

Abstract
Regulation and maintenance of cell water volume and intracellular pH (pHi) are vital functions that are interdependent; cell volume regulation affects, and is in turn affected by, changes in pHi. Disruption of either function underlies various pathologies. To study the interaction and kinetics of these two mechanisms, we developed and validated a quantitative fluorescence imaging microscopy method to measure simultaneous changes in pHi and volume in single cells loaded with the fluorescent probe BCECF. CWV is measured at the excitation isosbestic wavelength, whereas pHi is determined ratiometrically. The method has a time resolution of <1 s and sensitivity to osmotic changes of ∼1%. It can be applied in real time to virtually any cell type attached to a coverslip, independently of cellular shape and geometry. Calibration procedures and algorithms developed to transform fluorescence signals into changes in cell water volume (CWV) and examples of applications are presented.
INTRODUCTION
Mechanisms controlling cellular pH and water volume are among the evolutionarily oldest homeostatic functions. Presumably, these two vital mechanisms appeared when macromolecules were encased within a semipermeable plasma membrane creating an internal compartment in which metabolic reactions occurred (1,2). Mechanisms controlling cell water volume and intracellular pH (pHi) are tightly coupled; cell volume regulation affects, and is in turn affected by, changes in pHi. This interdependency becomes evident during the operation of membrane transporters involved in pHi regulation, such as 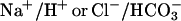 exchangers. Activation of these transporters requires transmembrane movement of acid equivalents in exchange for osmotically active ions (Na+ and Cl−). This offsets osmotic equilibrium resulting in unbalanced water fluxes across the plasma membrane with consequent changes in cell water volume (3–5). A number of metabolic disorders, such as hyperammonemia in acute liver failure (6) and lactacidosis in various pathologies (7), result in alterations of pHi and cell water volume control. A dreaded outcome of these disorders is the glial and neuronal swelling underlying often lethal brain edema. Thus, in view of the interdependency of cell water volume and pHi regulation and its relevance in cell physiology and pathology, development of methods for simultaneous measurement of these parameters in single cells in real time and with adequate sensitivity is of great importance. Here we present and validate a quantitative fluorescence imaging microscopy method that allows simultaneous measurement of changes in pHi and cell water volume (CWV) in single cells loaded with the fluorescent probe BCECF. The method is based on the spectral properties of this dye that has a pH-insensitive excitation isosbestic wavelength (IW) in which the fluorescence intensity varies with dye concentration and a peak excitation wavelength at 495 nm that is pH-sensitive. CWV changes are estimated from the fluorescence emitted by exciting the dye at its intracellular IW (∼438 nm), and pHi is measured by standard procedures from the ratio of the emitted fluorescence by exciting at 495 and 438 nm (8–10). The relatively high time resolution (<1 Hz) and sensitivity to osmotic changes (∼1%) of this technique permits detailed quantification of the monitored changes, thus allowing study of the kinetics and interactions between the two variables in single cells. The method can be applied in real time to virtually any cell type attached to a coverslip, independently of its shape and geometry. We present procedures to calibrate and transform the fluorescence signals into changes in CWV, as well as examples of applications of the method.
exchangers. Activation of these transporters requires transmembrane movement of acid equivalents in exchange for osmotically active ions (Na+ and Cl−). This offsets osmotic equilibrium resulting in unbalanced water fluxes across the plasma membrane with consequent changes in cell water volume (3–5). A number of metabolic disorders, such as hyperammonemia in acute liver failure (6) and lactacidosis in various pathologies (7), result in alterations of pHi and cell water volume control. A dreaded outcome of these disorders is the glial and neuronal swelling underlying often lethal brain edema. Thus, in view of the interdependency of cell water volume and pHi regulation and its relevance in cell physiology and pathology, development of methods for simultaneous measurement of these parameters in single cells in real time and with adequate sensitivity is of great importance. Here we present and validate a quantitative fluorescence imaging microscopy method that allows simultaneous measurement of changes in pHi and cell water volume (CWV) in single cells loaded with the fluorescent probe BCECF. The method is based on the spectral properties of this dye that has a pH-insensitive excitation isosbestic wavelength (IW) in which the fluorescence intensity varies with dye concentration and a peak excitation wavelength at 495 nm that is pH-sensitive. CWV changes are estimated from the fluorescence emitted by exciting the dye at its intracellular IW (∼438 nm), and pHi is measured by standard procedures from the ratio of the emitted fluorescence by exciting at 495 and 438 nm (8–10). The relatively high time resolution (<1 Hz) and sensitivity to osmotic changes (∼1%) of this technique permits detailed quantification of the monitored changes, thus allowing study of the kinetics and interactions between the two variables in single cells. The method can be applied in real time to virtually any cell type attached to a coverslip, independently of its shape and geometry. We present procedures to calibrate and transform the fluorescence signals into changes in CWV, as well as examples of applications of the method.
MATERIALS AND METHODS
Cell cultures
Neuroblastoma N1E-115 (11) and neuroblastoma × glioma NG108-15 (12) cell lines (kindly provided by Dr. Marshall Nirenberg, National Heart, Lung, and Blood Institute, Bethesda, MD) were cultured at 37°C in Dulbecco's modified eagle's medium supplemented with 10% fetal calf serum (FCS), 1% hypoxanthine-aminopterin-thymidine, and 1% l-glutamine, in a 5% CO2/95% air atmosphere. Cells from passages 16–25 were plated on 25-mm-diameter glass coverslips previously treated with poly-d-lysine. Differentiation was induced 24 h after plating by supplying the cells with a differentiation medium composed of 98% Dulbecco's modified eagle's medium, 2% FCS, 1% hypoxanthine-aminopterin-thymidine, 1% l-glutamine, 1 mM theophylline, and 10 μM prostaglandin E1. Cells were used 3 to 7 days after the differentiation treatment.
For primary cultures of neonatal rat cortical astrocytes, pups (P0-P4, Sprague Dawley) were used. Animals were decapitated, the meninges were removed, the brain cortices minced in Hank's balanced salt solution, incubated in Hank's balanced salt solution-trypsin 0.14%, and filtered through an 80-μm-diameter nylon mesh (13). The cell suspension was plated on 25-mm-diameter glass coverslips previously treated with poly-d-lysine. The plating density was ∼200,000 cells/coverslip. The maintenance medium consisted of minimum essential medium with Earle's salts, without l-glutamine, supplemented with fetal bovine serum (FBS) 20%, concentrated vitamin solution 3%, aminoacids 2%, sodium bicarbonate 2%, and penicillin-streptomycin 1% (v/v). Cells were incubated at 37°C in a 5% CO2/95% air atmosphere. After 2 days, the medium was changed to one in which FBS was reduced to 10%. Subsequent medium changes were made every 3 days. Ten days after the initial seeding, some cultures were differentiated by adding 0.25 mM dibutyryl cAMP to maintenance medium containing 3% FBS. The latter treatment yielded type 2 astrocytes. Astrocytic phenotype was confirmed by GFAP immunostaining. Experiments were done in astrocytes cultured for 12–30 days. All culture reagents were purchased from Gibco-Invitrogen (Carlsbad, CA). Animal-use protocols were approved by the Laboratory Animal Care and Use Committee of Wright State University, Dayton, OH.
Saline solutions
The control isosmotic solution (ISO) contained (in mM): NaCl, 120; KCl, 5.5; CaCl2, 2.5; MgCl2, 1.25; HEPES, 20; and glucose, 10. The pH was adjusted to 7.3 with NaOH. The osmolality was adjusted with sucrose to ∼312 mOsm/Kg water (for neuroblastoma cells) or ∼290 mosmol/Kg water (for astrocytes). Anisosmotic solutions were prepared by sucrose addition or removal keeping the ionic concentrations constant and at the value of the control ISO; they were expressed as percentage decrement or increment with respect to the control ISO. Isosmotic NH4Cl− or lactate-containing solutions were prepared from the ISO solution with mole-by-mole substitution of NaCl for NH4Cl or Na-lactate, respectively.
Dye loading
A coverslip containing the cells is mounted in an imaging chamber (RC-21BRW; Warner Instruments, Hamden, CT) and placed on the stage of an epifluorescence inverted microscope (see below). Cells are loaded with the acetoxymethyl esters of BCECF (5 μM) or calcein (2–4 μM) (Invitrogen-Molecular Probes) dissolved in control ISO and incubated (∼15–40 min) at room temperature. Dye loading progress is monitored until desired levels of fluorescence (∼600–2000 units with our system) are achieved. As with other fluorophores, the amount of dye loaded into cells is crucial. Excessive dye concentration can result in fluorescence self-quenching and the responses at the IW will have a sign opposite to that expected, i.e., fluorescence will increase with cell swelling (dye dilution) and decrease with cell shrinkage (14,15). The dye loading solution is washed out with ISO and the cells are equilibrated for 10–15 min in this solution. Experimental solutions are perfused (7 ml/min) by means of a valve system (VC-6; Warner Instruments). The chamber fluid is exchanged with a time constant of 3.6 ± 0.3 s (n = 6).
For spectral determinations of the IW of BCECF (see Fig. 6 F), BCECF free acid (5 μM) was dissolved in a solution containing (in mM): KCl, 130; NaCl, 10; MgCl2, 1; and MOPS, 10.
FIGURE 6.

Simultaneous measurement of changes in CWV and pHi in single cells. (A) Changes in CWV (Vt/V0) recorded in a BCECF-loaded N1E-115 cell exposed to two pulses of isosmotic solutions, the first containing 20 mM NH4Cl and the second 20 mM lactic acid, as indicated by bars. The concurrent changes in pHi are shown in B. (C) Changes in CWV recorded in a calcein-loaded cell upon exposure to osmotic calibration pulses (±10% anisosmotic) and a pulse of 10 mM NH4Cl, as indicated in the bars. Note that the volume responses are in every respect similar to those recorded with BCECF (compare with record in A). (D and E) Parallel CWV (D) and pHi (E) changes recorded in a BCECF-loaded N1E-115 cell exposed to anisosmotic calibration pulses applied before and during perfusion with a solution containing 20 mM NH4Cl. Bars indicate the percent change in external osmolality with respect to the isosmotic solution for each calibration pulse. (F) Effects of pH on fluorescence emitted upon exciting BCECF at or near its IW in a recording chamber without cells. There is negligible pH interference with the IW signal for excitation wavelengths of 438 or 436 ± 3 nm. However, cross talk was significant when exciting at wavelengths >438 nm. In each test solution, pH was adjusted with NaOH by reciprocal dilutions, keeping constant the BCECF concentration (5 μM free acid).
Imaging setup and fluorescence recording
Total fluorescence from a small circular digital pinhole region placed at the image plane of fluorophore-loaded single cells is measured with a system that includes an inverted, epifluorescence microscope Nikon Diaphot 300 (Nikon USA, Melville, NY) or Olympus IX-81 (Olympus America, Melville, NY) equipped with fluor oil-immersion objective lenses (Nikon, 40× nA 1.3 and 60× nA 1.4; Olympus, 40×, nA 1.35). The excitation light coming from a 75W xenon arc lamp passes through the input slit of a monochromator (Optoscan, Cairn Research Limited, Faversham, UK) that allows independent slit width control for each wavelength. BCECF is excited at 495 ± 2–5 nm and at 438 ± 3–6 nm. The delay between excitation light pulses is 15 ms. Calcein is excited at 495 ± 3 nm. The excitation light beam is transmitted through a liquid light guide and passes through neutral density filters (optional) before entering the microscope optical path. A dichroic mirror (515 nm) and a 535 ± 13-nm emission filter cube (Omega Optical, Brattleboro, VT, or Chroma Technology Corp, Rockingham, VT) is positioned underneath the objective lens in the filter holder of the microscope. In addition to neutral density filters, to further decrease light exposure to minimize photobleaching and photodynamic damage, fluorescence is collected at a frequency ≤0.2 Hz during shutter openings of 40–80 ms duration. Cell images are captured by a cooled digital CCD camera (ORCA-ER C4742-95, Hamamatsu; Hamamatsu City, Japan). Emitted fluorescence from selected digital pinholes and cell images is monitored on-line. Image acquisition, digital pinhole size and position, and fluorescence recording are done with MetaFluor imaging software (Molecular Devices, Sunnyvale, CA). A diagram of the general layout of the imaging setup is shown in Fig. 1.
FIGURE 1.

Imaging setup used to record CWV and pHi changes in single cells loaded with BCECF.
pHi measurements
The fluorescence excitation ratio (495/438 nm) for each recording point is translated into pHi using the high K+/nigericin in vivo calibration technique as described by Boyarsky and co-workers (8–10).
RESULTS
Basic principle
Changes in CWV are measured in single cells by introducing an impermeant volume marker (m) into cells and measuring its change in concentration (Fig. 2 A). If the intracellular content of the marker (Qm) remains constant throughout osmotic perturbations, fractional changes in its concentration (Cm) will reflect changes in CWV according to the following equation:
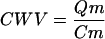 |
(1) |
With fluorescent probes, changes in CWV are measured from changes in the dye's fluorescence intensity (F) that result in turn from changes in its intracellular concentration (14,16,17). Using BCECF, CWV changes are inferred from changes in emitted F by excitation at the intracellular IW (∼438 nm). The changes in F are collected from a small region of a dye-loaded cell with either conventional wide-field fluorescence microscopy, or with confocal or multiphoton microscopy. The boundary of this region is delimited by a circular pinhole aperture located at the image plane (Fig. 2, B–D). This pinhole can be mechanical or digital and ideally should have a diameter of ∼3–10% of the total area of the cell for a given focal plane (see below).
FIGURE 2.

Basic principles of the method for measuring changes in CWV using intracellular fluorescent dyes as volume markers. Changes in CWV are inferred from changes in concentration of intracellular BCECF or any suitable dye. (A) Each large sphere represents a cell loaded with a volume marker (pink spheres). The cell in the middle (normalcellvolume) is at equilibrium with an isotonic solution. Exposure to a hypotonic (hypo) solution causes cell swelling and dilution of the volume marker. Exposure to a hypertonic solution (hyper) results in cell shrinkage and an increase in the marker concentration. If the marker is fluorescent, osmotically induced changes in its intracellular concentration will produce changes in relative F that can be converted to relative changes in CWV using Eq. 6. (B–D) Microphotographs of differentiated N1E-115 cells loaded with BCECF and observed with transmitted light (B); with fluorescence optics exciting at 495 nm (C); and with pseudocolor fluorescence intensity gradient (D). The circular regions (pinholes) located at the center of each cell indicate the area from which fluorescence is collected. (E) Changes in BCECF relative fluorescence (Ft/F0) recorded by exciting the dye at its intracellular IW (438 ± 2.5 nm) in response to two pulses of calibration solutions ±10% anisosmotic (yellow bar and gray bar, respectively). (F) Plot of the reciprocal of the apparent steady-state changes in fluorescence (F0/Ft) as a function of the reciprocal of the relative osmotic pressure of the medium (π0/πt) for the calibration pulses shown in E. The y intercept of a linear regression fitted to the observed data points (solid line) corresponds to the fraction of intracellularly trapped and osmotically insensitive fluorescence (Fbkg) that in this case was 0.62. Subtraction of Fbkg (Eqs. 5 and 6) yields the dotted line indicating ideal osmometric behavior. (G) Changes in CWV (Vt/V0) resulting from transformation of the fluorescence signals shown in E, using Eq. 6. (H) Plot of the apparent steady-state changes in Vt/V0 as a function of π0/πt. The solid line fitted to the data points has a slope ≈1, which corresponds to ideal osmometric behavior. (I) Repeatability test (intra-assay precision) of osmotic calibrations with BCECF. The value of Fbkg determined for the four pairs of calibration pulses was 0.55 ± 0.01 (mean ± SE). The variability (CV) of the changes in Vt/V0 was ∼2% for both hyperosmotic and hyposmotic calibration pulses.
As CWV changes, the concentration of the free diffusible fluorophore molecules (Cf) changes in inverse proportion (Eq. 1). As long as the relation between F and Cf is linear and has a positive slope (14,15), the emitted F collected through the pinhole region is directly proportional to Cf, and can be quantitatively described by the following simplified expression derived from the Beer-Lambert exponential law (18):
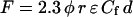 |
(2) |
where F (for a constant light input) and Cf have been defined, φ is the quantum efficiency of the fluorophore, r is a constant that represents optical instrumental factors, ɛ is the extinction coefficient (at ∼438 nm for the IW of BCECF), and d is the thickness (optical path length) of the sample. φ, r, and ɛ are constants, and d can also be considered constant when the depth of field of the objective is less than the cell height (see below). For measurements of CWV changes, it is critical to eliminate or minimize the effects of changes in d resulting from changes in cell geometry, particularly changes in cell height. With epi-illumination, the image of the intracellular region from which light is measured is a “volume element” of fixed size whose shape is determined by the pinhole aperture placed at the image plane and the numerical aperture of the objective (19). This volume element can be envisaged as formed by two inverted cones touching each other at the apex (19–22), roughly having the shape of an hourglass. The narrowest region, i.e., where the apexes touch each other, corresponds to the focal plane (Zo) along the z optical axis (depth) and is the point where F is maximal. The height of each cone will be Zo + DF and Zo − DF, and hence will be determined by the depth of field (DF) of the objective, defined in Young (19) and Piller (23) as
 |
(3) |
where nA is the numerical aperture of the objective lens, M the total magnification, and λ the wavelength of light (in micrometers). In other words, the regions above and below the focal plane contribute to the measurements made at Zo, depending on DF. Note that the DF “shrinks” inversely proportionally with the square of the nA. Hence the larger the nA, the shallower the DF will be. A shallow DF is crucial to assure that the recorded fluorescence signals come from the cell interior, thereby eliminating signal distortion resulting from out-of-focus fluorescence. This ensures that the focal volume element in which the dye molecules are dissolved is contained within the cell boundaries. In most cases, this is achieved by using oil-immersion objectives with high numerical apertures (e.g., 40×, nA 1.3, or 60×, nA 1.4), and by focusing at the point of maximal fluorescence in the optical z axis that roughly coincides with the center of the cell (24). For instance, if λ = 0.5 μm, M = 40×, and nA = 1.3 for an oil-immersion objective, DF will be ∼0.4 μm. Hence, if the cell height is of the order of several micrometers, say 5 μm or more, the “volume element” should remain contained within the cell boundaries. Note that objectives having large magnification and nA yield a shallow DF at the expense of fluorescence intensity. We have found that for most applications a 40×, nA 1.3 objective is adequate.
Calibration of fluorescence signals to measure water-volume changes in single cells
Fig. 2 E shows the osmotic behavior of a single BCECF-loaded neuroblastoma cell in which relative fluorescence changes in response to pulses of anisosmotic calibration solutions were measured through a circular pinhole region (Fig. 2 D) when exciting the dye at 438 ± 2.5 nm. Relative fluorescence is expressed as Ft/F0, where F0 is the fluorescence measured at the pinhole region in an isosmotic control solution having osmotic pressure π0, and Ft is the fluorescence of the same region upon exposure to a solution having osmotic pressure πt. The resulting changes in Ft/F0 reached a steady state shortly after the onset of each osmotic challenge. As expected with these modestly anisosmotic pulses the cells do not show regulatory volume responses. Plotting the reciprocal of the apparent steady-state changes in fluorescence (F0/Ft) as a function of the reciprocal of the relative osmotic pressure of the medium (π0/πt) yields a linear relationship (Fig. 2 F), indicating that the changes in fluorescence reflect changes in intracellular concentration of BCECF that in turn reflect changes in CWV. However, the slope of the regression line fitted to the data points is always <1, the slope expected for ideal osmometric behavior according to the equation:
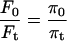 |
(4) |
Accordingly, if all the fluorophore molecules diffused freely in the cytosol and the emitted fluorescence was directly proportional to the concentration of BCECF, the following relation should hold:
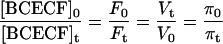 |
(5) |
where [BCECF]0 and [BCECF]t are the intracellular concentrations of BCECF at time 0 and at time t, respectively, Vt is the cell water volume at time t, and V0 is Vt when t = 0. The departure from ideal behavior (Fig. 2 F) is due to fluorophore trapped or bound to internal structures that is insensitive to changes in external osmolality (14). To transform the fluorescence signals into CWV changes, Fbkg must be assessed and subtracted (see below). CWV changes (Vt/V0) are computed from monitored changes in Ft/F0 resulting from exciting BCECF at the intracellular IW (∼438 nm) according to Eq. 6.
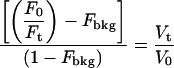 |
(6) |
To assess Fbkg, BCECF-loaded cells are treated with the pore-forming α-toxin from Staphylococcus aureus to produce selective plasma membrane permeabilization while monitoring Ft/F0. The steady-state fluorescence remaining after controlled membrane permeabilization is Fbkg. Detergents such as digitonin underestimate Fbkg. This is because even at low concentrations (e.g., 10 μM) digitonin permeabilizes not only the plasma membrane but also internal membranes, and may even lead to the release of intracellular structures (25). We found that Fbkg determined by controlled permeabilization with α-toxin corresponds to the value determined from the y intercept of a plot of F0/Ft versus π0/πt (Fig. 2 F). Hence, we routinely use this intercept as Fbkg, which is determined in every experiment from the osmotic calibration plot for each cell. Fbkg is fairly constant within cells of the same type. For example, in N1E-115 cells (n = 47) used here, Fbkg was 0.61 ± 0.01 (mean ± SE) and the coefficient of variation (CV) was 12.4%.
Fig. 2 G shows changes in Vt/V0 computed for the whole Ft/F0 trace (Fig. 2 E) applying Eq. 6. Fig. 2 H shows the corresponding plot of steady-state changes in Vt/V0 versus π0/πt. The solid line fitted to the calibration points has a slope ≈1, indicating that the cell behaves as an ideal osmometer within this range of external osmolalities. The accuracy of the osmotic calibration expressed as a percentage (100 × (observed value/theoretical (ideal) value)) was determined in 20 cells and was found to be 99.4 ± 1.2% (mean ± CV) for the ∼10% hyposmotic pulses, and 99.5 ± 1.0% for the ∼10% hyperosmotic pulses.
In a usual experimental protocol only one set of calibration pulses is applied before experimental manipulations, and the Fbkg value obtained from these pulses is used to calculate the whole volume transient for each cell. This procedure assumes that Fbkg remains constant for the duration of the experiment. To test the validity of this assumption, the degree of variability (expressed as CV) of Fbkg upon repeated calibrations (intra-assay precision) over periods of up to 1.5 h was determined in single cells, in experiments like the one shown in Fig. 2 I in which four pairs of calibration pulses were applied. The CV determined for each cell (n = 4) ranged between 2 and 4%, indicating that Fbkg is fairly constant for the duration of a typical experiment. Hence, a single pair of calibration pulses is sufficient to compute the whole Vt/V0 transient. Moreover, as is shown in Fig. 6, D and E, and discussed below, Fbkg remains constant during displacements of cell water volume and pHi.
Intracellular isosbestic wavelength of BCECF
Fluorescent dyes may change their spectral properties in an intracellular environment. Although this does not appear to be a problem with BCECF (10,26), knowledge of the precise value of its intracellular IW is critical to avoid pH interference with the volume signal. We determined the intracellular (in vivo) IW of BCECF and compared it with that measured in vitro by running BCECF spectra with a monochromator (SLM-Aminco, DMX 1100; excitation slit, 8 nm; scan, 400–516 nm) through the optical pathway of the microscope under two conditions: 1), in the experimental chamber without cells; and 2), in the cytosol of single neuroblastoma cells (NG108-15). The IW in vitro (437.5 ± 0.2 nm, n = 44) was not different from that in vivo (437 ± 0.3 nm, n = 151). There was negligible interference of pH with IW when using a wavelength excitation of 438 nm with bandwidths up to ±4 nm (see Fig. 6 F).
Effect of pinhole size and position on cell volume signals
The relative size and position of the pinhole through which F signals are collected is critical. Ideally, the F signal used to monitor CWV changes is emitted from osmotically active fluorophore molecules contained within a defined volume element and optical path length (d in Eq. 2). The volume element should remain contained within the cell boundaries upon changes in cell volume or cell shape. However, in most animal cells, height is not uniform, gradually decreasing from a maximum at the nuclear region to minimum values toward the cell periphery. Thus, as the cell periphery is approached more of the effective DF of the objective lens tends to lie outside the cell boundaries in the z optical axis. Also, as the cell periphery is approached, cell height rather than DF of the objective lens will determine the effective path length. The result is that F signals are distorted in these relatively thin regions of the cells, reflecting changes in path length that interfere with the dye-concentration-dependent signal used to monitor CWV. The influence of cell-height variations on recorded signals can be minimized by sampling at the center of the cell. Given that the volume of a spherical cell changes with the cube of the cell radius, in this region osmotically-induced changes in cell diameter are relatively small with respect to the total cell height (Figs. 3 and 4). Also, the pinhole size should be small enough to sample from a region having relatively uniform cell height (Fig. 3). Many of these parameters are difficult to quantify but can be analyzed empirically. To determine the optimal signal/noise ratio (S/N) we studied the effect of varying pinhole size (Fig. 3) and location (Fig. 4) in cells loaded with BCECF challenged with anisosmotic calibration pulses. As the pinhole size increased (Fig. 3 C) the monitored changes in Ft/F0 decreased in amplitude and the signals became distorted (Fig. 3 A, R1–R4). The two pinholes confined within the cell boundary (Fig. 3 C, regions 1 and 2) yielded the largest Ft/F0 signals, but the best S/N ratio (∼30) was obtained for region 1 (Fig. 3 E). This region had an area of 31 μm2, corresponding to ∼3% of the total cell area at the focal plane. For the pinholes that were larger than the cell and outside its boundaries (Fig. 3 C, regions 3 and 4) the Ft/F0 signals were distorted and considerably reduced in amplitude (Fig. 3 A, R3 and R4). The corresponding Vt/V0 for each Ft/F0 trace is shown in Fig. 3 B. Clearly the cleanest signal was obtained from region 1 (Fig. 3 B). The noise and distortion of the signals at region 3 (R3 in Fig. 3 A) were considerable and at region 4 the Ft/F0 signal (R4 in Fig. 3 A) prevented calculation of Vt/V0.
FIGURE 3.

Effect of pinhole size on fluorescence responses to osmotic calibration pulses applied to a cell loaded with BCECF. (A) Fluorescence changes (Ft/F0) in response to a 10% hyperosmotic and a 10% hyposmotic pulse (as indicated by boxes) recorded through pinholes of various sizes (R1–R4), as shown in C. (B) Transformation of Ft/F0 signals from each region into Vt/V0. (D) Plot of the apparent steady-state changes in CWV (Vt/V0) as a function of the reciprocal of the relative osmotic pressure of the medium (π0/πt) for regions R1–R3. The solid straight line denotes ideal osmometric behavior. (E) S/N calculated for Ft/F0 records obtained from R1–R4. S/N was calculated following Srinivas and Bonanno (33). Signal distortion increases and S/N decreases as the pinhole size increases. The trace having minimal distortion and maximal S/N corresponds to R1. BCECF was excited at 438 ± 2 nm (N1E-115 neuroblastoma cell). The pinhole sizes expressed as a percentage of total cell area were: R1, ≈3%; R2, ≈30%; R3, ≈83%; and R4, ≈163%.
FIGURE 4.

Effect of pinhole position on fluorescence responses to osmotic calibration pulses applied to a cell loaded with BCECF. (A) Fluorescence changes (Ft/F0) in response to a 10% hyperosmotic and a 10% hyposmotic calibration pulse applied as indicated by the yellow and gray bars, respectively. The pinhole from which Ft/F0 was recorded had an area of 31μm2 (∼3% of total cell area at the image plane). The pinhole was positioned in different cellular regions (R1–R4), as shown in E (1–4). (B) Ft/F0 signals from each region converted into Vt/V0. (C) Plot of the apparent steady-state changes in Vt/V0 as a function of the reciprocal of the relative osmotic pressure of the medium (π0/πt) for regions R1–R4. The solid line fitted to the data points denotes ideal osmometric behavior. (D) S/N calculated for Ft/F0 records obtained from regions R1–R4. The trace having minimal distortion and maximal S/N was obtained from R2, where the fluorescence intensity and the cell height are maximal (E, 2).
The effect of pinhole position on cell volume calibration pulses and S/N ratio are shown in Fig. 4. As expected, the maximal S/N ratio (∼35–40; Fig. 4 D) was obtained for the signals recorded in the region of maximal fluorescence (R2 in Fig. 4 A) corresponding to the region of maximal cell height (Fig. 4 E, region 2).
The method can be applied to cells of different height and shape
One of the main advantages of this method is that CWV changes can be measured in single cells independently of their geometry. The limiting factor is cell height, but with the appropriate combination of objective and nA it is possible to measure volume changes in cells having heights as small as 5 μm, such as cultured type 1 astrocytes (Fig. 5, A–D).
FIGURE 5.

Changes in CWV measured in BCECF-loaded cells of different height and shape. Changes in Ft/F0 in response to osmotic calibration pulses (A and E) recorded from a single type 1 astrocyte (C, pinhole region 8), and from a star-shaped type 2 astrocyte (G, pinhole region 1). The corresponding calibration plots for type 1 and type 2 astrocytes are shown in B–D and F–H, respectively. Type 1 astrocytes are characteristically “flat” (cell heights ∼4–5 μm); type 2 astrocytes have a more complex geometry and their height in the nuclear region is ≥10 μm (34,35). BCECF was excited at 438 ± 3 nm in C and G.
Simultaneous recording of CWV and intracellular pH changes in single cells exposed to NH3/NH4+ or lactic acid
Accuracy of CWV measurements requires lack of crosstalk between the fluorescence signals recorded at the intracellular IW and those arising from pH-sensitive spectral regions of BCECF. Fig. 6 (A, B, D, and E) shows recordings from individual cells in which cell water volume and pHi were altered by various experimental maneuvers to test signal independence and capabilities of the method. Fig. 6, A and B, shows the changes in Vt/V0 and pHi recorded in a single neuroblastoma (N1E-115) cell exposed to two pulses of isosmotic solutions containing 20 mM of NH4Cl and 20 mM Na+-lactate, respectively. Note that, although both treatments produced cell swelling (A), the concurrent pHi changes occurred, as expected, in opposite directions (B), demonstrating the independence of the BCECF fluorescence signals denoting CWV and pHi. Also, the kinetics of each swelling was different, reflecting differences in the mechanisms of solute permeation and/or intracellular accumulation. When the cell is exposed to NH4Cl, the neutral base NH3 rapidly diffuses into the cell and becomes protonated, forming 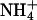 (27). The osmotically active
(27). The osmotically active 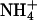 is accumulated in the cell interior, producing water influx and a relatively rapid cell swelling. In contrast, lactic acid crosses the plasma membrane through slower saturable transport mechanisms (7,28) and accumulates more slowly, producing a slower swelling than that produced by NH3.
is accumulated in the cell interior, producing water influx and a relatively rapid cell swelling. In contrast, lactic acid crosses the plasma membrane through slower saturable transport mechanisms (7,28) and accumulates more slowly, producing a slower swelling than that produced by NH3.
The changes in CWV measured with BCECF can be reproduced in cells loaded with calcein, a single excitation-emission fluorescent dye (Fig. 6 C), demonstrating that such changes are independent of the dye used as an intracellular volume marker. Another stringent test showing that the signals from the IW reflect osmotic volume changes is shown in Fig. 6 D. In this experiment the swelling induced by  exposure can be counteracted or increased in a predictable manner by applying anisosmotic pulses during exposure. These experiments provide further evidence of the independence of volume and pHi signals, inasmuch as the application of the anisosmotic pulses does not alter the time course and direction of the simultaneously recorded pHi changes shown in Fig. 6 E. These experiments also served to further test the constancy of Fbkg during experimental perturbations of CWV and pHi. The values of Fbkg estimated from each set of calibration pulses before and during NH4Cl exposure did not differ from each other (0.55 ± 0.2 and 0.55 ± 0.2; n = 12).
exposure can be counteracted or increased in a predictable manner by applying anisosmotic pulses during exposure. These experiments provide further evidence of the independence of volume and pHi signals, inasmuch as the application of the anisosmotic pulses does not alter the time course and direction of the simultaneously recorded pHi changes shown in Fig. 6 E. These experiments also served to further test the constancy of Fbkg during experimental perturbations of CWV and pHi. The values of Fbkg estimated from each set of calibration pulses before and during NH4Cl exposure did not differ from each other (0.55 ± 0.2 and 0.55 ± 0.2; n = 12).
In many applications, dye excitation is performed using commercial filters with relatively wide bandwidths (e.g., 440 ± 10 nm to excite at the IW of BCECF). We studied the effects of pH on the emitted fluorescence upon exciting BCECF at several wavelengths at and near the IW, using narrower slits (6 nm) than those commonly used. As shown in Fig. 6 F, for the range of pH values tested (5.40–7.47), minimal interference of pH with the IW fluorescence is achieved when BCECF is excited at 436 or 438 ± 3 nm, which corresponds to the intracellular IW. With longer wavelengths (e.g., 442 nm), the fluorescence increases with the pH. Thus, appropriate choice of the wavelength to excite at the IW and use of narrow excitation bandwidths assure negligible cross talk between signals and so reliable measurements.
DISCUSSION
We show that BCECF can be used to measure parallel changes in CWV and pHi in single cells. The advantages of this method are:
It is possible to perform simultaneous on-line measurement of changes in CWV and pHi in single cells with unique sensitivity and time resolution.
The fluorescence signals can be calibrated for defined changes in extracellular osmolality for each individual cell, allowing each cell to be used as its own osmometer.
The fluorescence at the IW is independent of changes in concentration of native intracellular ions, including Na+, H+, Ca2+, and Mg2+, within biologically meaningful ranges.
The spectral properties of BCECF are well known and, like other esterified dyes, it can be loaded into cells noninvasively.
Because of the quantum yield of BCECF, it is possible to reduce the intensity of the excitation light-producing minimal photobleaching and photodynamic damage.
Measurements are independent of changes in cell shape or geometry within the range of interest.
It can be used with any widefield fluorescence microscope for cells having heights of ≥5 μm with the appropriate choice of slits, excitation wavelengths, and objectives.
Although the possibility of using BCECF to measure CWV and pHi changes had been suggested (29), the method had not been calibrated and validated for quantitative analysis, and the value used for the IW (450 nm) was incorrect; at this wavelength the dye is significantly sensitive to pH.
In this method, the nucleus and the cytoplasm are considered as a single compartment given that small molecules such as BCECF free acid (556.5 mol wt) diffuse freely across the nuclear envelope. Although BCECF translational diffusion in the cytoplasm is approximately four times slower than in water (30), the dye equilibrates very rapidly, with a t1/2 recovery time (from fluorescence-recovery-after-photobleaching experiments) in the range of 2.2–4.8 ms (31), whereas the time course of the osmotic water fluxes that result in volume changes are in the range of seconds.
The method described here can be applied to in vitro models of pathologies associated with cell water volume and pHi imbalances such as hyperammonemia and lactacidosis (6,7), using cells (e.g., astrocytes) attached to coverslips. The principles of the method can also be applied in two-photon excitation imaging microscopy for more demanding situations involving shallower cells or cellular structures such as growth cones and neuronal dendritic spines. For instance, activity-dependent changes in dendritic spine volume have recently been shown to underlie long-term potentiation, a plastic change in synaptic transmission thought to play a key role in memory formation (32). This method may be applicable for studying the mechanisms underlying such changes.
Acknowledgments
We thank Dr. R. W. Putnam for comments on the manuscript, Dr. J. E. Olson for showing us how to culture astrocytes, and Mr. R. Garduño for technical assistance. Part of this work was done while F.J.A.-L. was a faculty member of the Dept. of Pharmacobiology, Cinvestav-IPN, Mexico, and of the Dept. of Neurobiology, National Institute of Psychiatry, Mexico.
This research was supported by the National Institute of Neurological Disorders and Stroke grant NS29227 to F.J.A.-L.
References
- 1.Morris, C. E. How did cells get their size? 2002. Anat. Rec. 268:239–251. [DOI] [PubMed] [Google Scholar]
- 2.Chen, I. A., and J. W. Szostak. 2004. Membrane growth can generate a transmembrane pH gradient in fatty acid vesicles. Proc. Natl. Acad. Sci. USA. 101:7965–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
3.Cala, P. M., and S. Grinstein. 1988. Coupling between Na+/H+ exchange and
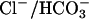 exchange in pH and volume regulation. In Na+/H+ exchange. S. M. Grinstein, editor. CRC Press, Boca Raton, FL. 201–208.
exchange in pH and volume regulation. In Na+/H+ exchange. S. M. Grinstein, editor. CRC Press, Boca Raton, FL. 201–208.
- 4.Heming, T. A., G. Boyarsky, D. Tuazon, and A. Bidani. 2000. pHi responses to osmotic cell shrinkage in the presence of open-system buffers. J. Appl. Physiol. 89:1543–1552. [DOI] [PubMed] [Google Scholar]
- 5.Jakab, M., J. Fürst, M. Gschwentner, G. Bottà, M. L. Garavaglia, C. Bazzini, S. Rodighiero, G. Meyer, S. Eichmüeller, E. Wöll, S. Chwatal, M. Ritter, and M. Paulmichl. 2002. Mechanisms sensing and modulating signals arising from cell swelling. Cell. Physiol. Biochem. 12:235–258. [DOI] [PubMed] [Google Scholar]
- 6.Felipo, V., and R. F. Butterworth. 2002. Neurobiology of ammonia. Prog. Neurobiol. 67:259–279. [DOI] [PubMed] [Google Scholar]
- 7.Ringel, F., R. C. Chang, F. Staub, A. Baethmann, and N. Plesnila. 2000. Contribution of anion transporters to the acidosis-induced swelling and intracellular acidification of glial cells. J. Neurochem. 75:125–132. [DOI] [PubMed] [Google Scholar]
- 8.Boyarsky, G., C. Hanssen, and L. A. Clyne. 1996. Inadequacy of high K+/nigericin for calibrating BCECF I. Estimating steady-state intracellular pH. Am. J. Physiol. 271:C1131–C1145. [DOI] [PubMed] [Google Scholar]
- 9.Boyarsky, G., C. Hanssen, and L. A. Clyne. 1996. Inadequacy of high K+/nigericin for calibrating BCECF II. Intracellular pH dependence of the correction. Am. J. Physiol. 271:C1146–C1156. [DOI] [PubMed] [Google Scholar]
- 10.Boyarsky, G., C. Hanssen, and L. A. Clyne. 1996. Superiority of in vitro over in vivo calibrations of BCECF in vascular smooth muscle cells. FASEB J. 10:1205–1212. [DOI] [PubMed] [Google Scholar]
- 11.Amano, T., E. Richelson, and M. Nirenberg. 1972. Neurotransmitter synthesis by neuroblastoma clones. Proc. Natl. Acad. Sci. USA. 69:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamprecht, B. 1977. Structural, electrophysiological, biochemical and pharmacological properties of neuroblastoma-glioma hybrid cells in culture. Int. Rev. Cytol. 49:99–170. [DOI] [PubMed] [Google Scholar]
- 13.Olson, J. E., and D. Holtzman. 1980. Respiration in rat cerebral astrocytes from primary culture. J. Neurosci. Res. 5:497–506. [DOI] [PubMed] [Google Scholar]
- 14.Crowe, W. E., J. Altamirano, L. Huerto, and F. J. Alvarez-Leefmans. 1995. Volume changes in single N1E–115 neuroblastoma cells measured with a fluorescent probe. Neuroscience. 69:283–296. [DOI] [PubMed] [Google Scholar]
- 15.Hamann, S., J. F. Kiilgaard, T. Litman, F. J. Alvarez-Leefmans, B. R. Winther, and T. Zeuthen. 2002. Measurement of cell volume changes by fluorescence self-quenching. J. Fluoresc. 12:139–145. [Google Scholar]
- 16.Alvarez-Leefmans, F. J., J. Altamirano, and W. E. Crowe. 1995. Use of ion-selective microelectrodes and fluorescent probes to measure cell volume. In Methods in Neurosciences, Vol. 27. J. Kraicer and S. J. Dixon, editors. Academic Press, New York. 361–391.
- 17.Altamirano, J., M. S. Brodwick, and F. J. Alvarez-Leefmans. 1998. Regulatory volume decrease and intracellular Ca2+ in murine neuroblastoma cells studied with fluorescent probes. J. Gen. Physiol. 112:145–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rost, F. W. D. 1992. Fluorescence Microscopy, Vol. 1. Cambridge University Press, New York.
- 19.Young, I. T. 1989. Image fidelity: characterizing the imaging transfer function. In Methods in Cell Biology, Vol. 30. D. L. Taylor and Y.-L. Wang, editors. Academic Press, New York. 1–45. [DOI] [PubMed]
- 20.Born, M., and E. Wolf. 1980. Principles of Optics, 6th ed. Pergamon Press, Oxford, UK.
- 21.Keller, H. E. 1995. Objective lenses for confocal microscopy. In Handbook of Biological Confocal Microscopy. J. B. Pawley, editor. Plenum Press, New York. 111–137.
- 22.Shaw, P. J. 1995. Comparison of wide-field/deconvolution and confocal microscopy for 3D imaging. In Handbook of Biological Confocal Microscopy. J. B. Pawley, editor. Plenum Press, New York. 373–387.
- 23.Piller, H. 1977. Microscope Photometry. Springer-Verlag, Berlin.
- 24.Weinlich, M., C. Theiss, C. T. Lin, and R. K. Kinne. 1998. BCECF in single cultured cells: inhomogeneous distribution but homogeneous response. J. Exp. Biol. 201:57–62. [DOI] [PubMed] [Google Scholar]
- 25.Ahnert-Hilger, G., and M. Gratzl. 1988. Controlled manipulation of the cell interior by pore-forming proteins. Trends Pharmacol. Sci. 9:195–197. [DOI] [PubMed] [Google Scholar]
- 26.Owen, C. S. 1992. Comparison of spectrum-shifting intracellular pH probes 5′(and 6′)-carboxy-10-dimethylamino-3-hydroxyspiro[7H-benzo[c]xanthene-7, 1′(3′H)-isobenzofuran]-3′-one and 2′,7′-biscarboxyethyl-5(and 6)-carboxyfluorescein. Anal. Biochem. 204:65–71. [DOI] [PubMed] [Google Scholar]
- 27.Boron, W. F., and P. De Weer. 1976. Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J. Gen. Physiol. 67:91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nedergaard, M., and S. A. Goldman. 1993. Carrier-mediated transport of lactic acid in cultured neurons and astrocytes. Am. J. Physiol. 265:R282–R289. [DOI] [PubMed] [Google Scholar]
- 29.Muallem, S., B. X. Zhang, P. A. Loessberg, and R. A. Star. 1992. Simultaneous recording of cell volume changes and intracellular pH or Ca2+ concentration in single osteosarcoma cells UMR-106–01. J. Biol. Chem. 267:17658–17664. [PubMed] [Google Scholar]
- 30.Kao, H. P., J. R. Abney, and A. S. Verkman. 1993. Determinants of the translational mobility of a small solute in cell cytoplasm. J. Cell Biol. 120:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swaminathan, R., S. Bicknese, N. Periasamy, and A. S. Verkman. 1996. Cytoplasmic viscosity near the cell plasma membrane: translational diffusion of a small fluorescent solute measured by total internal reflection-fluorescence photobleaching recovery. Biophys. J. 71:1140–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuzaki, M., N. Honkura, G. C. Ellis-Davies, and H. Kasai. 2004. Structural basis of long-term potentiation in single dendritic spines. Nature. 429:761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srinivas, S. P., and J. A. Bonanno. 1997. Measurement of changes in cell volume based on fluorescence quenching. Am. J. Physiol. 272:C1405–C1414. [DOI] [PubMed] [Google Scholar]
- 34.Shain, W., D. Bausback, A. Fiero, V. Madelian, and J. N. Turner. 1992. Regulation of receptor-mediated shape change in astroglial cells. Glia. 5:223–238. [DOI] [PubMed] [Google Scholar]
- 35.O'Connor, E. R., H. K. Kimelberg, C. R. Keese, and I. Giaever. 1993. Electrical resistance method for measuring volume changes in monolayer cultures applied to primary astrocyte cultures. Am. J. Physiol. 264:C471–C478. [DOI] [PubMed] [Google Scholar]