

Abstract
Living cells may be considered noisy or stochastic biochemical reactors. In eukaryotic cells, in which the number of protein or mRNA molecules is relatively large, the stochastic effects originate primarily in regulation of gene activity. Transcriptional activity of a gene can be initiated by transactivator molecules binding to the specific regulatory site(s) in the target gene. The stochasticity of activator binding and dissociation is amplified by transcription and translation, since target gene activation results in a burst of mRNAs molecules, and each copy of mRNA then serves as a template for numerous protein molecules. In this article, we reformulate our model of the NF-κB regulatory module to analyze a single cell regulation. Ordinary differential equations, used for description of fast reaction channels of processes involving a large number of molecules, are combined with a stochastic switch to account for the activity of the genes involved. The stochasticity in gene transcription causes simulated cells to exhibit large variability. Moreover, none of them behaves like an average cell. Although the average mRNA and protein levels remain constant before tumor necrosis factor (TNF) stimulation, and stabilize after a prolonged TNF stimulation, in any single cell these levels oscillate stochastically in the absence of TNF and keep oscillating under the prolonged TNF stimulation. However, in a short period of ∼90 min, most cells are synchronized by the TNF signal, and exhibit similar kinetics. We hypothesize that this synchronization is crucial for proper activation of early genes controlling inflammation. Our theoretical predictions of single cell kinetics are supported by recent experimental studies of oscillations in NF-κB signaling made on single cells.
INTRODUCTION
Typically, in mammalian cells, there are tens or hundreds of mRNA molecules of a given species and thousands of corresponding protein molecules, but typically only two homologous gene copies. This implies the discrete regulation of mRNA transcription, governed by the stochastic events of binding and dissociation of transcription factors to the regulatory site (1–6). Stochastic effects due to gene activation and inactivation are amplified by mRNA synthesis and protein translation. As a result, single gene activation may result in production of thousands of protein molecules, influencing the cell kinetics much more strongly than production and degradation of single mRNA or protein molecules. It was often assumed that the transcription rate of an inducible gene is a function of the nuclear concentration of a transcription factor. Such an approach is justified when the binding and dissociation events are very frequent, which may be assumed for prokaryotes, but is not justified for higher organisms. On the other hand, the large number of mRNA and protein molecules allows for a deterministic reaction-rate approximation in description of the time evolution of mRNA and protein levels, an approximation which is much more computationally efficient than the discrete model. Therefore, processes such as mRNA translation, formation, and degradation of protein complexes, and catalytic and spontaneous degradation, which involve a large number of molecules, may be modeled by ordinary differential equations (ODEs). As a contrast in prokaryotes, in which the mRNA molecules are very unstable (half-life time is of order of 1 min) and much less abundant, the stochasticity of formation, degradation, and translation of single mRNAs is of great importance (7–11). As a result, in prokaryotes there is a competition between stochastic effects caused by gene activation and mRNA processing.
Stochasticity in gene activation considerably influences cell kinetics and potentially leads to large cell-to-cell variability in mRNA and protein levels. Since each cell reacts to its own mRNA and protein levels, and not to the average levels in the population, the information about cell-to-cell variability could be crucial in the analysis of kinetics of single cell events like apoptosis. This is why, in this article on regulation of early immune response, we modify our previous simplified approach and model the transcriptional part of the regulatory network using a stochastic switch.
Nuclear factor κB (NF-κB) regulates numerous genes important for pathogen or cytokine inflammation, immune response, cell proliferation, and survival (reviewed in (12)). In mammals, the NF-κB family of transcription factors contains five members but the ubiquitously expressed p50 and RelA heterodimer constitutes the most common inducible NF-κB binding activity. In resting cells, p50-RelA heterodimers (referred to herein as NF-κB) are sequestered in the cytoplasm by association with the members of another family of proteins called IκB. This family includes several proteins but most of the IκB-family inhibitory potential is carried by IκBα, whose synthesis is controlled by a highly NF-κB-responsive promoter, generating autoregulation of NF-κB signaling (13). Activation of NF-κB requires degradation of IκBα, which allows NF-κB to translocate into the nucleus, bind to κB motifs present in promoters of numerous genes, and upregulate their transcription. NF-κB activating signals converge on the cytoplasmic IκB kinase (IKK), a multiprotein complex that phosphorylates IκBα, leading to its ubiquitination and then to its rapid degradation by the proteasome (reviewed in (14)). Activation of IKK kinase is induced by various extracellular signals including tumor necrosis factor-α (TNF) and interleukin-1 (IL-1) through complicated, not fully resolved, transduction pathways. IKK inactivation is controlled by the zinc finger protein termed A20, which, like IκBα, is strongly NF-κB responsive and generates a second autoregulatory loop in NF-κB signaling (15). Mice deficient in A20 develop severe inflammation and cachexia, are hypersensitive to TNF, and die prematurely (16). Nuclear NF-κB activates groups of genes through a process initiated by its binding to high affinity DNA binding sites in regulatory regions of their promoters. Although NF-κB binding to some genes results in rate-limiting complex formation of co-activators, pre-initiation factors, and RNA polymerase II, the mode of regulation for A20 and IκBα inhibitors appears to be distinct. Interestingly, chromatin immunoprecipitation assays have shown that the A20 and IκBα promoters are already bound by general transcription factors, co-activators, and RNA Pol II, waiting for the presence of NF-κB binding to activate them (17,18). In these promoters, RNA polymerase II is stalled in an activated state; upon NF-κB binding, RNA polymerase enters a competent elongation mode and thus is able to rapidly respond to NF-κB binding. In this manner, these inhibitor members of the NF-κB feedback loop are rapidly poised to respond to the presence of nuclear NF-κB.
The first attempt to mathematically analyze the IκB-NF-κB signaling module was a one-feedback loop model that concentrated on the interplay between three IκB isoforms (19). In contrast, our two feedback loop model (20) incorporates inhibitors operating at two levels in the signaling pathway: IκBα—the single species from IκB family whose knockout is lethal (21), and which binds the majority of cytoplasmic RelA; and A20—which inhibits activity of IKK whose action leads to the second negative feedback loop in NF-κB regulation. Incorporating the second inhibitor, A20, this latter model accurately predicts the profile of IKK activity.
In this article, we apply a stochastic switch together with deterministic reaction rate description by ODEs to model the NF-κB regulatory network at the single cell level.
MOTIVATION, MODEL FORMULATION, AND NUMERICAL IMPLEMENTATION
In the last few years, several important studies of NF-κB regulation at the single cell level have been performed (22–25). Using fluorescently tagged RelA and IκBα proteins, these experiments enable observation of intercompartment translocations of both proteins, showing a large heterogeneity in kinetics of cell responses to TNF or IL-1 stimulation. Nelson et al. (23) showed that the rate of nuclear RelA accumulation in response to TNF stimulation depends on the initial RelA/IκBα ratio. In cells with enhanced initial concentration of IκBα, the nuclear import is considerably slower. In accordance with Nelson et al. (23), Schooley et al. (25) found a broad distribution of nuclear RelA levels in the cell population at 10–90 min after IL-1 stimulation. Although these cited experiments indicate a large variability in cell kinetics, it is not straightforward to determine to which extent this variability is caused by stochastic regulation of gene expression, rather than being introduced by cell-to-cell variation in the amount of transfected plasmids (or their expression levels). In a recent experiment, Nelson et al. (26) proved the existence of long persisting single cell oscillations in NF-κB signaling. Since cell-to-cell synchronization decreases with time, these oscillations either do not appear or appear strongly damped when observed at the population level, but as concluded by the authors they are important in control of expression of numerous NF-κB-inducible genes. The point is that NF-κB must circulate between nucleus and cytoplasm where it may become activated by a post-translational modification, such as phosphorylation. Inhibition of its nuclear export results both in NF-κB dephosphorylation at Ser-536 after ∼3 h and in transitory κB-dependent luciferase reporter gene expression that peaked after ∼5 h (26).
The model involves two-compartment kinetics of the activators IKK and NF-κB, the inhibitors A20 and IκBα, and their complexes (Fig. 1). It is biologically justified to assume that the cytoplasmic complex IKK may exist in one of three forms (20):
Neutral (denoted by IKKn), synthesized de novo and specific to resting cells without an extracellular stimulus like TNF or IL-1.
Active (denoted by IKKa), arising from IKKn upon the TNF or IL-1 stimulation.
Inactive, but different from the neutral form, arising from IKKa possibly due to over-phosphorylation (denoted by IKKi).
In resting cells, the unphosphorylated IκBα binds to NF-κB and sequesters it in an inactive form in the cytoplasm. In response to extracellular signals such as TNF, IKK is transformed from its neutral form (IKKn) into its active form (IKKa), capable of phosphorylating IκBα, leading to IκBα degradation (Fig. 1 A). Degradation of IκBα releases the main activator NF-κB, which then enters the nucleus and triggers transcription of the two inhibitors and numerous other genes. The newly synthesized IκBα leads NF-κB out of the nucleus and sequesters it in the cytoplasm (Fig. 1 B), while A20 inhibits IKK by converting IKKa into the inactive form IKKi (Fig. 1 C), a form different from IKKn, but also not capable of phosphorylating IκBα. Considering IKK, we assume that each form of IKK undergoes degradation with the same degradation rate, and that IKKa can form transient complexes with IκBα proteins or (IκBα|NF-κB) complexes. Formation of these complexes leads to IκBα phosphorylation, ubiquitination, and degradation in the proteasome. At the same time, NF-κB is activated by RelA phosphorylation (27). Degradation of IκBα enables activated NF-κB to enter the nucleus, where it rapidly upregulates the transcription of mRNAs of inhibitory proteins A20 and IκBα. The A20 and IκBα mRNA transcripts are then translated into protein.
FIGURE 1.

Two-component feedback model of NF-κ B regulatory module. (A) Activation of NF-κB module. NF-κB is found in an inactivated complex in the cytoplasm with its inhibitor IκBα. TNF ligand acts as a switch to convert neutral IKK (IKKn) to the activated form, IKKa. IKKa, in turn, phosphorylates and degrades both free and NF-κB complexed IκBα. Liberated NF-κB enters the nucleus to induce transcription of IκBα and A20 genes. (B) NF-κB-IκBα autoregulatory loop. The IκBα protein is rapidly resynthesized, enters the nucleus, and recaptures NF-κB back into the cytoplasm. In the continued presence of IKKa, however, the resynthesized IκBα is continuously degraded, resulting in continued nuclear NF-κB translocation. (C) The NF-κB-A20 autoregulatory loop. A second level of negative autoregulation occurs with the resynthesis of A20. A20 is a ubiquitin ligase that degrades signaling intermediates coupling the TNF receptor with continuous IKK activation and directly associates with IKKa, converting it to catalytically inactive IKKi.
The inhibitor IκBα migrates between the nucleus and cytoplasm and forms complexes with IKKa and NF-κB molecules. The nuclear (IκBα|NF-κB) complexes quickly migrate into the cytoplasm. The second inhibitory protein A20 is considered only in the cytoplasm where it triggers the inactivation of IKK. It is assumed that the transformation rate from IKKa into IKKi is a sum of a constant term and a term proportional to the amount of A20. The total amount of NF-κB is kept constant, i.e., it is assumed that the degradation is balanced by synthesis, but the synthesis and degradation terms are omitted. For IKK we have synthesis and degradation terms, but since all IKK forms degrade with the same degradation rate, after the equilibrium is reached the total amount of IKK (i.e., IKKn + IKKa + IKKi) remains roughly constant.
The transcriptional regulation of A20 and IκBα genes is governed by the same rapid elongation regulatory mechanism with rapid coupling between NF-κB binding and transcription. The mechanisms for NF-κB-dependent regulation of IκBα and A20 are based on control of transcriptional elongation, described earlier. In this situation, stalled RNA polymerase II is rapidly activated by NF-κB binding to enter a functional elongation mode, and requires continued NF-κB binding for reinitiation. This is represented in our model by tight coupling of NF-κB binding to mRNA transcription. In fact, our experimental analysis of the kinetics of IκBα and A20 gene expression indicates that the patterns of mRNA expression are tightly coupled with NF-κB presence in the nucleus, without appreciable time delay (28). This model cannot be extended to other NF-κB-dependent genes that show distinct kinetics of induction. In this situation, the so-called late genes, like Naf-1 or NF-κB2, show a peak of induction 6 h after NF-κB binding. Activation of the late genes apparently requires the activation or binding of other rate-limiting regulatory factors (6). We assume that all cells are diploid, and both A20 and IκBα genes have two potentially active homologous copies, each of which is independently activated due to binding of the NF-κB molecule to a specific regulatory site in the gene promoter. Following the literature (1–5,29), we made the simplifying assumption that each gene copy may exist only in two states, active and inactive. When the copy is active, the transcription is initiated at a high rate; and when the copy is inactive, transcription is inhibited, or it is initiated with some low basal rate. The gene copy inactivates when the NF-κB molecule is removed from its regulatory site. We expect that this is due to the action of the IκBα molecules that bind to DNA-associated NF-κB, exporting it out from the nucleus, rather than to thermal dissociation. The last would be very sensitive to temperature. The assumed mechanism of gene activation and inactivation is simplified, but it is also limited by our current knowledge of the subject. Fortunately, the details of activation mechanisms are not crucial for the model. The most important is the single assumption that transcription of A20 and IκBα genes turns on and off with probabilities determined by regulatory factors. This distinguishes the applied approach from the deterministic one in which the transcription speed is a function of concentrations of these factors.
The stochastic simulation algorithm (30) is an essentially exact procedure for simulation of the time evolution of processes like the ones described above. This exact algorithm becomes computationally intensive when the number of reacting molecules is large as in our case, i.e., ∼105. In the last few years, several methods were proposed to accelerate the Gillespie algorithm. One is the τ-leap method (31,32), an approximate acceleration procedure, in which time is divided into τ-long intervals. It is required that τ is short enough so that the propensity functions for all reactions remain almost unchanged. Then, assuming that this condition is satisfied, the number of reactions in each reaction channel is a Poisson random variable, with parameter equal to the τ-interval propensity for the reaction channel considered. Another method is devised for the case when it is possible to separate system into slow and fast reaction channels (33,34). The idea is to approximate fast reactions either deterministically or as chemical Langevin equations, and to treat the slow reactions as stochastic events with time-varying reaction rates. Recently this idea has been improved by Cao et al. (35), who introduce a virtual fast system, being Markovian, which makes its analysis simpler. This improves the analysis in the cases when fast reactions are described by Langevin equations. In the case in which the fast reactions are fast enough to be described by the deterministic-rate equations, the method of Cao et al. (35) is reduced to that of Haseltine and Rawlings (33).
In this work, as in our recent article (29), we follow the method proposed by Haseltine and Rawlings (33) and split the reaction channels into fast and slow. Namely, we consider all reactions involving mRNA and protein molecules as fast and the reactions of gene activation and inactivation as slow. Fast reactions are approximated by the deterministic reaction-rate equations. The justification of this approximation will be given in the Appendices, where we compare it with the exact stochastic simulation algorithm in a simple example involving gene activation and inactivation, mRNA transcription, and protein translation. In this example, we will use the same reaction rates as appearing in our model.
According to the above, the mathematical representation of the model (see the Appendices) consists of 14 ordinary differential equations (ODEs) accounting for formation of complexes and their degradation, transport between nucleus and cytoplasm, transcription and translation, and of four algebraic equations accounting for propensity functions of binding and dissociation of NF-κB molecules to regulatory sites in A20 and IκBα promoters. We assume that both A20 and IκBα genes have two homologous copies independently activated due to NF-κB binding. It is assumed that in an infinitesimal time interval Δt, the probability Pb of NF-κB binding to regulatory sites in each allele is proportional to the nuclear amount of NF-κB,
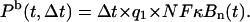 |
(1) |
NF-κB dissociation probability, Pd, is a sum of a constant term and a term proportional to nuclear concentration of IκBα, which is capable of removing NF-κB from regulatory sites in both A20 and IκBα genes,
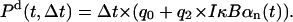 |
(2) |
It will be assumed that NF-κB binding and dissociation proceed independently in each homologous gene copy and that binding and dissociation propensities rb(t) = Pb(t, Δt)/Δt and rd(t) = Pd(t, Δt)/Δt are equal for each copy. The state of gene copy Gi (i = 1, 2) is Gi = 1 whenever NF-κB is bound to the promoter regulatory site, and Gi = 0 when the site is unoccupied. As a result, the gene state G = G1 + G2 can be equal to 0, 1, or 2. In this approximation, the stochasticity of single cell kinetics solely results from discrete regulation of transcription of A20 and IκBα genes.
We assume that the transcription rate Trate is the sum of a steady term and of an inducible term,
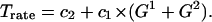 |
(3) |
Note that Eq. 3 naturally produces saturation in transcription speed. When the nuclear amount of regulatory factor NF-κB is very large, then the binding probability is much larger than the dissociation probability, and the gene state would be G = 2 for most of the time. In such a case, the transcription would proceed at a maximum rate, c2 + 2c1. This rate has been measured for β-actin by single RNA transcript visualization as 4 mRNA molecules per minute per one allele (2). In our calculations we assumed c1 = 0.075 mRNA/s, which corresponds to 4.5 mRNA molecules per minute. It is worth noting that the protein production rate is proportional to the product of transcription and translation efficiencies. Therefore having solely information about the amount of protein one may not determine these two coefficients. When fitting the model, we found that even if c2 = 0, transcription is regulated satisfactorily, and therefore to minimize the number of free parameters we assume c2 = 0. For the same reason we set q0 = 0 in Eq. 2.
In model computations, the amounts of all the substrates are specified in the numbers of molecules. Since we use the ODEs to describe most of the model kinetics, amounts of molecules are not integer numbers, but since these numbers are, in most cases, ≫1, such a description is reasonable.
The implemented numerical scheme follows that of Haseltine and Rawlings (33), namely:
- At simulation time t, for given states
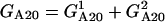 and
and 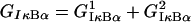 of the A20 and IκBα genes, we calculate the total propensity function r(t) of occurrence of any of the binding or dissociation reactions,
of the A20 and IκBα genes, we calculate the total propensity function r(t) of occurrence of any of the binding or dissociation reactions,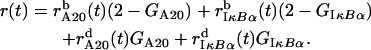
(4) We select two random numbers p1 and p2 from the uniform distribution on (0, 1).
- Using the fourth-order MATLAB solver we evaluate the system of 14 ODEs accounting for fast reactions, until time t + τ such that
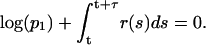
(5) - There are eight potentially possible reactions (NF-κB may bind/dissociate to/from any of two alleles of A20 and IκBα genes), but at any given time, only four of them have nonzero propensities. In this step, we determine which one of eight potentially possible reactions occurs at time t + τ using the inequality
where ri(t + τ), i = 1, …, 8 are individual reaction propensities and k is the index of the reaction to occur.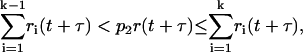
(6) Finally time t + τ is replaced by t, and we go back to item 1.
Because of a large number of undetermined parameters, model fitting is not straightforward. We decided to carry out the fit manually, rather than to try to digitize the blot data and then to apply one of the fitting engines available. The first reason is that such quantification is by no means unique; the second is that, when fitting, we have to take into account diverse, and usually not precise, information coming from different researchers as well as our own intuitive understanding of the process. We first performed the fitting for the deterministic model (20), then for the stochastic model, leaving most of the parameters unchanged. Fitting of the deterministic model is considerably faster because it is not necessary to average the outcome over many runs. We applied the following fitting method:
Start from a reasonable set of parameters, which produces a correct steady state in the absence of TNF signal.
Proceed with the signal initiated by TNF along the autoregulatory loops.
Iterate item 2 until the fit to all the data is satisfactory.
The TNF signal first causes transformation of IKKn into IKKa. IKKa catalyses degradation of cytoplasmic (IκBα|NF-κB), enabling the free NF-κB to enter the nucleus. Once NF-κB builds up in the nucleus it upregulates the transcription of the A20 and IκBα genes. After being translated, A20 facilitates transformation of IKKa into IKKi, while IκBα enters the nucleus, binds to NF-κB, and leads it into cytoplasm. As stated in item 2, we first fit the coefficients regulating IKK activation (using data on IKK activity), then the coefficients regulating degradation of the cytoplasmic (IκBα|NF-κB) and IκBα degradation, and so forth. If there were no feedback loops in the pathway, the proposed method would be quite efficient, but, since they exist, it is necessary to iterate the signal tracing several times until the fit is satisfactory. Once a satisfactory fit is found, we observe that the set of parameters chosen to fit the data is by far not unique. This ambiguity is mainly caused by the lack of measurements of absolute values of protein or mRNA amounts. The action exerted by some components of the model onto the rest of the pathway is determined by their amounts multiplied by undetermined coupling coefficients. Hence, once we have a good fit, we may obtain another one using a smaller coupling coefficient and by proportionately enlarging the absolute level of the component. There also exist single parameters, the value of which can be changed over a very broad range without a significant influence on the time behavior of any substrate or complex for which the data are at our disposal. Values of all parameters are listed in the Appendices.
RESULTS
Stochasticity of the model implies that simulations performed with the same model parameters and the same initial conditions, are different. Since each simulation corresponds to the evolution in a single cell, therefore only the outcome averaged over many simulations corresponds to experimental data obtained for a population of cells in culture. As it will be seen in the Appendices, with parameters fitted, the proposed model is able to faithfully reproduce time-behavior of all variables for which the data is available: nuclear NF-κB, cytoplasmic IκBα, A20, and IκBα mRNA transcripts, and total IKK and IKK kinase catalytic activity (IKKa) in both wild-type and A20-deficient cells.
In this section, we discuss the solutions of the model using the parameter set fitted to the experimental data from the experiments of Lee et al. (16) and Hoffmann et al. (19) on wild-type and A20-deficient mouse fibroblast cells.
Let us focus first on resting cell regulation. In the absence of TNF signal, IKK remains in its neutral (inactive) form, IKKn. This implies that it may not phosphorylate IκBα, which itself degrades at a relatively slow rate due to natural degradation. Until the amount of IκBα exceeds that of NF-κB, which is assumed to be constant, equal to 60,000 molecules (see Appendices), all NF-κB remains in cytoplasmic complexes with IκBα. In Fig. 2, we may observe that, because at t = 0 the number of cytoplasmic complexes of IκBα exceed 60,000 molecules, almost no NF-κB is in the nucleus. When, due to degradation, the amount of IκBα falls below 60,000, some NF-κB (typically a small fraction of cytoplasmic protein) enters the nucleus—where it may bind in a stochastic way to regulatory sites of IκBα and A20 promoters. Binding to IκBα results in a burst of IκBα transcription followed by an increase in IκBα protein level. In turn, free IκBα enters the nucleus, takes NF-κB out of its regulatory site, and leads almost all NF-κB back into the cytoplasm. Binding to A20 promoter results in a burst of A20 transcription, but A20 may not terminate the NF-κB binding. As a result, each time NF-κB enters the nucleus it causes a burst of IκBα, but not necessarily of A20. This implies that even if the (conditional) binding probabilities to IκBα and A20 promoters are equal, NF-κB binds more frequently to IκBα promoters. If, however, NF-κB binds to the A20 promoter before it binds to IκBα, it may remain there longer (since in this case binding does not lead to feedback reaction), and therefore may cause a larger burst of mRNA molecules. Although NF-κB may bind to both homologous gene copies, in a resting cell (in which there are relatively few NF-κB molecules in the nucleus), both copies are seldom simultaneously active. Let us note that since all cells in the population (in the absence of an external signal like TNF) behave asynchronously, the average IκBα and A20 transcript and protein levels in the cell population remain constant (plot not shown). The relatively low transcript level observed for IκBα and A20 in the population of unstimulated cells is usually explained by the existence of some basal transcription, independent of NF-κB. However, our model shows that, even if the basal transcription rate is zero, the average IκBα and A20 mRNA levels may be positive and constant in time for unstimulated cells.
FIGURE 2.

Numerical solution corresponding to a single (wild-type) resting cell. The amounts of substrates are given in numbers of molecules. D shows total IκBα, i.e., the sum of free and NF-κB bound substrate in both the cytoplasm and nucleus.
The time behavior of the main variables resulting from simulation of a single, TNF-stimulated cell is presented in Fig. 3. The simulation was performed for wild-type cells, in which all genes were potentially active. At t = 1 h, the rectangular TNF signal is turned on for 6 h, i.e., until the end of the simulation time. Under the TNF signal, IKK is promptly transformed into the active state IKKa and then into the inactive state IKKi. As a result, the persistent TNF stimulation causes a pulse activation of IKK, followed by a low tail. The pulse of IKKa initiates the cascade. First, the free cytoplasmic IκBα and cytoplasmic complexes (IκBα|NF-κB) are degraded (Fig. 3 B). The released NF-κB builds up in the nucleus, where it binds in a stochastic way to the regulatory sites in IκBα and A20 promoters (Fig. 3, D and E). We assume the same binding and dissociation probabilities for the IκBα and A20 genes. However, due to stochasticity, their activity in a single run may be considerably different, although in general it coincides with high nuclear levels of NF-κB. Due to the discrete regulation of transcriptional efficiency, the IκBα and A20 transcript levels (Fig. 3, F and G) look sawlike, with kinks corresponding to binding and dissociation events. The newly synthesized IκBα enters the nucleus and leads almost all NF-κB out of it (Fig. 3 C), whereas A20 (Fig. 3 H) triggers IKK inactivation. Let us note that since some NF-κB molecules are present in the nucleus even in the absence of TNF stimulation and may bind to regulatory sites, the system (single cell) does not have a steady state, either in the presence or absence of the TNF stimulation.
FIGURE 3.

Numerical solution corresponding to a single wild-type cell. The persistent TNF activation starts at 1 h. The amounts of substrates are given in numbers of molecules. B shows total IκBα, i.e., the sum of free and NF-κB-bound substrate in both the cytoplasm and the nucleus.
In Fig. 4, we compare oscillations in nuclear NF-κB level predicted by our model with those measured in single cell experiments by Nelson et al. (26). Since our model was fitted based on data (16,19) collected in experiments on mouse embryonic fibroblast, whereas Nelson et al. (26) did their experiments on human S-type neuroblastoma (SK-N-AS) cells (Fig. 4, B and C) and HeLa cells (Fig. 4 D), only a qualitative comparison is possible. The cells analyzed were transfected with plasmids expressing RelA fused to the red fluorescent protein (RelA-DsRed) and IκBα fused to the enhanced green fluorescent protein (Iκ Bα-EGFP). In the experiment shown in Fig. 4 C, the cells express control EGFP instead of IκBα-EGFP. As reported, 91% of these control cells (expressing control EGFP instead of IκBα-EGFP) showed prolonged oscillations in RelA nuclear-cytoplasmic localization (N-C oscillations). These oscillations appeared quite synchronous between cells in the first few cycles, but then they became out-of-phase—explaining why they appear to be strongly damped when observed at the population level (Fig. 7). We compare the kinetics of these control cells (Fig. 4 C) with our theoretical prediction (Fig. 4 A). In this simulation (Fig. 4 A), the level of total NF-κB was elevated two times (with respect to the value measured by (22,36) for fibroblast) to 120,000 to mimic transfection. Nelson et al. (26) estimates that the average overexpression of RelA fusion protein was 3–5 times that of the endogenous RelA level. We found that the persistence, amplitude, and period of these oscillations is not very sensitive to the total amount of total NF-κB molecules (Fig. 5). This can explain why the first oscillations in cells having possibly different levels of RelA-DsRed, shown in Fig. 4 C, are so well synchronized. In Fig. 5, the sixfold decrease and threefold increase in total NF-κB, with respect to its normal level in fibroblasts, is analyzed. Further increases of NF-κB level results in cessation of oscillations. This, together with the fact that we get the most convincing similarity to the Nelson et al. (26) profiles (Fig. 4 A versus C) elevating NF-κB molecules twofold (not 3–5-fold), could be because our model was fitted to fibroblast cells, and may suggest that the endogenous NF-κB is lower for SK-N-AS cells. The difference between various cell types becomes evident when SK-N-AS cells are compared with HeLa cells (Fig. 4, C versus D). As reported by Nelson et al. (26), only 30% of HeLa cells exhibited up to three detectable N-C oscillations that were markedly damped. In Fig. 6, we compare the current model prediction of the correlation of the peak-to-peak timing with the NF-κB level to the data of Nelson et al. ((26), Fig. 1), where there is little (first-to-second peak) or no correlation between the RelA-DsRed expression level and subsequent peak timing for SK-N-AS cells. According to the model, the 18-fold increase in total NF-κB causes ∼1.6-fold change in first-to-second peak timing, but <20% change in subsequent peak timing. Similarly, timing of the first peak is not sensitive to the level of NF-κB (Fig. 6).
FIGURE 4.

Model simulations and measurements of Nelson et al. (26). Four single cell simulations (or experiments) are represented by differently colored lines. (A) Model simulations: nuclear NF-κB after a persistent TNF activation starting at t = 0. The total amount of NF-κB was elevated to 120,000 for this simulation. (B–D) Nuclear to cytoplasmic RelA-DsRed fluorescence normalized to highest peak intensity. Cells were treated with continual 10 ng/ml TNFα. (B) SK-N-AS cells co-expressing RelA-DsRed and IκBα-EGFP. (C) SK-N-AS cells expressing RelA-DsRed and EGFP control. (D) HeLa cells expressing both RelA-DsRed and IκBα-EGFP.
FIGURE 7.

Numerical solution averaged over a population of 500 wild-type cells; all parameters as in Fig. 3. The amounts of substrates are given in numbers of molecules per cell.
FIGURE 5.

Oscillations of nuclear NF-κB after a persistent TNF treatment for six different levels of total NF-κB: 10,000 / 20,000 / 30,000 / 60,000 / 120,000 / 180,000.
FIGURE 6.

Correlation of the total NF-κB level with the peak-to-peak timing of NF-κB oscillations. (Left column) Nelson et al. (38) experiments on SK-N-As cells. (Right column) Model predictions. The line represents the average calculated based on 100 simulations performed at each point, corresponding to the total amount of NF-κB, equal to six different levels: 10,000 / 20,000 / 30,000 / 60,000 / 120,000 / 180,000. The dots corresponds to 500 single simulations, each done for the amount of total NF-κB randomly selected from uniform distribution on 10,000 and 180,000.
Recently there was an interesting discussion between Barken et al. (37) and Nelson et al. (38) concerning the dependence of oscillation period and oscillation persistence to the amount of NF-κB. Barken et al. (37), based on the model of Hoffman et al. (19), showed that oscillation period strongly depends on the assumed amount of total NF-κB, concluding that oscillations recorded in the overexpressed feedback system do not allow one to conclude that oscillations of the same persistence, amplitude, and period occur in normal genetically unaltered cells. In fact, according to the model of Hoffman et al. (19), the fourfold NF-κB overexpression causes the second peak in nuclear NF-κB, to appear not at 2 h (as normally), but at 5.5 h of persistent TNF stimulation ((37), Fig. 1 A). Even the relatively small 1.5-fold NF-κB overexpression causes substantial alternations in the NF-κB nuclear profile. This prediction is not confirmed by experiment ((38), Fig. 1). Following Nelson et al. (38), we expect that the substantial delay of the second peak caused by the NF-κB overexpression is an artifact of the Hoffmann model resulting from the assumption that the inducible IκBα expression is a second-order polynomial in nuclear NF-κB. As a result the fourfold change in NF-κB level causes a 16-fold increase in IκBα expression, which becomes so abundant that it would inhibit NF-κB nuclear re-entry for ∼5 h. Such large increase in IκBα expression seems to us to be biologically unjustified: There is a maximal physiological expression efficiency of ∼4 mRNAs/min from a single gene copy (2), which cannot be exceeded even if the regulatory factor is fairly abundant. Our current model shows that even the normal amount of NF-κB is capable of turning the IκBα gene on (Fig. 3) for ∼1/2 h during the first pulse, which implies that further growth of NF-κB nuclear level does not cause a significant rise in IκBα expression. Equation 3 in our model provides the natural saturation in transcription speed, but even the simple modification in which the second-order polynomial is replaced by a linear term results in reduced sensitivity of period to the NF-κB concentration ((20), Fig. 9) and ((38), Fig. 2).
FIGURE 9.

Scatter plots of total A20 versus nuclear NF-κB at six times after start of TNF stimulation.
Finally, let us also note that the oscillations predicted by any of the deterministic models (19,20,38) have essentially a different character than those in single cells (as shown in Fig. 4, B and C). Oscillation amplitude in deterministic models decreases exponentially to zero, or some positive value with time, as opposed to the single cell oscillations for which the amplitude is not a monotonous function of the peak number. For example, in Fig. 4 C, one can observe that the third peak of a blue trajectory is higher than the second one, and that fourth peak of a green trajectory is higher than the third one. In Fig. 4 B, the peak amplitude of red trajectory is growing starting from the second peak, and the green trajectory has third and fourth peaks higher than the second one. In the latter case, the fluctuations in peak amplitude can be also due to the different kinetics of IκBα-EGFP and of endogenous IκBα.
In a cell co-expressing IκBα-EGFP and RelA-DsRed (Fig. 4 B), one can observe large differences in duration of the first peak. As stated by Nelson et al. (26), these differences are due to the sensitivity of the system to variations in IκBα-expression efficiency. Cells with higher expression of IκBα-EGFP show statistically prolonged NF-κB nuclear activity followed by a longer period in which NF-κB is absent in the nucleus. We do not observe, however, such strong sensitivity of the first peak duration to the IκBα expression efficiency in our model (data not shown). It is possible that the strong extension of the first peak duration and generally more-complex pattern of peak amplitude is because the kinetics of IκBα-EGFP is slower than the kinetics of endogenous IκBα. One might expect that the fusion protein is degraded at a lower rate (under the TNF stimulation) and it is not as strongly induced by NF-κB. In Fig. 1 (26), we can observe that in cells not expressing IκBα-EGFP (initially pure red), the endogenous IκBα is degraded in the first 6 min (and as a result RelA-DsRed enters the nucleus), but in cells expressing IκBα-EGFP, it remains fairly abundant even after 60 min. Similarly, in cells ((26); see Fig. 6S, in that article's Supplementary Data) which, at t = 0, co-express IκBα-EGFP and RelA-DsRed, the endogenous IκBα rebuilds more quickly than IκBα-EGFP. At 300 min, RelA-DsRed is taken out from the nucleus—suggesting that the endogenous IκBα is rebuilt, but that the IκBα-EGFP is still absent (cells are purely red).
In Fig. 7, we show the same kinetics as in Fig. 3, but averaged over 500 simulation runs or over a population of 500 identical cells. The averaged kinetics is considerably different from kinetics of a particular cell. Despite the fact that each cell oscillates under extended TNF stimulation, all quantities averaged over many cells converge to steady-state levels. This effect is caused by a growing desynchronization of cells. Before the TNF signal, the cells are desynchronized (the single cell simulations are started at 20–30 h before the TNF signal), and it is the TNF signal that induces cell synchronization. The sharp decrease in total IκBα, followed by buildup of nuclear NF-κB, is observed in all simulations (Fig. 8). Thereafter, the stochastic nature of NF-κB binding causes that the peaks of gene activity to not-match, and this desynchronizes cell kinetics. The activity of the IκBα and A20 genes, averaged over a relatively large population, corresponds much better to the nuclear NF-κB level than it does in the single cell case.
FIGURE 8.

Scatter plots of total IκBα versus nuclear NF-κB at six times after start of TNF stimulation.
In Fig. 8, we present the scatter plots showing the relationship between nuclear NF-κB and total IκBα. In resting cells, t = 0, there is almost no nuclear NF-κB, which is sequestered by IκBα in the cytoplasm. In most of cells, the amount of total IκBα molecules is larger than the amount of total NF-κB molecules, which is kept constant, equal to 60,000, during the time course of the simulation. At t = 15 min and 30 min, almost all IκBα is degraded and most of the NF-κB is present in the nucleus. Then at 90 min, IκBα rebuilds and again leads most of the NF-κB out of the nucleus. Finally, 6 h after the beginning of TNF stimulation, the cell population is at apparent equilibrium, characterized by a relatively broad distribution of NF-κB and IκBα levels, an effect caused by desynchronization of the cell population. The inhibitory potential of IκBα and of the second NF-κB inhibitor, A20, is clearly visible in Figs. 8 and 9. At the crucial time, 30 min from the beginning of TNF stimulation, the amount of nuclear NF-κB is smaller in cells with larger amounts of any of the two inhibitors. In fact, levels of the two inhibitors are strongly positively correlated (data not shown) since the higher level of A20, implies lower level of (active) IKKa, and thus a higher level of IκBα.
The scatter plots in Fig. 10 show abundance of Iκ Bα and A20 proteins compared to their mRNA at three time points. We observe that initially there are relatively few (<100) IκBα mRNA molecules per cell, although the IκBα protein (which is mostly complexed with NF-κB) is abundant. Then at 30 min most of the IκBα protein is degraded, but the number of message molecules is large due to NF-κB-induced transcription. The kinetics of A20 transcript and protein is different. Initially there is a small amount of both protein and mRNA, then a growing amount of transcript is followed by the growing amount of protein. We observe broadening of the distribution in time, caused by desynchronization of cells due to stochasticity. Despite the fact that the scatter plots presented in Figs. 7–9 reveal that variability among cells grows in time, one may expect that there is a sizable fraction of cells, the evolution of which is close to the averaged evolution. In fact, however, none of the cells behave like the average. In Fig. 11, we show that single cell trajectories keep oscillating when the equilibrium distribution is reached, whereas the average trajectory resulting from averaging over 500 simulated cells stabilizes.
FIGURE 10.

(Scatter plots, left) Total IκBα protein versus IκBα message. (Scatter plots, right) A20 protein versus A20 message at three times after start of TNF stimulation.
FIGURE 11.

Trajectories projected on the (total IκBα, nuclear NF-κB, time) hyperplane. (Thin lines) Three single cell trajectories; (thick line) average trajectory.
DISCUSSION
This article, based on our earlier model (20), is the first to analyze mathematically the NF-κB regulatory module at a single cell level. We make use of an approach to gene expression in which stochasticity originates in binding of regulatory molecules. This idea was proposed over a decade ago by Ko (1), but then it was abandoned for several years to return recently (3–6,29). Other possible sources of stochasticity are the production and degradation of single mRNA molecules (8,39,40) or oligomerization reactions (41). These sources are of great importance in prokaryotes in which the number of mRNA and proteins molecules is small. However, in our case of higher eukaryotes, in which the number of mRNA and protein molecules is fairly large, the stochasticity due to relatively slow switching of the gene status is dominating.
The method of modeling molecular pathways applied by us follows that of Haseltine and Rawlings (33) and splits the reaction channels into fast and slow. It combines deterministic reaction-rate description (ODEs) for fast reaction channels (in this case, processes involving large number of molecules) with a stochastic switch for gene activity. This approach is computationally much more efficient than the stochastic simulation algorithm (30), but still accurate enough for our purposes (see Fig. 12).
FIGURE 12.

Exact stochastic simulation (thin line) compared with stochastic-deterministic approximation (thick line). (A) mRNA profile; (B) protein profile. The transcription, translation, and degradation rates are the same as for A20 gene; see Table 2.
After parameter fitting and averaging over population of simulated cells the model properly reproduces data of Lee et al. (16) and Hoffmann et al. (19) (see Fig. 13). The model shows, however, that the dynamics of any single cell could be much different than the average dynamics—a construct resulting from averaging over entire cell population. In other words, there is no such cell as an average cell that would follow the average dynamics.
FIGURE 13.

Model predictions based on average over 500 runs versus the measurements of Hoffman et al. (19) and Lee et al. (16) on wild-type cells and A20-deficient cells. (A and B) Correspondence between an active pool of IKK, i.e., IKKa, and the data of Lee et al. (16) on IKK kinase activity for wild-type and A20−/– cells, assessed using the GST-IκBα substrate. (C) Level of total IKK (16) for wild-type (upper blots) and A20−/− cells (lower blots) remains almost constant—which, in our model, results from the assumption that degradation of all IKK forms proceeds at the same rate. (D and E) Total cytoplasmic IκBα, respectively, for wild-type cells and A20−/− cells, from the experiments of Hoffman et al. (19) and Lee et al. (16). (F and G) Nuclear NF-κB (EMSA assay), respectively for wild-type cells and A20−/− cells (16,19). (H and I) IκBα mRNA from Lee et al. (16), for, respectively, wild-type and A20−/− cells. (J) A20 mRNA level (16). (K and L) nuclear NF-κB in wild-type cells for, respectively, 60-min- and 15-min-long TNF stimulations (19).
The difference between single cell dynamics and average dynamics is already visible and is arguably important in the absence of external stimuli. The average level of IκBα protein is ∼50% larger than the level of NF-κB, which is kept constant at 60,000 molecules (see Fig. 7 B, t = 0). This may suggest that the entire NF-κB is sequestered in cytoplasm; however, the single cell oscillations cause some NF-κB genes to occasionally enter the nucleus and regulate transcription of both A20 and IκBα, but also of other NF-κB-inducible genes (12). This implies that stochasticity in NF-κB kinetics could be necessary in the regulation of numerous genes in resting cells (compare to (42)). To verify this hypothesis, one should measure the distribution of basal activity (with no stimuli) of NF-κB-dependent genes. The broad distribution would imply that the activity of these genes is intermittent, and that the low constant mRNA level observed in microarray experiments (or in Northern blots) results from averaging over cell population. The result presented in Fig. 11 is partly similar to that obtained for resting cells; we show that although the equilibrium distribution is reached, and the average trajectory stabilizes, single cell trajectories keep oscillating. These single cell oscillations, observed under extended TNF stimulation are, as shown by Nelson et al. (26), crucial in maintaining NF-κB transcriptional activity and thus are necessary for proper regulation of late genes, which have their peak of activity several hours after the beginning of the TNF signal.
Theoretical predictions of our model are confirmed by experiments of Nelson et al. (26) on single cell oscillations in NF-κB signaling. There is a qualitative agreement between our simulations and oscillatory kinetics of SK-N-AS cells expressing RelA-DsRed and control EGFP (Fig. 4, A versus C). Behavior of these single cell oscillations is qualitatively different from that predicted by any of the deterministic models (19,20,38). According to the deterministic models, the peak amplitude is a monotonous function of peak number, which is in contrast to the single cell data ((26), Fig. 2), where one can observe that, for some cells, the next peak is higher than the previous one. The important property of the model is the low sensitivity of the oscillation period to the level of total NF-κB, which is in agreement with Nelson et al. (38). Previous models suggested the strong elongation of the oscillation period with the NF-κB level. This was due to the assumed quadratic (19,37) or linear (20,38) dependence of IκBα transcription speed to the NF-κB nuclear concentration. The current model has saturation naturally imbedded in transcriptional efficiency (Eq. 3), which prevents it exceeding the physiological limit. Obviously, such a saturation can be introduced to the deterministic models as well, but here it appears in the most natural way.
According to Nelson et al. ((26), Fig. 2) SK-N-AS cells expressing both RelA-DsRed and IκBα -EGFP show faster desynchronization, due to cell-to-cell variability in the number IκBα-EGFP gene copies. Cells with higher expression of IκBα-EGFP show statistically prolonged NF-κB nuclear activity followed by a longer period in which NF-κB is absent in the nucleus. We do not observe such strong sensitivity of the first peak duration to the IκBα expression efficiency in our model. One can expect, based on Nelson et al. (26) experimental data, that the strong extension of the first peak duration is due to the fact that the kinetics of IκBα-EGFP are slower than those of endogenous IκBα, i.e., that it is degraded with smaller rate and is not as NF-κB-inducible as endogenous IκBα. The combination of slower IκBα-EGFP and faster endogenous IκBα could explain the generally more complex pattern of N:C NF-κB oscillations for cells co-expressing RelA-DsRed and IκBα-EGFP (Fig. 4 B).
Contrary to the large variability in cell kinetics observed for resting cells and cells under extended TNF stimulation, application of the TNF signal causes cell synchronization. Also, most (but not all) cells behave similarly in the first 90 min of TNF stimulation (Figs. 4, 5, and 11). All analyzed cells (26, Fig. 2) show this TNF-induced synchronization, which is then lost in the course of time. It is only due to this TNF-induced synchronization that the averaged trajectory in Fig. 11 exhibits some oscillations. Since desynchronizing stochastic effects are ubiquitous, it appears to be a general rule that oscillations of the averaged trajectory must imply both the oscillatory character of regulation and the existence of some extracellular signal, which synchronizes the whole population. From the evolutionary perspective, the TNF-induced cell synchronization may result from the fact that all the cells follow the only path in which they could survive under the potentially lethal stimulation. The most critical period in this response is the first 90 min, in which the transcription of early genes like A20, IκBα, IL-6, or IL-8 must be initiated and then terminated (by NF-κB leaving the nucleus) to prevent an unabated inflammatory response.
Acknowledgments
The MATLAB program written to perform the simulations will be available at the website: http://www.stat.rice.edu/126mathbio/stochastic_immunity after the article is published.
This work was supported by National Heart, Lung and Blood Institute contract No. N01-HV-28184, Proteomic Technologies in Airway Inflammation (A. Kurosky, Principal Investigator) and by Polish Committee for Scientific Research grant No. 8T07A-045-20.
APPENDIX I: JUSTIFICATION OF DETERMINISTIC APPROXIMATION FOR TRANSCRIPTION AND TRANSLATION PROCESSES
Let us consider a simple example of a single gene having two copies independently activated and inactivated with equal and constant propensities, rb and rd, respectively. Let us also assume that rb = rd = 5 × 10−4, which results in the expected allele activation/inactivation time of order of 0.5 h. Let the transcription, mRNA degradation, translation, and protein degradation rates, c1, c3, c4, and c5, be equal to those of the A20 gene (Table 2). In Fig. 12 we compare the time evolution of mRNA and protein, simulated using exact stochastic simulation algorithm (thin line) with the stochastic-deterministic approximation (bold line). To compare these two evolutions in the stochastic-deterministic simulation the gene state was changed in the time points following from stochastic simulation algorithm. One can observe that the stochastic-deterministic approximation gives a satisfactory mRNA profile, while the protein profile is almost exactly followed. This latter is the most important to us since the rest of reactions in our model involve proteins only.
APPENDIX II: MODEL EQUATIONS
The mathematical representation of the model (see (20) for derivation) consists of 14 ODEs and four equations accounting for binding and dissociation probabilities of NF-κB molecules to regulatory sites in A20 and IκBα promoters. All the substrates are quantified by the numbers of molecules. The upper-case letters denote substrates or their complexes. Nuclear amount is represented by subscript n, while subscript c denoting amount of substrate in the cytoplasm is now omitted, to simplify the notation. Amounts of the mRNA transcript of A20 and IκBα are denoted by subscript t.
Notation guide
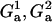 |
Logical variables, status of IκBα promoter in first and second homologous gene copy, G1 = 1, NF-κB attached and G1 = 0, NF-κB absent. |
| G1, G2 | Logical variable, status of A20 promoter in first and second homologous gene copy. |
| IKKa | Cytoplasmic amount of active form of IKK. |
| IKKi | Cytoplasmic amount of inactive form of IKK. |
| IKKn | Cytoplasmic amount of neutral form of IKK kinase. |
| IκBα | Cytoplasmic amount of IκBα. |
| IκBαn | Nuclear amount of IκBα. |
| IκBαt | Amount of IκBα mRNA transcript. |
| (IKKa|IκBα) | Cytoplasmic amount of complexes of IKKa and IκBα. |
| kv = V/U | The ratio of cytoplasmic to nuclear volume. |
| (NFκBn|IκBαn) | Nuclear amount of complexes of NF-κB and IκBα, notation for the remaining complexes is analogous (we will write NFκB instead of NF-κB in equations). |
| TR | Logical variable, TR = 1 if signal (e.g., TNF or IL-1) is present, TR = 0 if no signal is present. |
IKK in the natural state IKKn
The first term describes IKKn synthesis de novo; the second describes degradation; and the last one describes depletion of IKKn due to its signal-induced transformation into active form IKKa,
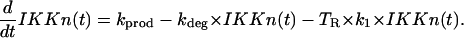 |
(7) |
IKK in the active state IKKa
The first line represents formation of IKKa from IKKi upon the signal activation, depletion of IKKa due to its transformation (spontaneous and induced by A20) into inactive form IKKi, and its degradation. We assumed that the signal (TR = 1) is needed for the catalytic action of A20 and that this action is proportional to the amount of A20, without any saturation. The second line describes depletion of free IKKa due to formation of complexes (IKKa|IκBα) and its recovery after catalytic degradation of these complexes. The third line represents changes in IKKa due to formation and catalytic degradation of trimolecular complexes,
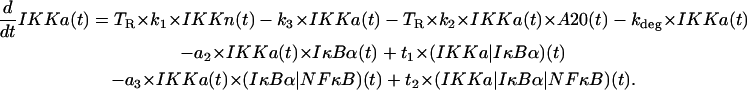 |
(8) |
IKK in the inactive state IKKi
The first two terms correspond to the formation of inactive IKKi from IKKa upon spontaneous inactivation and the catalytic action of A20, while the third term describes degradation,
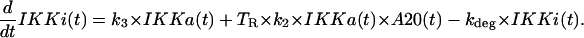 |
(9) |
IKKa|IκBα complexes
This equation represents formation of (IKKa|IκBα) complexes and their degradation. Protein IκBα is catalytically degraded due to phosphorylation caused by IKKa, while free IKKa is recovered (see the last term in the second line of Eq. 8),
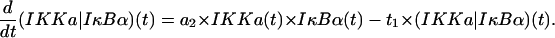 |
(10) |
(IKKa|IκBα|NF-κB) complexes
Described by their formation due to association of IKKa and (IκBα|NF-κB) and degradation due to catalytic activity of IKKa,
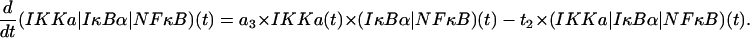 |
(11) |
Free cytoplasmic NF-κB
The first line represents liberation of free NF-κB due to dissociation of (IκBα|NF-κB) complexes and its depletion due to formation of these complexes. The first complete term in the second line accounts for liberation of NF-κB due to the catalytic activity of IKKa. The term in the third line describes transport of free cytoplasmic NF-κB to the nucleus,
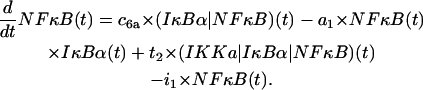 |
(12) |
Free nuclear NF-κB
The first term describes transport into the nucleus. The second term represents depletion of free nuclear NF-κB due to the association with nuclear IκBα and is adjusted, by multiplying the synthesis coefficient a1 by kv = V/U, to the smaller nuclear volume resulting in a larger concentration,
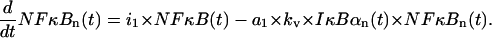 |
(13) |
A20 protein
Described by its mRNA synthesis and constitutive degradation,
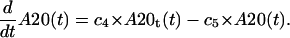 |
(14) |
A20 transcript
The first term stands for constitutive A20 mRNA synthesis and the second term for NF-κB inducible synthesis, while the last term describes degradation of the transcript,
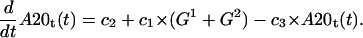 |
(15) |
Free cytoplasmic IκBα protein
The first line accounts for association with IKKa and NF-B, respectively. The second line describes formation of the IκBα protein due to the mRNA synthesis and the constitutive degradation of IκBα. The last line represents transport into and out of the nucleus,
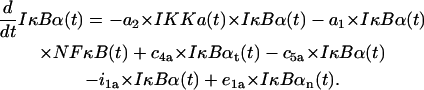 |
(16) |
Free nuclear IκBα protein
The first term corresponds to the association with nuclear NF-κB (adjusted, by multiplying the synthesis coefficient a1 by kv, for the smaller nuclear volume resulting in larger concentration), and the other terms represent the transport into and out of the nucleus,
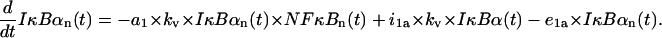 |
(17) |
IκBα transcript
The first term stands for constitutive IκBα mRNA synthesis and the second term for NF-κB inducible synthesis, while the last term describes degradation of transcript,
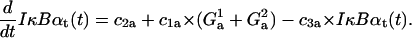 |
(18) |
Cytoplasmic (IκBα|NF-κB) complexes
The first line describes formation of the complexes due to IκBα and NF-κB association and their subsequent degradation. The first term in the second line represents depletion of the (IκBα|NF-κB) complexes due to the catalytic activity of IKKa. The last term represents transport of the complex from the nucleus,
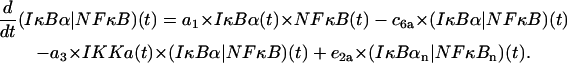 |
(19) |
Nuclear (IκBα|NF-κB) complexes
Described by their formation due to IκBα and NF-κB association (adjusted, by multiplying the synthesis coefficient a1 by kv, to the smaller nuclear volume resulting in larger concentration) and their transport out of the nucleus,
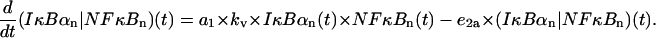 |
(20) |
The total amount of NF-κB (60,000 molecules; see Table 2) is kept constant in the course of simulation, and it is set by assuming the initial concentration of cytoplasmic complexes (IκBα|NF-κB).
The occupancy of IκBα and A20 regulatory sites (in each of the two homologous gene copies) changes in a stochastic way according to the probabilities calculated, based on time-dependent nuclear amounts of NF-κB (binding probabilities) and of IκBα (dissociation probabilities).
Probability of binding of the NF-κB molecule to the IκBα regulatory site in an infinitesimal time interval Δt,
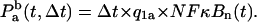 |
(21) |
Probability that NF-κB dissociate from the IκBα site in an infinitesimal time interval Δt,
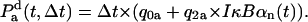 |
(22) |
Probability of binding of the NF-B molecule to the A20 regulatory site in an infinitesimal time interval Δt,
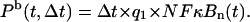 |
(23) |
Probability that NF-κB dissociate from the A20 site in an infinitesimal time interval Δt,
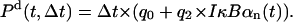 |
(24) |
APPENDIX III: MODEL PARAMETERS
The average volume of a fibroblast cell is V + U = 2000 μm3 = 2 × 10−12 l (22), and we adopted this value to convert concentrations of substrates (used previously in (20)) into numbers of molecules. We will also assume that the ratio of cytoplasmic to nuclear volume is kv = V/U = 5. Therefore the cytoplasmic concentration of 1 μM corresponds to N = 10−6 (mol/l) × V × Av ≈ 106 molecules, where Av = 6.022 × 1023 molecules/mol is the Avogadro number. Respectively, the same nuclear concentration corresponds to 2 × 105 molecules.
In Tables 1–4, we collect values of the measured, assumed, and fitted parameters (see (20) for discussion). When fitting the model one can find that not all parameters may be determined by fitting to the existing data. This is why we restrict our freedom and assume values of part of parameters (in accordance to available data and our intuition). For example, we assumed that most of the parameters accounting for IκBα and A20 kinetics are equal. We also assumed that coefficients c2a, c2, q0a, and q0 equal zero, and one may expect that they are small but positive. It is not, however, possible to deduce their value from the data at our disposal. By such an approach we show how much information can be inferred from available experimental data. All but three parameters in Tables 1–3 are adopted (after translation the concentrations into the numbers of molecules) from our previous article (20). The IκBα transport coefficients i1a and e1a are now twice larger, while IKK inactivation rate coefficient k2 is twice smaller. Coefficients c1a and c1 account for mRNA synthesis per one active allele, correspondingly for IκBα and A20 genes.
TABLE 1.
Two- and three-component interactions between IκBα, NF-κB, and IKKa
| Symbol | Values | Units | Description | Comments |
|---|---|---|---|---|
| a1 | 0.0000005 | s−1 | IκBα-NFκB association | (19) |
| a2 | 0.0000002 | s−1 | IKKa-IκBα association | Assumed |
| t1 | 0.1 | s−1 | IKKa|IκBα catalysis | Any large |
| a3 | 0.000001 | s−1 | IKKa-(IκBα|NF-κB) association | Assumed |
| t2 | 0.1 | s−1 | (IKK|IκBα|NF-κB) catalysis | Any large |
TABLE 2.
A20 and IκBα synthesis and degradation, IKK dynamics, and total amount of free and complexed NF-κB
| Symbol | Values | Units | Description | Comments |
|---|---|---|---|---|
| c1a | 0.075 | s−1 | IκBα -inducible mRNA synthesis | Assumed |
| c2a | 0.0 | s−1 | IκBα -constitutive mRNA synthesis | Assumed, any small |
| c3a | 0.0004 | s−1 | IκBα mRNA degradation | Fitted, (43) |
| c4a | 0.5 | s−1 | IκBα translation rate | Fitted |
| c5a | 0.0001 | s−1 | Spontaneous IκBα (free) protein degradation | (44) |
| c6a | 0.00002 | s−1 | IκBα degradation (complexed to NF-κB) | (44) |
| c1 | 0.075 | s−1 | A20-inducible mRNA synthesis | Assumed |
| c2 | 0.0 | s−1 | A20-constitutive mRNA synthesis | Assumed |
| c3 | 0.0004 | s−1 | A20 mRNA degradation | Assumed |
| c4 | 0.5 | s−1 | A20 translation rate | Assumed |
| c5 | 0.0003 | s−1 | A20 protein degradation | Fitted |
| k1 | 0.0025 | s−1 | IKK activation rate caused by TNF | Fitted |
| k2 | 5 × 10−8 | s−1 | IKK inactivation rate caused by A20 | Fitted |
| k3 | 0.0015 | s−1 | IKK spontaneous inactivation rate | Fitted |
| kprod | 25 | s−1 | IKKn production rate | Fitted |
| kdeg | 0.000125 | s−1 | IKKa, IKKn and IKKi degradation | Fitted |
| NF | 60,000 | Total amount of free and complexed NF-κB | Assumed (22,36) |
TABLE 3.
Transport between compartments, and assumed kv = V/U, ratio of cytoplasmic and nuclear volumes
| Symbol | Value | Units | Description | Comments |
|---|---|---|---|---|
| kv = V/U | 5 | Cytoplasmic to nuclear volume | Assumed | |
| i1 | 0.0025 | s−1 | NF-κB nuclear import | Fitted |
| e2a | 0.05 | s−1 | (IκBα|NF-κB) nuclear export | Fitted |
| i1a | 0.002 | s−1 | IκBα nuclear import | Fitted |
| e1a | 0.005 | s−1 | IκBα nuclear export | Assumed |
TABLE 4.
Parameters of NF-κB binding at A20 and IκBα sites
| Symbol | Value | Units | Description | Comments |
|---|---|---|---|---|
| q1a | 1.5 × 10−7 | s−1 | Binding at IκBα site | Fitted |
| q0a | 0 | s−1 | Constitutive dissociation at IκBα site | Assumed, any small |
| q2a | 10−6 | s−1 | IκBα inducible dissociation at IκBα site | Fitted |
| q1 | 1.5 × 10−7 | s−1 | Binding at A20 site | Assumed |
| q0 | 0 | s−1 | Constitutive dissociation at A20 site | Assumed |
| q2 | 10−6 | s−1 | IκBα inducible dissociation at A20 site | Assumed |
APPENDIX IV: MODEL VALIDATION
To validate the proposed model, we analyzed its ability to reproduce the data from Lee et al. (16) and Hoffmann et al. (19), experiments on mouse fibroblasts (see Fig. 13). Lee et al. (16) measured the response of wild-type and A20-deficient cells to a persistent TNF signal. For both types of cells, Lee et al. (16) collected data on IκBα mRNA, cytoplasmic IκBα, nuclear NF-κB, IKKβ (the key component of IKK), and IKK kinase activity—this latter estimated in an immunoprecipitation-kinase assay using GST-IκBα as a substrate. Additionally, for wild-type cells, they measured A20 mRNA levels. Hoffmann et al. (19) measured NF-κB binding (EMSA assay) and cytoplasmic IκBα level in response to persistent and pulselike TNF activation in wild-type cells. The most important data we miss for mouse fibroblast is the evolution of the level of protein A20. Whereas additional data could remove the ambiguities of the model, the data already collected enable us to trace the time behavior of the majority of the most important variables in the pathway considered.
Typical experimental data at our disposal consist of measurements made at time points that are not uniformly distributed. Therefore, to compare our solutions to experimental data, we rescale the time coordinate, i.e., we calculate the values of corresponding functions in the same time points as in the experiment, and then to guide the eye we connect the resulting discrete points by straight-line segments, thus obtaining a sawlike graph. The IKK kinase activity time-profile, measured using the GST-IκBα substrate in Lee et al. (16), is compared with active pool of IKK, i.e., IKKa (see Fig. 13, A and B). The transient nature of IKK activity is not due to the change in the amount of total IKK (IKKn + IKKa + IKKi). The amount of total IKK remains constant, which, according to our model, results from the assumption that degradation of all IKK forms proceeds at the same rate. Let us note the inverse correspondence between the amounts of mRNA and protein: In wild-type cells the level of protein is high (Fig. 13 D) and level of message is low (Fig. 13 H), while in A20−/− cells there is much of IκBα mRNA (Fig. 13 I) resulting from high nuclear NF-κB activity (Fig. 13 G), but the IκBα protein is degraded due to the prolonged IKK activity (Fig. 13 B) and its level is low (Fig. 13 E). This inverse correspondence between message and protein shows that the assumption of proportionality between message and protein, which is often made in the analysis of microarray experiments, may not be valid.
References
- 1.Ko, M. S. H. 1991. Stochastic model for gene induction. J. Theor. Biol. 153:181–194. [DOI] [PubMed] [Google Scholar]
- 2.Femino, A. M., F. S. Fay, K. Fogarty, and R. H. Singer. 1998. Visualization of single RNA transcripts in situ. Science. 280:585–590. [DOI] [PubMed] [Google Scholar]
- 3.Kepler, T. B., and T. C. Elston. 2001. Stochasticity in transcriptional regulation: origins, consequences, and mathematical representations. Biophys. J. 81:3116–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blake, W. J., M. Kaern, C. R. Cantor, and J. J. Collins. 2003. Noise in eukaryotic gene expression. Nature. 422:633–637. [DOI] [PubMed] [Google Scholar]
- 5.Raser, J. M., and E. K. O'Shea. 2004. Control of stochasticity in eukaryotic gene expression. Science. 304:1811–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paszek, P., T. Lipniacki, A. R. Brasier, B. Tian, D. E. Novak, and M. Kimmel. 2005. Stochastic effects of multiple regulators on expression profiles in eukaryotes. J. Theor. Biol. 233:423–433. [DOI] [PubMed] [Google Scholar]
- 7.McAdams, H. H., and A. Arkin. 1997. Stochastic mechanisms in gene expression. Proc. Natl. Acad. Sci. USA. 94:814–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arkin, A., J. Ross, and H. H. McAdams. 1998. Stochastic kinetics analysis of developmental pathway bifurcation in λ-phage infected Escherichia coli cells. Genetics. 149:1633–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thattai, M., and A. Oudenaarden. 2001. Intrinsic noise in gene regulatory networks. Proc. Natl. Acad. Sci. USA. 98:8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kierzek, A. M., J. Zaim, and P. Zielenkiewicz. 2001. The effect of transcription and translation initiation frequencies on the stochastic fluctuations in prokaryotic gene expression. J. Biol. Chem. 276:8165–8172. [DOI] [PubMed] [Google Scholar]
- 11.Swain, P. S., M. B. Elowitz, and E. D. Siggia. 2002. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc. Natl. Acad. Sci. USA. 99:12795–12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian, B., and A. R. Brasier. 2003. Identification of a nuclear factorκ β-dependent gene network. Recent Prog. Horm. Res. 58:95–130. [DOI] [PubMed] [Google Scholar]
- 13.Sun, S.-C., P. A. Ganchi, D. W. Ballard, and W. C. Greene. 1993. NF-κB Controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science. 259:1912–1915. [DOI] [PubMed] [Google Scholar]
- 14.Karin, M. 1999. The beginning of the end: IκB kinase (IKK) and NF-κB activation. J. Biol. Chem. 274:27339–27342. [DOI] [PubMed] [Google Scholar]
- 15.Krikos, A., C. D. Laherty, and V. M. Dixit. 1992. Transcriptional activation of the tumor necrosis factor α-inducible zinc finger protein, A20, is mediated by κB elements. J. Biol. Chem. 267:17971–17976. [PubMed] [Google Scholar]
- 16.Lee, E. G., D. L. Boone, S. Chai, S. L. Libby, M. Chien, J. P. Lodolce, and A. Ma. 2000. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 289:2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ainbinder, E., M. Revach, O. Wolstein, S. Moshonov, N. Diamant, and R. Dikstein. 2002. Mechanism of rapid transcriptional induction of tumor necrosis factor α-responsive genes by NF-κB. Mol. Cell. Biol. 22:6354–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ainbinder, E., L. Amir-Zilberstein, Y. Yamaguchi, H. Handa, and R. Dikstein. 2004. Elongation inhibition by DRB sensitivity-inducing factor is regulated by the A20 promoter via a novel negative element and NF-κB. Mol. Cell. Biol. 24:2444–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann, A., A. Levchenko, M. L. Scott, and D. Baltimore. 2002. The IκB-NF-κB signaling module: temporal control and selective gene activation. Science. 298:1241–1245. [DOI] [PubMed] [Google Scholar]
- 20.Lipniacki, T., P. Paszek, A. R. Brasier, B. Luxon, and M. Kimmel. 2004. Mathematical model of NF-κB regulatory module. J. Theor. Biol. 228:195–215. [DOI] [PubMed] [Google Scholar]
- 21.Gerondakis, S., M. Grossmann, Y. Nakamura, T. Pohl, and R. Grumont. 1999. Genetic approaches in mice to understand Rel/NF-κB and IκB function: transgenics and knockouts. Oncogene. 18:6888–6895. [DOI] [PubMed] [Google Scholar]
- 22.Carlotti, F., R. Chapman, S. K. Dower, and E. E. Qwarnstrom. 1999. Activation of nuclear factor κB in single living cells. J. Biol. Chem. 274:37941–37949. [DOI] [PubMed] [Google Scholar]
- 23.Nelson, G., L. Paraoan, D. G. Spiller, G. J. C. Wilde, A. M. Browne, P. K. Djali, J. F. Unitt, E. Sullivan, E. Floettmann, and M. R. H. White. 2002. Multi-parameter analysis of the kinetics of NF-κB signalling and transcription in single living cells. J. Cell Sci. 115:1137–1148. [DOI] [PubMed] [Google Scholar]
- 24.Yang, L., K. Ross, and E. E. Qwarnstrom. 2003. RelA control of Iκ Bα phosphorylation. J. Biol. Chem. 278:30881–30888. [DOI] [PubMed] [Google Scholar]
- 25.Schooley, K., P. Zhu, S. K. Dower, and E. E. Qwarnstrom. 2003. Regulation of nuclear translocation of nuclear factor-κB RelA: evidence for complex dynamics at the single cell level. Biochem. J. 369:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson, D. E., A. E. C. Ihekwaba, M. Elliot, J. R. Johnson, C. A. Gibney, B. E. Foreman, G. Nelson, V. See, C. A. Horton, D. G. Spiller, S. W. Edwards, H. P. McDowell, J. F. Unitt, E. Sullivan, R. Grimley, N. Benson, D. Broomhead, D. B. Kell, and M. R. H. White. 2004. Oscillations in NF-κB signaling control the dynamics of gene expression. Science. 306:704–708. [DOI] [PubMed] [Google Scholar]
- 27.Jiang, X., N. Takahashi, N. Matsui, T. Tetsuka, and T. Okamoto. 2003. The NF-κB Activation in lymphotoxin receptor signaling depends on the phosphorylation of p65 at serine 536. J. Biol. Chem. 278:919–926. [DOI] [PubMed] [Google Scholar]
- 28.Tian, B., D. E. Nowak, M. Jamaluddin, S. Wang, and A. R. Brasier. 2005. Identification of direct genomic targets downstream of the NF-κB transcription factor mediating TNF signaling. J. Biol. Chem. 280:17435–17448. [DOI] [PubMed] [Google Scholar]
- 29.Lipniacki, T., P. Paszek, A. Marciniak-Czochra, A. R. Brasier, and M. Kimmel. 2005. Transcriptional stochasticity in gene expression. J. Theor. Biol. In press. [DOI] [PubMed]
- 30.Gillespie, D. T. 1977. Exact stochastic simulations of coupled chemical reactions. J. Phys. Chem. 81:2340–2361. [Google Scholar]
- 31.Gillespie, D. T. 2001. Approximate accelerated stochastic simulation of chemically reacting system. J. Phys. Chem. 115:1716–1733. [Google Scholar]
- 32.Cao, Y., L. R. Petzold, M. Rathinam, and D. T. Gillespie. 2004. The numerical stability of leaping methods for stochastic simulation of chemically reacting systems. J. Chem. Phys. 121:12169–12178. [DOI] [PubMed] [Google Scholar]
- 33.Haseltine, E. L., and J. B. Rawlings. 2002. Approximate simulation of coupled fast and slow reactions for stochastic chemical kinetics. J. Chem. Phys. 117:6959–6969. [Google Scholar]
- 34.Rao, Ch. V., and A. P. Arkin. 2003. Stochastic chemical kinetics and the quasi-steady-state assumption: application to the Gillespie algorithm. J. Chem. Phys. 118:4999–5010. [Google Scholar]
- 35.Cao, Y., D. T. Gillespie, and L. R. Petzold. 2005. The slow-scale stochastic algorithm. J. Chem. Phys. 122:014116. [DOI] [PubMed] [Google Scholar]
- 36.Carlotti, F., S. K. Dower, and E. E. Qwarnstrom. 2000. Dynamic shuttling of nuclear factor κB between the nucleus and cytoplasm as a consequence of inhibitor dissociation. J. Biol. Chem. 275:41028–41034. [DOI] [PubMed] [Google Scholar]
- 37.Barken, D., C. J. Wang, J. Kearns, R. Cheong, A. Hoffman, and A. Levchenko. 2005. Comment on “Oscillations in NF-κB signaling control the dynamics of gene expression”. Science. 308:52a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson, D. E., C. A. Horton, V. See, J. R. Johnson, G. Nelson, D. G. Spiller, D. B. Kell, and M. R. H. White. 2005. Response to comment on “Oscillations in NF-κB signaling control the dynamics of gene expression”. Science. 308:52b. [DOI] [PubMed] [Google Scholar]
- 39.Ackers, G. K., A. D. Johnson, and M. A. Shea. 1982. Quantitative model for gene regulation by λ-phage repressor. Proc. Natl. Acad. Sci. USA. 79:1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gilman, A., and A. P. Arkin. 2002. Genetic code: representation and dynamics models of genetic components and networks. Annu. Rev. Genomics Hum. Genet. 3:341–369. [DOI] [PubMed] [Google Scholar]
- 41.Pirone, J. R., and T. C. Elston. 2004. Fluctuations in transcription factor binding can best explain the graded and binary responses observed in inducible gene expression. J. Theor. Biol. 226:111–121. [DOI] [PubMed] [Google Scholar]
- 42.Rao, Ch. V., D. M. Wolf, and A. P. Arkin. 2002. Control, exploitation and tolerance of intracellular noise. Nature. 420:231–237. [DOI] [PubMed] [Google Scholar]
- 43.Blattner, C., P. Kannouche, M. Litfin, K. Bender, H. J. Rahmsdorf, J. F. Angulo, and P. Herrlich. 2000. UV-induced stabilization of c-Fos and other short-lived mRNAs. Mol. Cell. Biol. 20:3616–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pando, M. P., and I. M. Verma. 2000. Signal-dependent and -independent degradation of free and NF-κB-bound IκBα. J. Biol. Chem. 275:21278–21286. [DOI] [PubMed] [Google Scholar]