 2001. Three-dimensional structure of cyanobacterial Photosystem I at 2.5 Å resolution. Nature. 411:909–917. [DOI] [PubMed] [Google Scholar]
- 6.Krauss, N., W.-D. Schubert, O. Klukas, P. Fromme, H. T. Witt, and W. Saenger. 1996. Photosystem I at 4 A resolution represents the first structural model of a joint photosynthetic reaction centre and core antenna system. Nat. Struct. Biol. 3:965–973. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Shem, A., F. Frolow, and N. Nelson. 2003. Crystal structure of plant Photosystem I. Nature. 426:630–635. [DOI] [PubMed] [Google Scholar]
- 8.Müller, M. G., J. Niklas, W. Lubitz, and A. R. Holzwarth. 2003. Ultrafast transient absorption studies on Photosystem I reaction centers from Chlamydomonas reinhardtii. 1. A new interpretation of the energy trapping and early electron transfer steps in Photosystem I. Biophys. J. 85:3899–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holzwarth, A. R., M. G. Müller, J. Niklas, and W. Lubitz. 2005. Charge recombination fluorescence in Photosystem I reaction centers from Chlamydomonas reinhardtii. J. Phys. Chem. B. 109:5903–5911. [DOI] [PubMed] [Google Scholar]
- 10.Döring, G., J. L. Bailey, W. Kreutz, J. Weikard, and H. T. Witt. 1968. Some new results in photosynthesis. Naturwissenschaften. 55:219–220. [PubMed] [Google Scholar]
- 11.Stiehl, H. H., and H. T. Witt. 1968. Die kurzzeitigen ultravioletten Differenzspektren bei der Photosynthese. Z. Naturforsch. 23B:220–224. [PubMed] [Google Scholar]
- 12.Webber, A. N., and W. Lubitz. 2001. P700: the primary electron donor of Photosystem I. Biochim. Biophys. Acta. 1507:61–79. [DOI] [PubMed] [Google Scholar]
- 13.Melkozernov, A. N. 2001. Excitation energy transfer in Photosystem I from oxygenic organisms. Photosynth. Res. 70:129–153. [DOI] [PubMed] [Google Scholar]
- 14.Hastings, G., S. Hoshina, A. N. Webber, and R. E. Blankenship. 1995. Universality of energy and electron transfer processes in Photosystem I. Biochemistry. 34:15512–15522. [DOI] [PubMed] [Google Scholar]
- 15.Melkozernov, A. N., H. Su, S. Lin, S. Bingham, A. N. Webber, and R. E. Blankenship. 1997. Specific mutation near the primary donor in Photosystem I from Chlamydomonas reinhardtii alters the trapping time and spectroscopic properties of P700. Biochemistry. 36:2898–2907. [DOI] [PubMed] [Google Scholar]
- 16.Holzwarth, A. R., D. Dorra, M. G. Müller, and N. V. Karapetyan. 1998. Structure-function relationships and excitation dynamics in Photosystem I. In Photosynthesis: Mechanism and Effects. G. Garab, editor. Kluwer Academic Publishers, Dordrecht, The Netherlands. 497–502.
- 17.Schlodder, E., M. Cetin, M. Byrdin, I. V. Terekhova, and N. V. Karapetyan. 2005. P700+- and 3P700-induced quenching of the fluorescence at 760 nm in trimeric Photosystem I complexes from the cyanobacterium Arthrospira platensis. Biochim. Biophys. Acta. 1706:53–67. [DOI] [PubMed] [Google Scholar]
- 18.Holzwarth, A. R., G. H. Schatz, H. Brock, and E. Bittersmann. 1993. Energy transfer and charge separation kinetics in Photosystem I: 1. Picosecond transient absorption and fluorescence study of cyanobacterial Photosystem I particles. Biophys. J. 64:1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibasiewicz, K., V. M. Ramesh, A. N. Melkozernov, S. Lin, N. W. Woodbury, R. E. Blankenship, and A. N. Webber. 2001. Excitation dynamics in the core antenna of PS I from Chlamydomonas reinhardtii CC 2696 at room temperature. J. Phys. Chem. B. 105:11498–11506. [Google Scholar]
- 20.Melkozernov, A. N., S. Lin, and R. E. Blankenship. 2000. Femtosecond transient spectroscopy and excitonic interactions in Photosystem I. J. Phys. Chem. B. 104:1651–1656. [DOI] [PubMed] [Google Scholar]
- 21.Melkozernov, A. N., H. Su, A. N. Webber, and R. E. Blankenship. 1998. Excitation energy transfer in thylakoid membranes from Chlamydomonas reinhardtii lacking chlorophyll b and with mutant Photosystem I. Photosynth. Res. 56:197–207. [Google Scholar]
- 22.Trinkunas, G., and A. R. Holzwarth. 1996. Kinetic modeling of exciton migration in photosynthetic systems. 3. Application of genetic algorithms to simulations of excitation dynamics in three-dimensional Photosystem I core antenna/reaction center complexes. Biophys. J. 71:351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gobets, B., and R. van Grondelle. 2001. Energy transfer and trapping in Photosystem I. Review. Biochim. Biophys. Acta. 1507:80–99. [DOI] [PubMed] [Google Scholar]
- 24.Gobets, B., J. T. M. Kennis, J. A. Ihalainen, M. Brazzoli, R. Croce, I. H. M. van Stokkum, R. Bassi, J. P. Dekker, H. van Amerongen, G. R. Fleming, and R. van Grondelle. 2001. Excitation energy transfer in dimeric light harvesting complex I: A combined streak-camera/fluorescence upconversion study. J. Phys. Chem. B. 105:10132–10139. [Google Scholar]
- 25.Gobets, B., I. H. M. van Stokkum, M. Rögner, J. Kruip, E. Schlodder, N. V. Karapetyan, J. P. Dekker, and R. van Grondelle. 2001. Time-resolved fluorescence emission measurements of Photosystem I particles of various cyanobacteria: a unified compartmental model. Biophys. J. 81:407–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Käss, H., P. Fromme, H. T. Witt, and W. Lubitz. 2001. Orientation and electronic structure of the primary donor radical cation P-700*• in Photosystem I: a single crystals EPR and ENDOR study. J. Phys. Chem. B. 105:1225–1239. [Google Scholar]
- 27.Plato, M., N. Krauss, P. Fromme, and W. Lubitz. 2003. Molecular orbital study of the primary electron donor P700 of Photosystem I based on a recent x-ray single crystal structure analysis. Chem. Phys. 294:483–499. [Google Scholar]
- 28.Breton, J. 2001. Fourier transform infrared spectroscopy of primary electron donors in type I photosynthetic reaction centers. Biochim. Biophys. Acta. 1507:180–193. [DOI] [PubMed] [Google Scholar]
- 29.Prokhorenko, V. I., and A. R. Holzwarth. 2000. Primary processes and structure of the Photosystem II reaction center: a photon echo study. J. Phys. Chem. B. 104:11563–11578. [Google Scholar]
- 30.Zouni, A., H. T.Witt, J. Kern, P. Fromme, N. Krauβ, W. Saenger, and P. Orth. 2001. Crystal structure of Photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 409:739–743. [DOI] [PubMed] [Google Scholar]
- 31.Diner, B. A., E. Schlodder, P. J. Nixon, W. J. Coleman, F. Rappaport, J. Lavergne, W. F. J. Vermaas, and D. A. Chisholm. 2001. Site-directed mutations at D1-His198 and D2-His197 of Photosystem II in Synechocystis PCC 6803: sites of primary charge separation and cation and triplet stabilization. Biochemistry. 40:9265–9281. [DOI] [PubMed] [Google Scholar]
- 32.Barter, L. M. C., J. R. Durrant, and D. R. Klug. 2003. A quantitative structure-function relationship for the Photosystem II reaction center: supermolecular behavior in natural photosynthesis. Proc. Natl. Acad. Sci. USA. 100:946–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Brederode, M. E., I. H. M. van Stokkum, E. Katilius, F. van Mourik, M. R. Jones, and R. van Grondelle. 1999. Primary charge separation routes in the BChl:BPhe heterodimer reaction centers of Rhodobacter sphaeroides. Biochemistry. 38:7545–7555. [DOI] [PubMed] [Google Scholar]
- 34.van Brederode, M. E., and R. van Grondelle. 1999. New and unexpected routes for ultrafast electron transfer in photosynthetic reaction centers. FEBS Lett. 455:1–7. [DOI] [PubMed] [Google Scholar]
- 35.Diner, B. A., and F. Rappaport. 2002. Structure, dynamics, and energetics of the primary photochemistry of Photosystem II of oxygenic photosynthesis. Annu. Rev. Plant Biol. 53:551–580. [DOI] [PubMed] [Google Scholar]
- 36.Angerhofer, A., and R. Bittl. 1996. Radicals and radical pairs in photosynthesis. Photochem. Photobiol. 63:11–38. [Google Scholar]
-
37.Muhiuddin, I. P., P. Heathcote, S. Carter, S. Purton, S. E. J. Rigby, and M. C. W. Evans. 2001. Evidence from time resolved studies of the
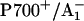 radical pair for photosynthetic electron transfer on both the PsaA and PsaB branches of the Photosystem I reaction centre. FEBS Lett. 503:56–60. [DOI] [PubMed] [Google Scholar]
radical pair for photosynthetic electron transfer on both the PsaA and PsaB branches of the Photosystem I reaction centre. FEBS Lett. 503:56–60. [DOI] [PubMed] [Google Scholar] - 38.Guergova-Kuras, M., B. Boudreaux, A. Joliot, P. Joliot, and K. Redding. 2001. Evidence for two active branches for electron transfer in Photosystem I. Proc. Natl. Acad. Sci. USA. 98:4437–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fairclough, W. V., A. Forsyth, M. C. W. Evans, S. E. J. Rigby, S. Purton, and P. Heathcote. 2003. Bidirectional electron transfer in Photosystem I: electron transfer on the PsaA side is not essential for phototrophic growth in Chlamydomonas. Biochim. Biophys. Acta. 1606:43–55. [DOI] [PubMed] [Google Scholar]
- 40.Cohen, R. O., G. Z. Shen, J. H. Golbeck, W. Xu, P. R. Chitnis, A. I. Valieva, A. J. van der Est, Y. Pushkar, and D. Stehlik. 2004. Evidence for asymmetric electron transfer in cyanobacterial Photosystem I: analysis of a methionine-to-leucine mutation of the ligand to the primary electron acceptor A(0). Biochemistry. 43:4741–4754. [DOI] [PubMed] [Google Scholar]
- 41.Krabben, L., E. Schlodder, R. Jordan, D. Carbonera, G. Giacometti, H. Lee, A. N. Webber, and W. Lubitz. 2000. Influence of the axial ligands on the spectral properties of P700 of Photosystem I: a study of site-directed mutants. Biochemistry. 39:13012–13025. [DOI] [PubMed] [Google Scholar]
- 42.Webber, A. N., H. Su, S. E. Bingham, H. Käss, L. Krabben, M. Kuhn, R. Jordan, E. Schlodder, and W. Lubitz. 1996. Site-directed mutations affecting the spectroscopic characteristics and midpoint potential of the primary donor in Photosystem I. Biochemistry. 35:12857–12863. [DOI] [PubMed] [Google Scholar]
- 43.Witt, H., E. Bordignon, D. Carbonera, J. P. Dekker, N. V. Karapetyan, C. Teutloff, A. Webber, W. Lubitz, and E. Schlodder. 2003. Species-specific differences of the spectroscopic properties of P700: analysis of the influence of non-conserved amino acid residues by site-directed mutagenesis of Photosystem I from Chlamydomonas reinhardtii. J. Biol. Chem. 278:46760–46771. [DOI] [PubMed] [Google Scholar]
- 44.Witt, H., E. Schlodder, C. Teutloff, J. Niklas, E. Bordignon, D. Carbonera, S. Kohler, A. Labahn, and W. Lubitz. 2002. Hydrogen bonding to P700: site-directed mutagenesis of threonine A739 of Photosystem I in Chlamydomonas reinhardtii. Biochemistry. 41:8557–8569. [DOI] [PubMed] [Google Scholar]
- 45.Gibasiewicz, K., V. M. Ramesh, S. Lin, K. Redding, N. W. Woodbury, and A. N. Webber. 2003. Excitonic interactions in wild-type and mutant PSI reaction centers. Biophys. J. 85:2547–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petrenko, A., A. L. Maniero, J. van Tol, F. MacMillan, Y. J. Li, L.-C. Brunel, and K. Redding. 2004. A high-field EPR study of P700ñ in wild-type and mutant Photosystem I from Chlamydomonas reinhardtii. Biochemistry. 43:1781–1786. [DOI] [PubMed] [Google Scholar]
- 47.Wang, R. L., V. Sivakumar, Y. J. Li, K. Redding, and G. Hastings. 2003. Mutation induced modulation of hydrogen bonding to P700 studied using FTIR difference spectroscopy. Biochemistry. 42:9889–9897. [DOI] [PubMed] [Google Scholar]
- 48.Owens, T. G., S. P. Webb, L. Mets, R. S. Alberte, and G. R. Fleming. 1989. Antenna structure and excitation dynamics in Photosystem I. II. Studies with photosynthetic mutants of Chlamydomonas reinhardtii lacking Photosystem II. Biophys. J. 56:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hippler, M., F. Drepper, J. Farah, and J.-D. Rochaix. 1997. Fast electron transfer from cytochrome c6 and plastocyanin to Photosystem I of Chlamydomonas reinhardtii requires PsaF. Biochemistry. 36:6343–6349. [DOI] [PubMed] [Google Scholar]
- 50.Porra, R. J., W. A. Thompson, and P. E. Kriedemann. 1989. Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim. Biophys. Acta. 975:384–394. [Google Scholar]
- 51.Croce, R., M. G. Müller, R. Bassi, and A. R. Holzwarth. 2001. Carotenoid-to-chlorophyll energy transfer in recombinant major light-harvesting complex (LHC II) of higher plants. I. Femtosecond transient absorption measurements. Biophys. J. 80:901–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holzwarth, A. R. 1996. Data analysis of time-resolved measurements. In Biophysical Techniques in Photosynthesis. Advances in Photosynthesis Research. J. Amesz and A. J. Hoff, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 75–92.
- 53.Gibasiewicz, K., V. M. Ramesh, S. Lin, N. W. Woodbury, and A. N. Webber. 2002. Excitation dynamics in eukaryotic PS I from Chlamydomonas reinhardtii CC 2696 at 10 K. Direct detection of the reaction center exciton states. J. Phys. Chem. B. 106:6322–6330. [Google Scholar]
-
54.Tang, C.-K., J. C. Williams, A. K. W. Taguchi, J. P. Allen, and N. W. Woodbury. 1999. The
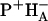 charge recombination reaction rate constant in Rhodobacter sphaeroides reaction centers is independent of the P/P+ midpoint potential. Biochemistry. 38:8794–8799. [DOI] [PubMed] [Google Scholar]
charge recombination reaction rate constant in Rhodobacter sphaeroides reaction centers is independent of the P/P+ midpoint potential. Biochemistry. 38:8794–8799. [DOI] [PubMed] [Google Scholar] - 55.Kammel, M., J. Kern, W. Lubitz, and R. Bittl. 2003. Photosystem II single crystals studied by transient EPR: the light-induced triplet state. Biochim. Biophys. Acta. 1605:47–54. [DOI] [PubMed] [Google Scholar]
- 56.Lubitz, W., F. Lendzian, and R. Bittl. 2002. Radicals, radical pairs and triplet states in photosynthesis. Acc. Chem. Res. 35:313–320. [DOI] [PubMed] [Google Scholar]
- 57.Breton, J., E. Nabedryk, and W. Leibl. 1999. FTIR study of the primary electron donor of Photosystem I (P700) revealing delocalization of the charge in P700+ and localization of the triplet character in 3P700. Biochemistry. 38:11585–11592. [DOI] [PubMed] [Google Scholar]
-
58.Savikhin, S., W. Xu, P. Martinsson, P. R. Chitnis, and W. S. Struve. 2001. Kinetics of charge separation and
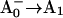 electron transfer in Photosystem I reaction centers. Biochemistry. 40:9282–9290. [DOI] [PubMed] [Google Scholar]
electron transfer in Photosystem I reaction centers. Biochemistry. 40:9282–9290. [DOI] [PubMed] [Google Scholar] - 59.Trinkunas, G., and A. R. Holzwarth. 1994. Kinetic modeling of exciton migration in photosynthetic systems. 2. Simulations of excitation dynamics in two-dimensional Photosystem I core antenna/reaction center complexes. Biophys. J. 66:415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gobets, B., I. H. M. van Stokkum, F. van Mourik, J. P. Dekker, and R. van Grondelle. 2003. Excitation wavelength dependence of the fluorescence kinetics in Photosystem I particles from Synechocystis PCC 6803 and Synechococcus elongatus. Biophys. J. 85:3883–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holzwarth, A. R., M. G. Müller, G. Gatzen, M. Hucke, and K. Griebenow. 1994. Ultrafast spectroscopy of the primary electron and energy transfer processes in the reaction center of Photosystem II. J. Luminesc. 60, 61:497–502. [Google Scholar]
- 62.Konermann, L., G. Gatzen, and A. R. Holzwarth. 1997. Primary processes and structure of the Photosystem II reaction center. V. Modeling of the fluorescence kinetics of the D1–D2-Cyt-b559 complex at 77 K. J. Phys. Chem. B. 101:2933–2944. [Google Scholar]
- 63.Gatzen, G., M. G. Müller, K. Griebenow, and A. R. Holzwarth. 1996. Primary processes and structure of the Photosystem II reaction center: III. Kinetic analysis of picosecond energy transfer and charge separation processes in the D1–D2-cyt-b559 complex measured by time-resolved fluorescence. J. Phys. Chem. 100:7269–7278. [Google Scholar]
- 64.Holzwarth, A. R., and M. G. Müller. 1996. Energetics and kinetics of radical pairs in reaction centers from Rhodobacter sphaeroides. A femtosecond transient absorption study. Biochemistry. 35:11820–11831. [DOI] [PubMed] [Google Scholar]
- 65.Lin, S., J. Jackson, A. K. W. Taguchi, and N. W. Woodbury. 1998. Excitation wavelength dependent spectral evolution in Rhodobacter sphaeroides R-26 reaction centers at low temperatures: the Q(y) transition region. J. Phys. Chem. B. 102:4016–4022. [Google Scholar]
- 66.Peloquin, J. M., J. C. Williams, X. Lin, R. G. Alden, A. K. W. Taguchi, J. P. Allen, and N. W. Woodbury. 1994. Time-dependent thermodynamics during early transfer in reaction centers from Rhodobacter sphaeroides. Biochemistry. 33:8089–8100. [DOI] [PubMed] [Google Scholar]
- 67.Vassiliev, S., C.-I. Lee, G. W. Brudvig, and D. Bruce. 2002. Structure-based kinetic modeling of excited-state transfer and trapping in histidine-tagged Photosystem II core complexes from Synechocystis. Biochemistry. 41:12236–12243. [DOI] [PubMed] [Google Scholar]
- 68.Dashdorj, N., W. Xu, P. Martinsson, P. R. Chitnis, and S. Savikhin. 2004. Electrochromic shift of chlorophyll absorption in Photosystem I from Synechocystis sp PCC 6803: a probe of optical and dielectric properties around the secondary electron acceptor. Biophys. J. 86:3121–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]