

Abstract
Current methods for identifying neoplastic cells and discerning them from their normal counterparts are often nonspecific, slow, biologically perturbing, or a combination thereof. Here, we show that single-cell micro-Raman spectroscopy averts these shortcomings and can be used to discriminate between unfixed normal human lymphocytes and transformed Jurkat and Raji lymphocyte cell lines based on their biomolecular Raman signatures. We demonstrate that single-cell Raman spectra provide a highly reproducible biomolecular fingerprint of each cell type. Characteristic peaks, mostly due to different DNA and protein concentrations, allow for discerning normal lymphocytes from transformed lymphocytes with high confidence (p ≪ 0.05). Spectra are also compared and analyzed by principal component analysis to demonstrate that normal and transformed cells form distinct clusters that can be defined using just two principal components. The method is shown to have a sensitivity of 98.3% for cancer detection, with 97.2% of the cells being correctly classified as belonging to the normal or transformed type. These results demonstrate the potential application of confocal micro-Raman spectroscopy as a clinical tool for single cancer cell detection based on intrinsic biomolecular signatures, therefore eliminating the need for exogenous fluorescent labeling.
INTRODUCTION
Many biomedical technologies are used to detect, identify, quantify, and sort both normal and neoplastic cells. Current radiological, pharmacological, histological, cellular, and molecular techniques, however, are destructive, compromised by imperfect specificity, perturb cellular biology, or a combination thereof. Technologies that avoid these shortfalls rely on cell-induced alterations in either light scattering or electrical conductivity. Unfortunately, these less invasive methods are applicable only to the identification or quantification of a minority of cell types and often require confirmatory evaluation that is subject to the aforementioned shortfalls. There is a need for new techniques that overcome such limitations to provide an easily automated, accurate method for cancer detection for both research and clinical environments.
Raman spectroscopy is a laser-based analytical technique that enables chemical characterization of molecules in a sample. It is based on the inelastic scattering of photons by molecular bond vibrations. A small portion of the incident photons are scattered by interaction with the bonds resulting in a shift toward lower frequencies. The energy difference between the incident and scattered photons corresponds to the vibrational energy of the specific molecular bond interrogated. A Raman spectrum obtained from cells or tissues is an intrinsic molecular fingerprint of the sample, revealing detailed information about DNA, protein, and lipid content as well as macromolecular conformations. This technique is rapid, noninvasive, and nondestructive. Recently, Raman spectroscopy has emerged as a novel nondestructive diagnostic tool for cancer detection (1–10) and identification of malignancy at different stages of the evolution of neoplasia in cells and tissue. For example, Utzinger et al. (11) studied squamous dysplasia and delineated high-grade dysplasia from all others based on observed alterations in peaks assigned to collagen, phospholipids, and DNA. Stone et al. (12) observed optical markers that discriminate cancer in laryngeal tissue using the relative intensity of the nucleic acid (DNA)/collagen mode at 1336 cm−1 and the amide III mode at 1250 cm−1. Omberg et al. (13) also studied the differences in the Raman signature of normal rat embryo fibroblast cells with those transfected with an oncogene that causes rapid, invasive tumor formation.
Confocal Raman spectroscopy allows femtoliter volumes within a sample to be probed with a diffraction-limited spatial resolution of <1 μm and permits microspectroscopy of a single cell. In combination with optical trapping (14,15) (also called laser tweezers) where a single laser beam is used for both manipulation and spectral interrogation, Raman spectroscopy of a single, “trapped” cell in suspension over an extended period of time is feasible, provided that the cell is nonadherent and the trapping forces can overcome the size and weight of the cell. This method, known as laser tweezers Raman spectroscopy (LTRS), has been used to study the dynamics of individual cells (15). For Raman spectroscopy to be developed as a clinical tool for single-cell cancer screening in a flow system, it is envisioned that optical trapping of the individual floating cells would be an essential component of such a Raman cytometry system.
In this work, we demonstrate the use of LTRS to obtain the Raman spectra of unfixed, normal lymphocytes in suspension. These spectra are compared to those of transformed Jurkat T- and Raji B- lymphocytes from cell lines to identify biochemical changes associated with neoplasia at the single-cell level. Micro-Raman spectroscopy is used to acquire the spectra of the transformed cells due to their larger cell size. To our knowledge, no cancer studies using Raman spectroscopy have so far been performed on individual, live cells in suspension and chemical changes between single normal and transformed hematopoietic cells have not been previously characterized using Raman spectroscopy.
MATERIALS AND METHODS
Cell preparation
Human peripheral blood mononuclear cells (PBMC) were obtained by Ficoll-Hypaque density gradient of heparinized venous blood obtained from healthy volunteers, as described elsewhere (16). The cells were washed three times in RPMI 1640 medium before purification. T-cells were separated by rosette formation with activated sheep red blood cells followed by Ficoll-Hypaque density gradient centrifugation. The lymphocytes in the B-enriched fraction are routinely >95% surface immunoglobulin positive as assessed by flow cytometry (17). T-cells were recovered from the rosetted pellet after twice lysing the sheep red blood cells with ammonium chloride buffer and thrice washing with RPMI 1640 medium. Both T- and B-cell populations were suspended in RPMI 1640 medium supplemented with 10% fetal calf serum at a density of ∼106 cells/ml. Cell preparations were >99% viable as assessed by trypan blue dye exclusion. Immediately before Raman spectra analysis the cells were washed and suspended in phosphate-buffered saline (PBS). Raji B cells and Jurkat T cells were obtained from American Type Culture Collection (ATCC, Rockville, MD) and maintained in culture with RPMI 1640 medium supplemented with 10% fetal calf serum. The cells were also washed and suspended in phosphate-buffered saline immediately before the Raman spectroscopic analysis. The use of fresh human samples for this study was in accordance with the University of California Institutional Review Board practice guidelines.
Raman spectroscopy of single cells
Raman spectra of individual cells are acquired using a confocal Raman microscope system that has been described previously (18). Briefly, a continuous wave (CW) 30-mW He-Ne laser beam emitting monochromatic 633 nm light (Spectra Physics, Mountain View, CA) is directed through a 633-nm bandpass filter (632.8NB3, Omega Filters, Brattleboro, VT) to remove plasma emission generated within the laser tube from the laser beam. The beam is expanded to 6 mm and delivered into an inverted optical microscope (Zeiss Axiovert 200, Göttingen, Germany). The microscope is equipped with a dichroic longpass beamsplitter to reflect the laser beam into a 100× 1.3 numerical aperture oil immersion objective (Zeiss Plan-NEOFLUAR), resulting in a diffraction limited spot of ∼0.5 μm diameter at the laser focus. Typical laser powers at the laser focus are 8–10 mW. The beam is focused through a glass coverslip of thickness 0.17 mm, which rests on a computer-controlled nanopositioning stage capable of scanning samples over a 100 μm × 100 μm transverse region and 20 μm axially. Cells in buffer solution placed on the coverslip are probed by the laser and spectroscopic signals generated at the focus are collected by the same objective, passed through the dichroic beamsplitter, and focused through a 100-μm pinhole for background signal rejection. A 633-nm holographic notch filter (Kaiser Optical) rejects residual backscattered laser light and the signal is directed into a spectrometer (Triax 320, Jobin-Yvon SPEX, Edison, NJ) equipped with a 1200 grooves/mm grating blazed at 500 nm and a liquid nitrogen cooled charge-coupled device (CCD) camera (Roper Scientific, Trenton, NJ) with a 1340 × 100 pixel chip. An acquisition time of 3 min for each cell is sufficient to yield Raman spectra with well-defined peaks. A flip mirror can alternatively direct the signal to a photomultiplier tube for fluorescence/Raman imaging. A separate CCD camera is used to collect white light microscope images of the cells being probed.
Acquisition of Raman spectra of single optically trapped cells
The same laser beam and oil immersion objective can be used for simultaneous optical trapping of individual cells and Raman interrogation (18). A 30-μl drop of suspended live cells in PBS solution is placed on the coverslip. A cell is positioned near the focus of the laser beam by moving the manual stage and becomes stably trapped and immobilized in three dimensions, ∼15 μm above the coverslip surface. Human T- and B-cells of ∼6-μm diameter are easily trapped and isolated away from the substrate and other cells. Larger cells could not be trapped and were interrogated after settling onto the coverslip surface.
Acquisition of Raman spectra of cells adhered to aglass surface
The size of the Jurkat and Raji cultured cells, ∼10–15 μm in diameter, prevented their manipulation by the laser trap. Instead, these larger cells are allowed to settle and become immobilized on a poly-L-lysine coated coverslip. The laser is then focused into the center of the cell, as determined by the overlap of the backscattered laser light at the focus and the white light image of the cell on the CCD camera. It should be noted that the size of these Jurkat and Raji neoplastic cultured cells is not representative of the size of cells from leukemia patients, which have a smaller diameter similar to that of normal T-and B-cells and can be optically trapped. For the purposes of this study, neoplastic cultured cells are used as model systems of leukemia cells for spectral characterization.
Spatial variation of Raman spectra within individual cells
The focal volume from which Raman signals are acquired within the cell is ∼1 μm3. To determine the homogeneity of the spectral signal in different regions of the cell, different locations within a single cell are probed. The cells (Jurkat, Raji, T-, B-) are allowed to adhere to the poly-L-lysine coated coverslip and then located using autofluorescence and Raman signals as the image contrast. The sample was raster scanned with respect to the laser focus to build the image pixel by pixel using the photomultiplier tube. Typical parameters of the imaging technique were 256 × 256 pixels with a 2-ms/pixel acquisition time. After acquiring an image of the cells, the laser spot was repositioned onto 4–5 different regions within the cell and Raman spectra were acquired at each location.
Data analysis
An individual Raman spectrum of a cell is acquired on a CCD chip comprised of 1340 individual channels. All spectra are calibrated using a toluene solution at room temperature as standard. Raman spectra are collected within the spectral region from ∼600 to 1800 cm−1. This region is known as the molecular fingerprint region and provides the most information on the biological constituents of a cell. Spectra are background corrected by subtraction of a third-order polynomial fit via an automated routine written using Igor Pro (WaveMetrics, Portland, OR) software. Individual spectra are then normalized with respect to the total area under the Raman curve. The spectra from each cell type are averaged individually, by channel, to obtain a mean Raman spectrum of that particular cell type. The standard deviation is calculated for each of the 1340 channels to determine cell-to-cell variability within a particular cell type. Same-channel differences between different cell-type spectra are defined using Student's t-test statistics. Subsequently, a principal component analysis (PCA) is performed on all spectra to extract persistent features of spectra from individual cell classes and to compare different cell classes against each other.
PCA, in the physical sciences also referred to as Eigenvector analysis, has many applications, including data reduction, variable (channel) selection and outlier detection (19). Its main purpose is to explain the variance-covariance structure of the data using a linear combination of the original variables to form principal components (PC). By finding combinations of the original dimensions that describe the largest variance between the data sets, all of the information from the original data set can be accounted for by a smaller number of variables. Herein, PCA is used to reduce the Raman spectral data with 1340 channels (dimensions) to two primary PCs. In most cases, the first two PCs account for close to 50% of the variances. A PCA routine was written using MATLAB software (The MathWorks, Natick, MA).
RESULTS
Biomolecular characterization of single hematopoietic cells
A typical Raman spectrum can be obtained from an individual living T-cell with 3-min integration time using ∼8 mW of laser power at the focus of a 100× oil immersion objective. Fig. 1 shows the average Raman spectra of all four cell types that we studied, human T- and B-cells isolated from a volunteer's blood, and transformed Jurkat T and Raji B cells. Each spectrum represents an average of measurements from 45 T-cells, 36 B-cells, 16 Raji cells and 86 Jurkat cells (solid lines) ± 1 SD (shaded lines). All of the spectra exhibit similar peak structures and locations with variations between cell types occurring mainly in select peak intensities as discussed in detail below. To confirm that any spectral differences between normal and neoplastic cells cannot be attributed to the manner in which the signals were acquired (i.e., optical trapping versus surface immobilization), individual normal T- and B-cells were also immobilized and probed on a poly-L-lysine coated glass slide. No differences in the spectral signatures between these cells and the optically trapped cells were observed.
FIGURE 1.

Average Raman spectra of individual Raji, B-, Jurkat, and T-cells in the fingerprint range from 600 cm−1 to 1700 cm−1. Each spectrum was obtained by averaging the spectra of several (∼16–86) individual spectra. The solid lines indicate the average spectra and the shaded lines delineate one standard deviation.
Spatial variability within a single cell
Several regions within a single cell were probed for their Raman signature to evaluate the spatial variation within a single cell. To determine this spatial variation, single live cells are first imaged and then probed at random locations. Fig. 2, top shows an autofluorescence image of a typical live Jurkat cell in PBS adhered to a glass coverslip. The laser focus is moved to different locations within the cell, labeled 1 through 5 in the image, and Raman spectra are obtained (Fig. 2, bottom). Analogous experiments were performed on Raji cells and single live T- and B-cells adhered to poly-L-lysine coated glass coverslips, with equivalent results (data not shown). These spectra indicate that there are no large spatial variations in the Raman signals within a single living cell, particularly no major variations in the signal intensities of peaks assigned to proteins and DNA. This result is further supported by the previous results shown in Fig. 1 that indicate random sampling of different cells in arbitrary locations yields reproducible data with low standard deviations.
FIGURE 2.

(Top) Autofluorescence image of a Jurkat T-cell adhered to a glass coverslip. The cells were excited at 632.8 nm wavelength and scanned in confocal microscopy mode to provide a spatial map of the location and size of the cells. Raman spectra were then obtained by repositioning the laser beam to positions 1–5 as indicated in the image. (Bottom) Raman spectra obtained from the different locations within the Jurkat T-cell as shown in top panel. Each spectrum was acquired within 180 s integration time with a laser power of 10 mW at 632.8 nm wavelength.
Raman spectral differences between normal and transformed cells
Fig. 3 a compares the mean Raman spectra obtained from 45 normal human T-cells and the mean Raman spectra of 86 cells obtained from their transformed counterpart, Jurkat T-cells. Also shown in the figure are the mean difference spectra (below). Fig. 3 b is a scatter plot of the intensity value of select peaks from individual spectra and their average peak value with associated standard deviations. All indicated peaks have significantly different (p ≪ 0.05) mean intensities as determined by t-test analysis. Similarly, a comparison of normal human B-cells with their transformed, Raji B-cell, counterpart revealed similar peaks with significantly different mean intensities (Fig. 3, c and d). The most obvious differences between normal cells and transformed cell lines can be found in the peaks at 678 cm−1, 785 cm−1, 1093 cm−1, 1126 cm−1, 1337 cm−1, 1373 cm−1, 1447 cm−1, 1485 cm−1, 1510 cm−1, and 1575 cm−1. These differences will be discussed further in the next section.
FIGURE 3.

(a) Overlay plot of the Raman spectra obtained from T (black line) and Jurkat (red line) cells. Also shown below is the difference spectrum. (b) Scatter plot showing the intensity value of select peaks with the most distinguishable differences from individual T- and Jurkat cell spectra and their average peak strengths and standard deviations. (c) Overlay plot of the Raman spectra obtained from B- (black line) and Raji cells (red line). The difference spectrum is shown below. (d) Scatter plot showing the intensity value of select peaks from individual B- and Raji cell spectra and the average peak strengths and standard deviations.
Principal component analysis of Raman spectra
Principal component analysis was performed on the individual Raman spectra. Direct comparisons between normal T- and B-cells and their respective transformed counterparts, Jurkat and Raji, are presented in Fig. 4, a and b. The plot of the first PC versus the second PC of the Raman spectra for the normal and Jurkat T-cells is shown in Fig. 4 a. In this analysis, 45 normal T-cell spectra were compared with 45 Jurkat T-cells. The variance attributed to each of the first three PCs of the Raman spectra accounts for 51% of the variance between the Raman spectra of the two cell types. The two-dimensional transformation presented necessarily incorporates only the first two PCs. Hence, 43% (first PC 27.6% + second PC 15.7%) of the variance between normal and Jurkat T-cell Raman spectra is used to discriminate between these two cell types. An analogous comparison between 36 normal B-cells and 16 transformed Raji B-cells is presented in Fig. 4 b. The first two PCs used in this comparison account for 59% of the variance between the Raman spectra obtained from these cell types (first PC 46.8% + second PC 12.9%). Finally, the four cell types were compared simultaneously to determine the resolution afforded by using the first two principal components, which accounted for 48% (first PC 33.1 + second PC 14.4%) of the variance (Fig. 4 c). Fig. 5 compares the difference spectra between T-cell and Jurkat cell (line a) and B-cell and Raji cell (line b) with the contributions of each channel (loading value) to the first PC for each of the three comparisons presented in Fig. 4 (lines c–e). A large positive or negative deviation from the baseline corresponds to increased contributions to the PC.
FIGURE 4.

Principal component analysis of all individual spectra of Raji, Jurkat, T-, and B-cells. Plots of the first principal component (PC1) versus the second principal component (PC2) for (a) Jurkat cells and T-cells, (b) Rajicells and B-cells, and (c) Jurkat, Raji, T-, and B-cells.
FIGURE 5.

Comparison of the difference spectra between (a) T-Jurkat and (b) B-Raji with the first principal component for each comparison presented in Fig. 4. (c) The T-cell versus Jurkat cell. (d) B-cell versus Raji cell. (e) T-cell versus B-cell versus Jurkat cell versus Raji cell. Increased positive or negative deviation from baseline indicates greater contribution to the PC.
DISCUSSION
Spectral variations from cell to cell
LTRS is capable of providing reproducible Raman spectral data from individual living cells. The cell-to-cell spectral variation for a particular cell type is low as shown in the standard-deviation plots for each of the four spectra in Fig. 1 and the plots in Fig. 3. The data in Fig. 2 also indicate a very low intracellular variability. This low spatial variation is surprising since it is known that many different cellular components are compartmentalized and studies by Uzunbajakava et al. (20) have observed such spatial variations in the Raman spectra of single, fixed lymphocytes. We attribute the relatively low spatial variations in our study to several factors. Lymphocytes have a very large nuclear/cell ratio, with the nucleus occupying ∼80% of the cell volume as observed under histochemical staining (21). Therefore, it is likely that the large nucleus is constantly being probed. Furthermore, this study probed live (nonfixed) cells, whose intracellular organelles can be subjected to radiation forces from the focused laser beam. It is likely that within the 3 min spectral integration time, the large nucleus is consistently being optically trapped within the focus when different areas within the cell are being probed (22). Nevertheless, variations between spectra within a cell type are still present. The largest variations occur in channels related to Raman peaks representing biochemical constituents, with different peaks exhibiting different magnitudes in their standard deviations. These defining variations are not surprising since there is cell-to-cell variability in the concentration of chemical components that may be emphasized by cells in a different portion of the cell cycle or cells with higher metabolic activity. Most importantly, however, both cell-to-cell and spatial variability are insignificant when compared to the much larger differences that are observed between normal and transformed cell spectra (see Fig. 3) as is confirmed by spectral analysis, channel-by-channel t-test analysis, and PCA, discussed below.
Spectral assignments and analysis
Raman spectroscopy for the detection of cancers in whole tissue is an established technique, enabling, e.g., the characterization and discrimination of benign and malignant tumors or different types of tumors in a variety of tissues (6,23). Typically, in these cases a relatively large volume deep inside the tumor is probed, resulting in averaged information from a large number of tumor cells in very different metabolic states (e.g., surface cells versus interior cells). This aids the analysis of whole tumors, but the results obtained from such studies are generally not applicable to individual cancer cells. Here, we analyze and characterize differences between individual T- and B-cells and their transformed leukemic counterparts, Jurkat and Raji cells.
The Raman spectra that we obtained from all 4 types of cells are similar to spectra of most living cells. The results obtained using LTRS (see Fig. 1) are consistent with other Raman spectral data obtained from human lymphocytes (24). The spectra of the four cell types share many similar peaks that can be assigned to cellular constituents (DNA/RNA, proteins, lipids, carbohydrates), based on previous data as summarized in Table 1. Typical bands in these spectra (see Fig. 1) are indicative of nucleotide conformation (600–800 cm−1), backbone geometry and phosphate ion interactions (800–1200 cm−1), electronic structure of the nucleotides (1200–1600 cm−1), and C-C and C-H modes due to proteins and lipids. Also, amide vibrations, such as the amide I-band (due to C=O stretching) and amide III band (due to C-N stretching, and N-H bending) in proteins are easily identifiable. Specific assignments of individual peaks can be found in Table 1. Here, we focus on and discuss some of the most visually distinct peaks that exhibit the most significant differences between transformed and normal cells. These distinct peaks are highlighted in Fig. 3, both in the difference spectra between T- and B-cells and their transformed counterparts and the average peak intensity points and standard deviation bars for each of the 10 most distinct peaks.
TABLE 1.
Raman frequencies and their assignments
| Raman frequency in wavenumber units (cm−1) | Assignment* |
|---|---|
| 618 | p: C-C twist |
| 640 | p: C-S str., C-C twist Tyr |
| 666 | G, T, Tyr, G bk in RNA |
| 678 | G (C-2′-endo-anti) |
| 725 | A |
| 746 | T |
| 785 | U, T, C, bk: O-P-O |
| 831 | O-P-O asym. str., Tyr |
| 852 | Tyr. ring breath. |
| 893 | bk, p: C-C skeletal |
| 1003 | Phe, C-C skeletal |
| 1031 | Phe, p: C-N str. |
| 1053 | C-O str., p: C-N str. |
| 1093 | O-P-O sym. str., p: C-N |
| 1126 | p: C-N str. |
| 1156 | p: C-C, C-N str. |
| 1175 | Tyr, Phe, p: C-H bend |
| 1208 | A, T, p: amide III |
| 1257 | A, T, p: amide III |
| 1263 | T,A, p: C-H bend |
| 1302 | P: amide III |
| 1318 | G, p: C-H def. |
| 1337 | A, G, p: C-H def. |
| 1373 | T, A, G |
| 1421 | A, G |
| 1447 | p: C-H2 def. |
| 1485 | G, A |
| 1510 | A |
| 1575 | G, A |
| 1605 | Phe, Tyr, p: C=C |
| 1615 | Tyr, Trp, p: C=C |
| 1655–1680 | p: amide I, T, G, C |
Abbreviations: str, stretching; def, deformation vibration; sym, symmetric; asym, asymmetric; bk, vibration of the DNA backbone; U, C, T, A, G, ring breathing modes of the DNA/RNA bases; p, protein; Tyr, tyrosine; Trp, tryptophan; and Phe, phenylalanine. See text for more details. Assignments are based on studies 31–36 in References.
In general, most features of the Raman spectra of normal T- and B-cells are very similar, but quite distinct from those of Raji and Jurkat cells, which in turn are both quite similar if compared against each other. Peaks that are almost exclusively due to ring breathing modes in the DNA bases, such as the modes at 678 cm−1, 785 cm−1, 1337 cm−1, 1373 cm−1, 1485 cm−1, 1510 cm−1, and 1575 cm−1 are significantly reduced in intensity in Jurkat and Raji spectra. These reduced peak heights indicate that the overall DNA concentration in the probe volume of the laser beam is significantly lower in transformed cells than in normal cells. This is further confirmed by the similarly reduced intensity of the 1093 cm−1 mode of the symmetric 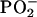 stretching vibration of the DNA backbone. Some peaks that are due to protein vibrations, however, are significantly stronger in intensity in transformed cells. This is the case for the 1126 cm−1 C-N stretching vibration, and the 1447 cm−1 CH2 deformation mode, which indicates a higher protein concentration in transformed cells than in normal cells. If we use the 1447 cm−1 mode as a marker mode for the protein concentration, and the 1093 cm−1 phosphate backbone vibration as a marker mode for the DNA concentration, then the relative protein/DNA ratio in Jurkat cells is 52% higher than in T-cells, whereas it is 28% higher in Raji than in B-cells. Between B- and T-cells, the ratio is almost identical (2% higher in B-cells), whereas there is still a significant difference between Jurkat and Raji cells (17% higher in Jurkat than Raji). These differences based on the CH2 protein deformation mode are all the more so remarkable if we compare these observations to the phenylalanine mode at 1003 cm−1. This mode stays nearly constant for all cell types, but interestingly, and as can be best seen from the difference spectra in Fig. 3, it shifts slightly to higher wavenumbers (1004 cm−1) in the transformed cells. It is not entirely clear what might cause such a consistent shift in the transformed cells, but it appears plausible that this could be due to a somewhat different protein composition (different amino acids right next to phenylalanine) for the majority of proteins in Jurkat and Raji cells. Another subtle difference that distinguishes transformed cells from normal cells is the presence of slight shoulders in their spectra at 813 cm−1 and 1240 cm−1. These are the positions of the two most distinct peaks for RNA and might indicate a slightly elevated concentration of RNA in the transformed cells versus the normal cells. Other distinct RNA modes, such as the ribose vibrations at 867 cm−1, 915 cm−1, and 974 cm−1 are too weak to contribute to our spectra. In summary, our Raman spectra of individual transformed and normal cells indicate significantly lower DNA concentrations and higher protein concentrations in transformed cells with potentially higher RNA concentrations.
stretching vibration of the DNA backbone. Some peaks that are due to protein vibrations, however, are significantly stronger in intensity in transformed cells. This is the case for the 1126 cm−1 C-N stretching vibration, and the 1447 cm−1 CH2 deformation mode, which indicates a higher protein concentration in transformed cells than in normal cells. If we use the 1447 cm−1 mode as a marker mode for the protein concentration, and the 1093 cm−1 phosphate backbone vibration as a marker mode for the DNA concentration, then the relative protein/DNA ratio in Jurkat cells is 52% higher than in T-cells, whereas it is 28% higher in Raji than in B-cells. Between B- and T-cells, the ratio is almost identical (2% higher in B-cells), whereas there is still a significant difference between Jurkat and Raji cells (17% higher in Jurkat than Raji). These differences based on the CH2 protein deformation mode are all the more so remarkable if we compare these observations to the phenylalanine mode at 1003 cm−1. This mode stays nearly constant for all cell types, but interestingly, and as can be best seen from the difference spectra in Fig. 3, it shifts slightly to higher wavenumbers (1004 cm−1) in the transformed cells. It is not entirely clear what might cause such a consistent shift in the transformed cells, but it appears plausible that this could be due to a somewhat different protein composition (different amino acids right next to phenylalanine) for the majority of proteins in Jurkat and Raji cells. Another subtle difference that distinguishes transformed cells from normal cells is the presence of slight shoulders in their spectra at 813 cm−1 and 1240 cm−1. These are the positions of the two most distinct peaks for RNA and might indicate a slightly elevated concentration of RNA in the transformed cells versus the normal cells. Other distinct RNA modes, such as the ribose vibrations at 867 cm−1, 915 cm−1, and 974 cm−1 are too weak to contribute to our spectra. In summary, our Raman spectra of individual transformed and normal cells indicate significantly lower DNA concentrations and higher protein concentrations in transformed cells with potentially higher RNA concentrations.
These main distinguishing spectral features between normal and transformed cells are very plausible because typical characteristics of transformed cells include increased levels of RNA, a much larger nucleus, and reduced cytoplasm (25). The larger nucleus in transformed cells likely affects the compactness of the chromatin and thus the concentration of DNA in the probe volume. In addition, whereas normal cells have a relatively low level of transcription and have chromatin that is highly condensed and inactive, the increased transcription and replication of transformed cells requires an open configuration of the chromatin. Transformed cells would also require a larger number of proteins to sustain the increased synthesis of RNA. This same trend has been previously observed in the Raman study of mature peripheral blood lymphocytes and eye lens epithelial cells (20).
Our findings that transformed cells have lower concentrations of nucleic acid and higher concentrations of protein are in agreement with a previous Raman study (26) comparing osteoblast-like human osteosarcoma derived cell lines and nontumor bone cells, which was attributed to the differences in the cellular activity between the two cell groups. Another study using rat embryo fibroblasts (13) also observed higher protein concentrations relative to DNA concentrations in cells with the oncogene gene. Interestingly, earlier studies (27–29) investigating the biochemical differences between normal and chronic lymphocytic leukemic cells using Fourier transform infrared microspectroscopy (FTIR) observed increases in the DNA content relative to proteins in neoplastic cells and increases in the DNA/RNA ratio. In addition, studies using flow cytometry to quantify DNA content in acute lymphocytic leukemica have also found higher DNA content in leukemia cells (30). The apparent discrepancy between their results and those using Raman spectroscopy likely results from the different technology employed. FTIR spectroscopy probes the entire cell or groups of cells and provides information about the overall cellular DNA content, whereas the LTRS method probes a confined area within an individual cell and yields information about the local DNA density. There are several plausible explanations to reconcile these results. Since the transformed cells in our study have a larger nucleus and overall diameter, it is possible that the DNA density would decrease whereas the overall DNA content of the entire cell would increase. In addition, the decrease in Raman signals attributed to DNA could also be a reflection of the higher level of transcription of neoplastic cells, which would require some decondensation of the chromatin structure, as stated previously (24,26). These explanations are consistent with both the results from the FTIR studies and the LTRS work presented, herein.
Principal component analysis
PCA was used to reduce the large amount of spectral information contained in the Raman spectra into 2–3 important parameters (principal components). A scatter plot generated from this data transformation shows clusters of points representing different cell groups, a graphical representation that is similar to results observed from flow cytometry.
PCA comparing normal and Jurkat T-cells (Fig. 4 a) uses only the first and second principal component values for the two axes and shows that the two cell types form distinct, separate clusters. Normal T-cells form one cluster and Jurkat T-cells form the second cluster. PCA reveals two outlier values from the normal T-cell population within the Jurkat cluster and a small border area between the two clusters. Application of the third principal component, which accounts for 8.3% of the variance, as a third axis of a 3-D plot does little to alter this result (data not shown). The sensitivity of this technique for identifying transformed cells is calculated to be 97.8%, with a Jurkat T-cell specificity of 95.5%. The total number of cells correctly classified is 96.7%. PCA analysis with the two B-cell groups (Fig. 4 b) using only the first two PCs yields a similar plot that also forms two distinct clusters. The analysis is able to correctly classify all cell types into their respective cluster, indicating a sensitivity of 100%. The simultaneous analysis of all four cell types (Fig. 4 c) shows that PCA can separate the cell types into two clusters corresponding to normal and transformed cells. The sensitivity for cancer detection is 98.3%, with specificity of 96.3%. Overall, 97.2% of the cells were correctly classified as being normal or transformed cells. Although the transformed cell lines, Jurkat and Raji, form a cluster different than the normal cells, it is not possible to positively delineate between the normal cells, or between the transformed cell lines.
Across all three PC plots, it is clear that principal component 1 (x axis) provides the majority of the cluster separation. Biochemical interpretation of the differences between normal and transformed cells can also be extracted/inferred from the PC1 loading values (Fig. 5, c–e). Irrespective of the comparison (T-cell versus Jurkat, B-cell versus Raji, or all four cell types) the major contribution to the first PC was from channels associated with DNA, RNA, and protein concentration differences that had been previously identified in the difference spectra (Fig. 5, a and b), as evidenced by their similar spectral profiles. This indicates that PCA is able to detect the same DNA, RNA, and protein differences that have been identified in the difference spectra and reduce this information into a single PC1 value.
The cell clusters identified using a two-component PCA plot remain imperfect. The PCA plot of T- versus Jurkat cells (Fig. 4 a) contains outlier cells that are not correctly classified. This observation is best explained by the purification procedure used to isolate the normal lymphocyte populations wherein there may be 2–5% contamination with other mononuclear cells. The positive identification of these outlier cells will require real-time analysis of the acquired Raman spectra data and micromanipulation of the individual cell after data acquisition. It is also possible that use of a greater number of principal components will better define the individual cell clusters. A more formal, mathematical cluster analysis of the data will be necessary to address this possibility. Alternatively, the outlier cells may represent individual T-cells that are activated and progressing through the cell cycle such that they appear similar biologically to a transformed Jurkat T-cell. Regardless, it is encouraging that these results provide strong evidence that single-cell Raman spectroscopy provides a novel, nondestructive means that allows for biological discrimination between normal and neoplastic/transformed cells.
SUMMARY
This is the first report to demonstrate that normal and transformed human hematopoietic cells in suspension can be discriminated at the single-cell level using micro-Raman spectroscopy and LTRS. The method is a useful tool capable of discerning cells via their biomolecular differences rather than physical attributes. Specific biomolecular differences observed include an increase in protein and RNA concentration and a decrease in the cellular DNA concentration. We attribute this observation to the different size of the nucleus and the different active state of the transformed cell. PCA is able to classify the cells into distinct clusters for highly sensitive cancer cell detection and discrimination. It remains to be demonstrated whether these differences are maintained for other neoplastic cells and whether this approach can discriminate between similar cells such as T- and B-cells. The transformed cells used in this study served as a model for neoplastic hematopoietic cells but, due to their large size, could not be manipulated in the laser trap. It should be noted that the size and shape of leukemic cells from real patients are closer to that of the normal T- and B-cells investigated in this study, so it will be possible to analyze real patient samples using LTRS. These results show tremendous promise for the development of LTRS as a clinical tool for the rapid identification and sorting of cells analogous to flow cytometry, but based on their biochemical signatures rather than surface phenotype or fluorescent labeling.
Acknowledgments
We thank Joseph Tuscano, MD, for providing laboratory space for the preparation of the human lymphocyte samples.
This work was supported by the Laboratory Directed Research and Development Program at Lawrence Livermore National Laboratory, the University of California, Davis, Cancer Center, funding from the National Science Foundation (Center for Biophotonics), and by the Children's Miracle Network, University of California, Davis. The Center for Biophotonics, a National Science Foundation Science and Technology Center, is managed by the University of California, Davis, under cooperative agreement No. PHY 0120999. Work at Lawrence Livermore National Laboratory was performed under the auspices of the U.S. Dept. of Energy by the University of California, Lawrence Livermore National Laboratory, under contract No. W-7405-Eng-48.
References
- 1.Huang, Z. W., A. McWilliams, H. Lui, D. I. McLean, S. Lam, and H. S. Zeng. 2003. Near-infrared Raman spectroscopy for optical diagnosis of lung cancer. Int. J. Cancer. 107:1047–1052. [DOI] [PubMed] [Google Scholar]
- 2.Schut, T. C. B., M. J. H. Witjes, H. Sterenborg, O. C. Speelman, J. L. N. Roodenburg, E. T. Marple, H. A. Bruining, and G. J. Puppels. 2000. In vivo detection of dysplastic tissue by Raman spectroscopy. Anal. Chem. 72:6010–6018. [DOI] [PubMed] [Google Scholar]
- 3.Krishna, C. M., G. D. Sockalingum, J. Kurien, L. Rao, L. Venteo, M. Pluot, M. Manfait, and V. B. Kartha. 2004. Micro-Raman spectroscopy for optical pathology of oral squamous cell carcinoma. Appl. Spectrosc. 58:1128–1135. [DOI] [PubMed] [Google Scholar]
- 4.Crow, P., J. S. Uff, J. A. Farmer, M. P. Wright, and N. Stone. 2004. The use of Raman spectroscopy to identify and characterize transitional cell carcinoma in vitro. BJU Int. 93:1232–1236. [DOI] [PubMed] [Google Scholar]
- 5.Frank, C. J., R. L. Mccreery, and D. C. B. Redd. 1995. Raman-spectroscopy of normal and diseased human breast tissues. Anal. Chem. 67:777–783. [DOI] [PubMed] [Google Scholar]
- 6.Mahadevan-Jansen, A., and R. Richards-Kortum. 1996. Raman Spectroscopy for the detection of cancers and precancers. J. Biomed. Opt. 1:31–70. [DOI] [PubMed] [Google Scholar]
- 7.Manoharan, R., Y. Wang, and M. S. Feld. 1996.Histochemical analysis of biological tissues using Raman spectroscopy. Spectrochim. Acta Part A Mol. Spectrosc. 52:215–249. [Google Scholar]
- 8.Kaminaka, S., T. Ito, H. Yamazaki, E. Kohda, and H. Hamaguchi. 2002. Near-infrared multichannel Raman spectroscopy toward real-time in vivo cancer diagnosis. J. Raman Spectrosc. 33:498–502. [Google Scholar]
- 9.Kaminaka, S., H. Yamazaki, T. Ito, E. Kohda, and H. O. Hamaguchi. 2001. Near-infrared Raman spectroscopy of human lung tissues: possibility of molecular-level cancer diagnosis. J. Raman Spectrosc. 32:139–141. [Google Scholar]
- 10.Min, Y. K., T. Yamamoto, E. Kohda, T. Ito, and H. Hamaguchi. 2005. 1064 nm near-infrared multichannel Raman spectroscopy of fresh human lung tissues. J. Raman Spectrosc. 36:73–76. [Google Scholar]
- 11.Utzinger, U., D. L. Heintzelman, A. Mahadevan-Jansen, A. Malpica, M. Follen, and R. Richards-Kortum. 2001. Near-infrared Raman spectroscopy for in vivo detection of cervical precancers. Appl. Spectrosc. 55:955–959. [Google Scholar]
- 12.Stone, N., P. Stavroulaki, C. Kendall, M. Birchall, and H. Barr. 2000. Raman spectroscopy for early detection of laryngeal malignancy: preliminary results. Laryngoscope. 110:1756–1763. [DOI] [PubMed] [Google Scholar]
- 13.Omberg, K. M., J. C. Osborn, S. L. L. Zhang, J. P. Freyer, J. R. Mourant, and J. R. Schoonover. 2002. Raman spectroscopy and factor analysis of tumorigenic and non-tumorigenic cells. Appl. Spectrosc. 56:813–819. [Google Scholar]
- 14.Xie, C. G., M. A. Dinno, and Y. Q. Li. 2002. Near-infrared Raman spectroscopy of single optically trapped biological cells. Opt. Lett. 27:249–251. [DOI] [PubMed] [Google Scholar]
- 15.Xie, C. G., Y. Q. Li, W. Tang, and R. J. Newton. 2003. Study of dynamical process of heat denaturation in optically trapped single microorganisms by near-infrared Raman spectroscopy. J. Appl. Phys. 94:6138–6142. [Google Scholar]
- 16.Boyum, A. 1968. Separation of leucocytes from blood and bone marrow. Scand. J. Clin. Lab. Invest. Suppl. 21:77–89. [PubMed] [Google Scholar]
- 17.Hannamharris, A. C., D. S. Taylor, and P. C. Nowell. 1985. Cyclosporin a directly inhibits human B-cell proliferation by more than a single mechanism. J. Leukoc. Biol. 38:231–239. [DOI] [PubMed] [Google Scholar]
- 18.Chan, J. W., A. P. Esposito, C. E. Talley, C. W. Hollars, S. M. Lane, and T. Huser. 2004. Reagentless identification of single bacterial spores in aqueous solution by confocal laser tweezers Raman spectroscopy. Anal. Chem. 76:599–603. [DOI] [PubMed] [Google Scholar]
- 19.Wold, S., K. Esbensen, and P. Geladi. 1987. Principal component analysis. Chemom. Intell. Lab. Syst. 2:37–52. [Google Scholar]
- 20.Uzunbajakava, N., A. Lenferink, Y. Kraan, E. Volokhina, G. Vrensen, J. Greve, and C. Otto. 2003. Nonresonant confocal Raman imaging of DNA and protein distribution in apoptotic cells. Biophys. J. 84:3968–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochiai, F., and M. Eguchi. 1987. Morphometrical evaluation of acute leukemic-cells by electron-microscopy—discrepancy between morphological-characteristics in fab classification and electron-microscopic morphometry. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 52:403–411. [PubMed] [Google Scholar]
- 22.Xie, C. G., C. Goodman, M. A. Dinno, and Y. Q. Li. 2004. Real-time Raman spectroscopy of optically trapped living cells and organelles. Opt. Express. 12:6208–6214. [DOI] [PubMed] [Google Scholar]
- 23.Richards-Kortum, R. and E. Sevick-Muraca. 1996. Quantitative optical spectroscopy for tissue diagnosis. Annu. Rev. Phys. Chem. 47:555–606. [DOI] [PubMed] [Google Scholar]
- 24.Uzunbajakava, N., A. Lenferink, Y. Kraan, B. Willekens, G. Vrensen, J. Greve, and C. Otto. 2003. Nonresonant Raman imaging of protein distribution in single human cells. Biopolymers. 72:1–9. [DOI] [PubMed] [Google Scholar]
- 25.Golub, T. R., and D. G. Gilliland. 1998. The molecular biology of cancer, chapter 28. In Hematology of Infancy and Childhood, 5th Ed. D. G. Nathan and S. H. Orkin, editors. Elsevier Science, Health Sciences Division, London. 1092–1146.
- 26.Notingher, I., G. Jell, U. Lohbauer, V. Salih, and L. L. Hench. 2004. In situ non-invasive spectral discrimination between bone cell phenotypes used in tissue engineering. J. Cell. Biochem. 92:1180–1192. [DOI] [PubMed] [Google Scholar]
- 27.Benedetti, E., E. Bramanti, F. Papineschi, I. Rossi, and E. Benedetti. 1997. Determination of the relative amount of nucleic acids and proteins in leukemic and normal lymphocytes by means of Fourier transform infrared microspectroscopy. Appl. Spectrosc. 51:792–797. [Google Scholar]
- 28.Schultz, C. P., K. Z. Liu, J. B. Johnston, and H. H. Mantsch. 1996. Study of chronic lymphocytic leukemia cells by FT-IR spectroscopy and cluster analysis. Leuk. Res. 20:649–655. [DOI] [PubMed] [Google Scholar]
- 29.Schultz, C. P., K. Z. Liu, J. B. Johnston, and H. H. Mantsch. 1997. Prognosis of chronic lymphocytic leukemia from infrared spectra of lymphocytes. J. Mol. Struct. 408:253–256. [Google Scholar]
- 30.Look, A. T., P. K. Roberson, D. L. Williams, G. Rivera, W. P. Bowman, C. H. Pui, J. Ochs, M. Abromowitch, D. Kalwinsky, G. V. Dahl, S. George, and S. B. Murphy. 1985. Prognostic importance of blast cell-DNA content in childhood acute lymphoblastic-leukemia. Blood. 65:1079–1086. [PubMed] [Google Scholar]
- 31.Puppels, G. J., F. F. M. Demul, C. Otto, J. Greve, M. Robertnicoud, D. J. Arndtjovin, and T. M. Jovin. 1990. Studying single living cells and chromosomes by confocal Raman microspectroscopy. Nature. V347:301–303. [DOI] [PubMed] [Google Scholar]
- 32.Peticolas, W. L. 1995. Raman spectroscopy of DNA and proteins. Methods Enzymol. 246:389–416. [DOI] [PubMed] [Google Scholar]
- 33.Hud, N. V., F. P. Milanovich, and R. Balhorn. 1994. Evidence of novel secondary structure in DNA-bound protamine is revealed by Raman spectroscopy. Biochemistry. V33:7528–7535. [DOI] [PubMed] [Google Scholar]
- 34.Deng, H., V. A. Bloomfield, J. M. Benevides, and G. J. Thomas. 1999. Dependence of the Raman signature of genomic B-DNA on nucleotide base sequence. Biopolymers. V50:656–666. [DOI] [PubMed] [Google Scholar]
- 35.Erfurth, S. C., and W. L. Peticolas. 1975. Melting and premelting phenomenon in DNA by laser Raman-scattering. Biopolymers. 14:247–264. [DOI] [PubMed] [Google Scholar]
- 36.Benevides, J. M., and G. J. Thomas. 1983. Characterization of DNA structures by Raman spectroscopy: high-salt and low-salt forms of double helical poly(dG-dC) in H2O and D2O solutions and application to B, Z and A-DNA. Nucleic Acids Res. 11:5747–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]