

Abstract
Neurodegenerative disease can originate from the misfolding and aggregation of proteins, such as Amyloid-β, SOD1, or Huntingtin. Fortunately, all cells possess protein quality control machinery that sequesters misfolded proteins, either refolding or degrading them, before they can self-associate into proteotoxic oligomers and aggregates. This activity is largely performed by the stress response chaperones (i.e., Hsp70). However, the expression level of molecular chaperones varies widely among cell types. To understand the potential consequence of this variation, we studied the process of protein aggregation in the presence of molecular chaperones using mathematical modeling. We demonstrate that protein aggregation, in the presence of molecular chaperones, is a bistable process. Bistability in protein aggregation offers an explanation for threshold transitions to high aggregate concentration, which are observed both in vitro and in vivo. Additionally, we show that slight variations in chaperone concentration, due to natural fluctuations, have important consequences in a bistable system for the onset of protein aggregation. Therefore, our results offer a possible theoretical explanation for neuronal vulnerability observed in vivo and the onset of neurodegenerative phenotypes in neurons lacking an effective heat-shock response.
INTRODUCTION
The proper folding of proteins is among the most fundamental and essential cellular processes. Given the importance of protein folding and quality control, all cells possess an elaborate network of molecular chaperones and other components for protein homeostasis that facilitate nascent polypeptides through this process (1). Chaperones are essential for guiding folding and protecting against self-association of misfolded species into protein oligomers and aggregates. These aggregates are thought to interfere with the normal functioning of the cell and promote cell dysfunction and death (2,3). For neurons, this leads to a variety of neurodegenerative diseases including Parkinson's, Huntington's, Alzheimer's, ALS, Scrapie, and others (4).
The primary stress response chaperone (Hsp70) binds to motifs rich in hydrophobic residues, thus holding intermediates and guiding folding reactions. Consequently, the sequestering activity of chaperones serves to prevent misfolding and aggregation (5). Hsp70 together with its co-chaperones is also capable of escorting substrates to the proteasome for degradation (6). This process of chaperones targeting misfolded substrates either toward the refolding or degradation machinery is called protein triage (7).
Threshold phenomena, where changes in a control parameter result in a sharp change in an output, have been observed in a variety of biological systems including the Cdc2-cyclin B cell-cycle network, the activation of the JNK cascade, and the lysis/lysogeny switch in the λ-phage (8–12). The nonlinear phenomenon common to these different systems is the presence of a bistable switch that generates these thresholds. In a bistable system, two stable steady states (and a third, unstable steady state) co-exist for a certain range of system parameters; through perturbations in the system parameters or environmental conditions, the system can switch between the two states, in a threshold-dependent manner.
Observations of protein aggregation or refolding often reveal a similar sharp transition in the system, when changing the intrinsic properties of the aggregating species or the concentration of molecular chaperones. For example, in Caenorhabditis elegans, the shift between nontoxic and toxic protein aggregates occurs when expression of a protein containing a 35-mer polyglutamine repeat was increased to a 40-mer polyglutamine repeat (13). A threshold response was also observed for the reactivation of denatured G6PDH in vitro in the presence of DnaK, where a twofold increase in DnaK concentration resulted in more than a fourfold increase in G6PDH reactivation (14). The formation of yeast prion aggregates also displayed threshold-dependent formation of aggregate fibrils at different concentrations of Hsp104 (15).
We employed mathematical modeling to study the origin of threshold behavior in protein misfolding and aggregation in the presence of molecular chaperones. We demonstrate that the introduction of molecular chaperones into a model of protein aggregation creates a threshold in the model through bistability, and that the range of bistability is a function of chaperone concentration and the propensity for self-association of the misfolded protein species. We further use the models to study the potential of the system for stochastic switching from low to high concentrations of aggregates in individual cells. These models focus on the initial formation of soluble oligomers of an aggregation-prone protein, which recent experimental studies have implicated as the toxic precursor species in neurons (16,17).
MATHEMATICAL MODELS
Models of protein aggregation, disaggregation, and folding
We considered an in vitro system where the substrate protein concentration was constant, and there was no chaperone-assisted degradation of misfolded proteins. Our models incorporated four intrinsic processes of protein damage and quality control (misfolding, aggregation, disaggregation, and refolding). We modeled the aggregation process using two different mechanistic descriptions of the process. First, we considered a lumped aggregate model, where one species described all aggregated proteins; this lumped aggregate also acted as a catalyst for further aggregation (18,19). Similar models of protein aggregation, which included explicit autocatalytic formation of the aggregates, have been used previously to describe the prion aggregation process (19,20). Second, we separated the growing aggregate into discrete aggregation states. In this formulation, we included new species for each additional monomer we allowed to incorporate into the aggregate nucleus. Common to both aggregate models was the inclusion of disaggregation and refolding steps, which we assumed were functions of the molecular chaperone concentration.
The lumped aggregate model
The lumped aggregate model (Fig. 1 A) simplified the in vitro experiments to three protein states: a folded state (F), an unfolded protein (U), and the lumped aggregate (A). Consistent with our model of an in vitro process, we assumed that the total protein concentration was constant,
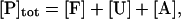 |
(1) |
where [P]tot is the total protein concentration, [F] is the concentration of folded/native proteins, [U] is the concentration of unfolded/misfolded pro teins, and [A] is the concentration of protein aggregates. In the lumped aggregate model, the aggregates are not true aggregates, and are actually individual disease-causing proteins. Molecular chaperones were considered to catalyze the fluxes from the aggregate to unfolded protein and from the unfolded protein to the folded protein. Therefore, the maximal rates of these fluxes were assumed to be proportional to the concentration of molecular chaperones in the system. The parameters and their dimensionless scaling of the lumped aggregate model are shown in Table 1. To reflect the fact that both the disaggregation and refolding were catalyzed by the molecular chaperones and aggregation was catalyzed by the aggregate, the lumped aggregate model was assumed to follow Michaelis-Menten kinetics,
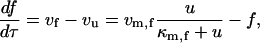 |
(2) |
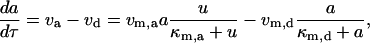 |
(3) |
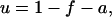 |
(4) |
where lowercase species indicates dimensionless species, all scaled by [P]tot.
FIGURE 1.

Lumped aggregate model. (A) The model contains three states: F, folded protein pool; U, unfolded, aggregation-prone intermediate species; and A, aggregated proteins. The value Vx represents the flux from one state to the next. For example, Vu is the unfolding flux from state F to state U. The values Vd (disaggregation flux) and Vf (folding flux) are assumed to be proportional to the concentration of molecular chaperones in the system. (B) Steady states of the system by graphical analysis of va and vd (dimensionless fluxes; see Table 1), and identification of parameter regimes that led to bistability. (I) Maximal rate of vd (vm,d) greater than the maximal rate of va (vm,a). (II) Same as I, except vm,d < vm,a. Three steady states are present in case II. (III) Same maximal rates as II, but the affinity of the aggregates for self-association was increased above the affinity of chaperones for the aggregates.
TABLE 1.
Mechanistic assumptions and parameters of the lumped aggregate model
| Flux* | Parameter | Interpretation | Dimensionless scaling |
|---|---|---|---|
| Vu | Flux from F to U (unfolding) | 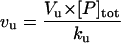 |
|
| ku | Rate constant of protein unfolding | Time scaling | |
| Vf | Flux from U to F (refolding) | 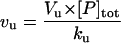 |
|
| Vm,f | Maximal rate of refolding | 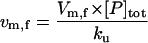 |
|
| Km,f | Affinity of chaperones for unfolded substrate | 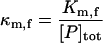 |
|
| Va | Flux from U to A (aggregation) | 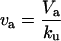 |
|
| Vm,a | Maximal rate of protein aggregation | 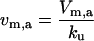 |
|
| Km,a | Affinity of misfolded substrates for preformed aggregates | 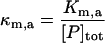 |
|
| Vd | Flux from A to U (disaggregation) |  |
|
| Vm,d | Maximal rate of disaggregation by molecular chaperones | 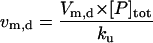 |
|
| Km,d | Affinity of chaperones for aggregated protein | 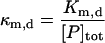 |
|
| t | Time | 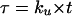 |
From Fig. 1 A.
The discrete aggregate model
A number of recent theoretical studies considered the protein aggregation mechanism in detail (21–24). Based on these studies, we included a growing aggregate nucleus in the model through the discrete addition of unfolded protein monomers to oligomers (Fig. 3; explanation of each species in Table 2). In contrast to the lumped aggregate model, the molecular chaperones were treated as individual, dynamic species in the discrete aggregate model. The total concentration of protein and molecular chaperones were assumed to be conserved in the discrete aggregate model,
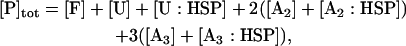 |
(5) |
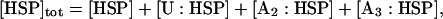 |
(6) |
where [P]tot is the total concentration of protein, and [HSP]tot is the total concentration of molecular chaperones (heat-shock proteins). In addition, we defined a dimensionless “total aggregate” quantity that reflected the percentage of total protein in the various aggregate species:
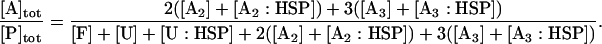 |
(7) |
We included oligomers up to a size of three monomer units because the formation of larger aggregates, more than three monomers, does not change the qualitative behavior of the system and the minimum size of the system provides insight on the origins of the observed threshold phenomena. The formation of the dimer species is distinct because of the association of two monomers versus the association of a small oligomer and a monomer. In addition, the inclusion of at least two oligomeric species allowed us to test explicitly the effect of the two different equilibrium constants (monomer-monomer versus aggregate-monomer association) on the bistability.
FIGURE 3.

Discrete aggregate model. State F represents the folded proteins; U represents the unfolded protein. The addition of two U monomers forms A2, the initial aggregate species. The addition of another U monomer to A2 creates A3. “X:HSP” indicates “X complexed with HSP”, where X is U, A2, or A3. The kinetic and thermodynamic parameters associated with each step are labeled on the diagram; ki is an irreversible rate constant for step ‘i’, 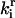 is the reverse or backward step for reversible reaction ‘i,’ and
is the reverse or backward step for reversible reaction ‘i,’ and 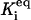 is the equilibrium constant for a set of reversible reactions ‘i’. For example,
is the equilibrium constant for a set of reversible reactions ‘i’. For example, 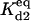 is the equilibrium constant associated with the reversible binding of HSP to A2 (forming A2:HSP),
is the equilibrium constant associated with the reversible binding of HSP to A2 (forming A2:HSP), 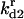 is the rate constant for dissociation of the A2:HSP complex back to A2 and HSP, and kd2 is the rate constant of the catalytic disaggregation of A2:HSP to U:HSP and U. For clarity, only the association is shown explicitly for each reversible reaction.
is the rate constant for dissociation of the A2:HSP complex back to A2 and HSP, and kd2 is the rate constant of the catalytic disaggregation of A2:HSP to U:HSP and U. For clarity, only the association is shown explicitly for each reversible reaction.
TABLE 2.
Species in the discrete aggregate model and their dimensionless scaling
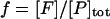
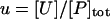
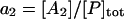
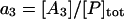
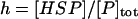
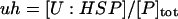
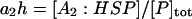
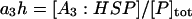
We used generalized mass-action kinetics for the mathematical formulation of the discrete aggregate model (explanation of the kinetic and thermodynamic parameters and their dimensionless scaling in Table 3),
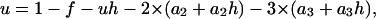 |
(8) |
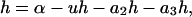 |
(9) |
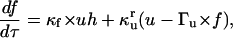 |
(10) |
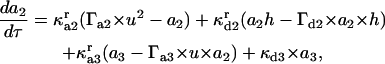 |
(11) |
 |
(12) |
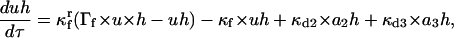 |
(13) |
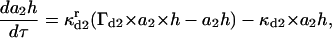 |
(14) |
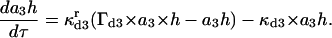 |
(15) |
TABLE 3.
Parameters of the discrete aggregate model, their dimensionless scaling, and their dimensionless numerical value
| Parameter* | Interpretation†‡ | Dimensionless parameter | Scaling¶ | Numerical value |
|---|---|---|---|---|
| Parameters associated with protein unfolding | ||||
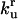 |
Rate constant for unassisted refolding of U to F | 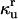 |
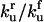 |
1 × 10−1 |
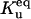 |
Equilibrium between U and F | Γu | 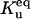 |
1 × 101 |
| Parameters associated with protein aggregation | ||||
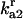 |
Rate constant for disassociation of A2 to 2U | 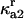 |
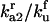 |
1 × 103 |
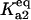 |
Equilibrium between A2 and U | Γa2 | 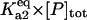 |
5 × 100 |
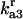 |
Rate constant for disassociation of A3 to A2, U | 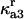 |
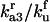 |
1 × 103 |
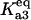 |
Equilibrium between A3 and A2, U | Γa3 | 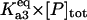 |
1 × 101 |
| Parameters associated with protein refolding | ||||
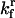 |
Rate constant for unfolded protein-HSP association | 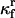 |
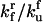 |
1 × 103 |
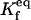 |
Equilibrium between the U:HSP and U, HSP | Γf | 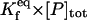 |
1 × 102 |
| kf | Rate constant of chaperone assisted refolding | κf | 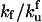 |
1 × 101 |
| Parameters associated with disaggregation by molecular chaperones | ||||
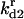 |
Rate constant for dissociation of A2:HSP to A2, HSP | 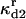 |
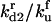 |
1 × 103 |
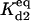 |
Equilibrium between A2:HSP and A2, HSP | Γd2 | 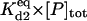 |
1 × 103 |
| kd2 | Rate constant for disaggregation of the dimers | κd2 | 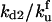 |
3 × 101 |
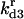 |
Rate constant for dissociation of A3:HSP to A3, HSP | 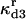 |
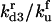 |
1 × 103 |
 |
Equilibrium between A3:HSP and A3, HSP | Γd3 | 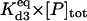 |
1 × 103 |
| kd3 | Rate constant for disaggregation of trimers | κd3 | 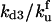 |
3 × 101 |
| Ratio between total chaperones and total protein | Concentration ratio between the total chaperones and total protein | α | 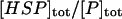 |
Varied |
From Fig. 3.
All equilibrium constants are defined as association constants. For example, 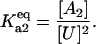
Forward rate constants were determined by multiplying the equilibrium constant by the reversible rate constant.
Time is scaled by the forward rate constant of protein unfolding, 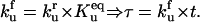
RESULTS
Bistability as a function of molecular chaperone concentration in the lumped aggregate model
The steady-state (time-invariant) behavior of low-dimension nonlinear systems is often analyzed using graphical analysis (25,26). The steady-state concentration of aggregates was determined from the condition that the dimensionless aggregation flux, va, equals the dimensionless disaggregation flux, vd:
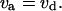 |
(16) |
Plotting va and vd versus the dimensionless concentration of aggregate showed that three steady states were possible in this system (Fig. 1 B, case II). Therefore, the system was capable of displaying bistability. However, two conditions should be satisfied for three steady states to exist; see below.
Condition 1
The maximal dimensionless rate of protein aggregation (vm,a) must be greater than the maximal dimensionless rate of disaggregation (vm,d). Otherwise, only one steady state will exist (Fig. 1 B, case I).
Condition 2
The slope of vd must be greater than the slope of va at [A] = 0, or only one or two steady states will be present. This condition can be satisfied through the combination of several parameters, including decreasing the relative affinities of the aggregates for self-association versus association with chaperones (Fig. 1 B, case III), or increasing the chaperone concentration or activity for disaggregation (see Appendix).
With the regime of bistability in the model established, we tested the effect of variations in the protein quality control machinery on the appearance of aggregation thresholds. Conditions 1 and 2 are functions of the maximal rates of disaggregation and refolding. Therefore, changes in the molecular chaperone concentration affected the number of steady states possible in the system. Varying these maximal rates in the model confirmed that above a critical threshold in molecular chaperone concentration there was only one steady state of the network, mostly folded proteins, and no aggregated protein (Fig. 2, A–C). However, once the chaperone concentration was lowered below this critical threshold, a new state emerged, which contained the proteins in aggregated form. Cells that operate at a chaperone concentration below this bifurcation point can effectively switch between the two states; in other words, cells with no aggregates could switch to the high aggregation state if an aggregate seed of sufficient size appeared in the system.
FIGURE 2.

Bistability in the lumped aggregate model as a function of molecular chaperone concentration. (A–C) Steady states of states F, U, and A versus vm,f and vm,d (dimensionless maximal rates). Dashed segments denote the unstable steady states. All concentrations scaled by total protein concentration ([P]tot).
Bistability in a discrete protein aggregate model
We used the deficiency theory to determine the existence of multiple steady states in the mass-action discrete aggregate model (27,28). The analysis also provided the values of dimensionless parameters that gave rise to bistability, which resulted in physiological ratios for the rates and affinities of the different biophysical processes (Table 3). For example, we chose the dimensionless affinity of the chaperones for their substrates to be significantly higher than the dimensionless affinity of the unfolded proteins for self-association to reflect the use of ATP to drive tight associations of Hsp70 with its substrates (29). Similarly, we chose the dimensionless rate constant for spontaneous refolding (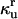 ) as several orders-of-magnitude slower than the dimensionless rate constant of chaperone-assisted refolding (κf) to reflect the established catalytic role of chaperone protein refolding. Finally, we used the common assumption of fast reversible rate constants by choosing the dimensionless reversible rate constants (
) as several orders-of-magnitude slower than the dimensionless rate constant of chaperone-assisted refolding (κf) to reflect the established catalytic role of chaperone protein refolding. Finally, we used the common assumption of fast reversible rate constants by choosing the dimensionless reversible rate constants ( ) to be several orders-of-magnitude greater than the catalytic rate constants (κx). Interestingly, similar pathways or processes had comparable orders of magnitude for their rate and equilibrium constants. Also interesting, the two Conditions of the lumped aggregate model for bistability also held for the dimensionless parameters of the discrete aggregate model. This demonstrated the utility of simple models, such as the lumped aggregate model, for elucidating the features or design of more complicated biological systems.
) to be several orders-of-magnitude greater than the catalytic rate constants (κx). Interestingly, similar pathways or processes had comparable orders of magnitude for their rate and equilibrium constants. Also interesting, the two Conditions of the lumped aggregate model for bistability also held for the dimensionless parameters of the discrete aggregate model. This demonstrated the utility of simple models, such as the lumped aggregate model, for elucidating the features or design of more complicated biological systems.
Using the parameters in Table 3, we simulated the discrete aggregate model with varying total molecular chaperone concentration. The discrete aggregate model displayed bistability similar to the lumped aggregate model (Fig. 4 A). However, this detailed mechanistic model provided better insight on the response of protein aggregation to changes on chaperone activity, and allowed us to investigate the impact of other physicochemical parameters on the bistable behavior of the system.
FIGURE 4.

Bistability in the discrete aggregate model. (A) Bistability of folded, unfolded, and aggregated proteins as a function of scaled molecular chaperone concentration. The steady states of folded (solid line), unfolded (dashed-dotted line), and total aggregated (dashed line) proteins are shown. (B) Two-dimensional bifurcation diagram of the bistable region as a function of 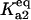 and scaled HSP concentration. Horizontal lines indicate
and scaled HSP concentration. Horizontal lines indicate 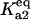 relative to
relative to 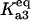 :
: 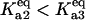 (solid line),
(solid line), 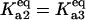 (dashed line), and
(dashed line), and 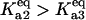 (dash-dot line). (C) Steady-states of state-scaled aggregated protein versus scaled HSP concentration with the three ratios of
(dash-dot line). (C) Steady-states of state-scaled aggregated protein versus scaled HSP concentration with the three ratios of 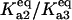 from B. (Solid line)
from B. (Solid line) 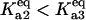 ; (dashed line)
; (dashed line) 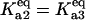 ; and (dash-dotted line)
; and (dash-dotted line) 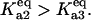 Thin-line segments denote the unstable steady state.
Thin-line segments denote the unstable steady state.
For these parameters, the range of the concentration of molecular chaperones to the concentration of the total protein where we observed bistability was between a ratio of 0.16 and 0.21. This concentration range was similar to the threshold observed in the disaggregation of amyloidlike structures of Sup35 by Hsp104, which occurred at a concentration ratio near 0.07 (15). However, the ratio of molecular chaperones to total substrate where the threshold transition to low or high aggregate concentrations occurs is dependent on both the type of chaperone and the substrate. For example, the in vitro threshold for reactivation of G6PDH by DnaK occurred between a ratio of concentrations of 3–6 for DnaK to G6PDH (14). In general, we would not expect an exact correspondence between the values of the model parameters and the experimentally observed threshold values because of unaccounted-for processes in vivo, such as chemical modifications of protein substrates, and an unknown relationship between the concentration of molecular chaperones and their effective activity for protein quality control.
In the discrete aggregate model, after the initial formation of the A2 dimer, subsequent aggregation steps were more thermodynamically favorable, a fact that effectively introduced the feedback activation of aggregate formation found in the lumped aggregate model. To assess the dependence of bistability on the relative equilibrium constants between unfolded protein and dimer/oligomer concentration, we performed a two-parameter bifurcation analysis (Fig. 4 B). We specifically investigated the behavior of the system for the different chaperone concentrations when the initial aggregate formation was thermodynamically less favorable 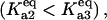 equally favorable
equally favorable  and more favorable
and more favorable 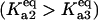 than the larger aggregate formation. In all three regimes, the model displayed bistability. However, the critical chaperone concentrations depended on the equilibrium constants of aggregate formation; the bistable switch occurred at higher chaperone concentrations if the formation of the first, small oligomers was more favorable that the larger ones (Fig. 4 C). This implied that the formation of small oligomers was the determining step for initiation of protein aggregation.
than the larger aggregate formation. In all three regimes, the model displayed bistability. However, the critical chaperone concentrations depended on the equilibrium constants of aggregate formation; the bistable switch occurred at higher chaperone concentrations if the formation of the first, small oligomers was more favorable that the larger ones (Fig. 4 C). This implied that the formation of small oligomers was the determining step for initiation of protein aggregation.
The molecular chaperones are necessary for bistability
The process of protein aggregation, without molecular chaperones, is a competition between the thermodynamic driving forces for aggregation versus spontaneous refolding. The driving force of aggregation is proportional to the equilibrium constant between the unfolded monomers and the small oligomers 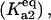 and spontaneous refolding is inversely proportional to the equilibrium constant of unfolding
and spontaneous refolding is inversely proportional to the equilibrium constant of unfolding 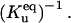 Therefore, we quantified the competition between refolding and protein aggregation by defining a new dimensionless parameter,
Therefore, we quantified the competition between refolding and protein aggregation by defining a new dimensionless parameter,
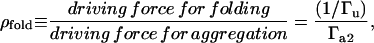 |
(17) |
which reflected how much spontaneous refolding was thermodynamically favored versus aggregation. For all values of ρfold, the presence of a bistable transition in the system was only observed for non-zero values of chaperone concentration (Fig. 5 A). In fact, using deficiency theory (27,28), we determined that the refolding and disaggregation activities of the molecular chaperones were necessary for a bistable threshold to exist in the discrete aggregate model. Below the bistable regime, the total aggregate concentration decreased monotonically as the ρfold was increased to favor refolding (Fig. 5 B). However, within the bistable regime, the appearance of a second stable steady state allowed the system to transition from relatively high aggregate concentration to low aggregate concentration, without any change in the thermodynamics of refolding and aggregation. These results suggested that even at low chaperone concentrations and high aggregation potential, the chaperone quality control mechanism is capable of preventing aggregation through the introduction of this bistable threshold.
FIGURE 5.

Competition between refolding and aggregation versus HSP concentration. (A) Two-parameter continuation of ρfold versus [HSP]tot/[P]tot. (B) [A]tot/[P]tot versus ρfold for three different chaperone concentrations ([HSP]tot/[P]tot = 0.001 (dashed line), 0.005 (dash-dotted line), and 0.01 (solid line)). Thin line segments denote the unstable steady state.
Intrinsic noise and the onset of aggregation
At the cellular level, low protein concentrations lead to significant fluctuations in the distribution of species and deviations from deterministically predicted time courses (30,31). The intrinsic noise in a bistable intracellular process has the potential for the fluctuations in the concentration of certain species to cross a critical threshold and switch between steady states. Such phenomena occur in several biological systems such as the Sonic-Hedgehog signaling and the lysis-lysogeny switch of the bacteriophage-λ (12,32,33).
We performed a stochastic simulation of the discrete aggregate model to investigate the effect of intrinsic noise in protein aggregation. We simulated the stochastic dynamics of our system using the Gibson algorithm, an efficient modification of the Gillespie algorithm (34–36), and we studied the effect of intrinsic noise on the formation of aggregates within the low-chaperone concentrations, high-aggregate regime (Fig. 6 A). The deterministic response reached 50% of its maximal value at time = 9.5 (all times are dimensionless); however, only 63% of the cells had reached their 50% maximal value at this time (Fig. 6 B). The most rapid aggregating cells achieved 50% maximal aggregation in approximately half the time of the deterministic simulation (time ≈5), and the slowest aggregating cells did not reach their 50% maximal value until time ≈25. Additionally, the wide distribution of transition times from low to high aggregated states led to a distribution of aggregation states (Fig. 6 C); in other words, the initially homogeneous population underwent a transition through which the population was distributed between a low and high protein aggregate concentration.
FIGURE 6.

Stochastic simulations of the discrete aggregate model ([HSP]tot/[P]tot = 0.16). (A) Plotted is a representative sample (33) of the stochastic dynamics of 482 stochastic simulations (solid lines); the equivalent deterministic result is also included (thick dashed line). Time is dimensionless, scaled by the rate constant of unfolding (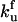 ). (B) Time of crossing to high aggregate concentration. The time [A]tot/[P]tot = 0.50 for each cell is summarized in the histogram. The deterministic time is included for reference (dashed line). (C) The scaled aggregate concentration in each cell at time = 9.5. The deterministic value is included for reference (dashed line).
). (B) Time of crossing to high aggregate concentration. The time [A]tot/[P]tot = 0.50 for each cell is summarized in the histogram. The deterministic time is included for reference (dashed line). (C) The scaled aggregate concentration in each cell at time = 9.5. The deterministic value is included for reference (dashed line).
Comparison of the transient dynamics to in vitro experiments showed that the timescale of this transient was 1–2 h (14,15). Thus, a perturbation to the protein quality control machinery that results in the system crossing the bifurcation point to high aggregate concentration will lead to rapid formation of protein aggregates. However, additional timescales influence the time of onset for protein aggregates in vivo. These processes include the time for expression and accumulation of disease-causing proteins, and the slow time for changes to occur in the activity or concentration of the protein quality control machinery.
DISCUSSION
We presented a theoretical analysis of the process of protein aggregation and rescue by molecular chaperones. Our model displayed a threshold for the formation of protein aggregates with respect to both intrinsic properties of the protein (equilibrium constants for self-association) and extrinsic properties of the system (concentration of molecular chaperones). Previous experimental studies showed that a threshold exists for the formation of protein aggregates with respect to both of these properties. In vitro studies showed thresholds with respect to the concentration of molecular chaperones for several different substrates and chaperones (14,15). Additionally, the expression of different length polyglutamine expansions in C. elegans showed a polyQ-dependent increase in the number of observed aggregates, and a dramatic decrease in the time of aggregation onset between a polyglutamine expansion of Q35 and Q40 (13). In vivo aggregation processes are far more complicated than the reactions in the model. For example, the model does not contain any explicit information about chemical modifications of polyQ proteins post-aggregation. However, the model does implicitly capture the effect of expanding the polyglutamine repeat, which in the model is equivalent to increasing the propensity of the proteins for self-association. Based on the analysis of our models, we proposed that a nonlinear phenomena known as bistability caused the threshold. The presence of bistability in the system has significant implications for the onset and potential prevention of protein aggregation. Potential therapies must account for hysteresis in bistable systems. For example, reduction of the concentration of molecular chaperones drives the system to the upper aggregate state. Restoration of the system to the low aggregate steady state requires an increase of the concentration of the molecular chaperones above the level at which bistability exists to overcome the hysteresis phenomena due to bistability.
The essential nature of the molecular chaperones machinery for generating the bistable switch revealed an interesting interplay between protein aggregation and molecular chaperone production and potential consequences for the failure of the heat-shock response. Our analysis showed that the molecular chaperones maintained the system in the low aggregate steady state, even as conditions increasingly favored the formation of protein aggregates. However, one of the unique aspects of many neuronal cell types is their inability to induce Hsp70 expression above basal levels under stress (37,38). Therefore, cellular stresses coupled with slow degradation of chaperones activity or concentration could cause the system to cross the threshold from the bistable region to the high-aggregate steady state.
In vivo, each individual biophysical process of the model is more complicated than simple self-association or chaperone binding; mechanistically, each process might require the binding of cofactors, hydrolysis of ATP, etc. However, the use of different models and the parametric analyses showed that the basic observation of bistability is robust to several mechanistic assumptions and parameter values. The finding that a simple mechanism generates the expected threshold is significant because there are many in vivo mechanisms for irreversible protein aggregation. For example, for the particular case of polyQ-associated disorders, the aggregates are cross-linked by transglutaminase (39). This process of cross-linking generates irreversible aggregates. Although this cross-linking contributes to the long-time formation of Huntingtin aggregates, the model demonstrates that this modification and other possible biochemical modifications of the protein aggregates are not necessary for the system to display a switch to high levels of protein aggregates. The origin of the bistable switch is in the feedback from the cellular protein quality control machinery, and its interactions (or failure to interact) with the misfolded substrates.
It is important to distinguish between the steady state of the protein aggregates and the timescale for the appearance and formation of protein aggregates. Steady-state analysis indicates whether aggregates will form, given certain intrinsic and extrinsic properties of the system. Many of these properties influence the timescale or dynamics for the appearance and formation of aggregates. The activity of the molecular chaperones is an extrinsic property of the system that may drift (become impaired) over years or decades, and this drift in activity may cause the system to cross the steady-state threshold to the high aggregate steady state and induce the formation of protein aggregates. In addition, we showed that intrinsic stochastic fluctuations influenced the timescale for formation of protein aggregates. These fluctuations led to the ensembles forming aggregates over a broad distribution of times. Thus, individuals with the same genotype can form aggregates at very different ages. However, the current state of the model does not address at what point protein aggregates become toxic to neurons and lead to the onset of neurodegenerative disorders, since, like most of the diseases, this depends on the function of many other cellular processes.
A whole genome RNAi screen in C. elegans identified 186 genes whose protein products act as modifiers of protein aggregation (40). This revealed that the prevention of protein aggregation is not the sole responsibility of the chaperone machinery, but involves the complex interaction of many essential intracellular processes. For example, of the 186 genes, 14 genes were associated with protein degradation pathways and the proteasome. Chaperones enhance the in vivo rate of dissociation and degradation of aggregates. Because enhanced clearance of aggregates also requires the degradative machinery, we can suggest that it is both the chaperone folding machinery and the degradative/clearance machinery of proteasomes, and that autophagy must be involved. Although chaperones are an essential component of these events, they are not the only components necessary for the proper dissociation of damaged proteins in the cell. Therefore, a full description of the in vivo process and identification of potential points of failure of protein homeostasis will require a more detailed description of this process and its interaction with the molecular chaperone machinery and protein aggregates (41). Mathematical models allow us to consider each essential piece of the complex biology individually and to begin to incorporate these components into a more complete theory for the design of protein quality control in the human cell.
APPENDIX: CONDITION 2 IN THE LUMPED AGGREGATE MODEL
Condition 2 required that the slope of vd was greater than the slope of va near a = 0:
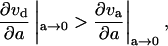 |
(18) |
where
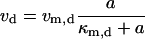 |
(19) |
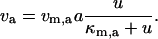 |
(20) |
Taking the derivative of vd and va with respect to a:
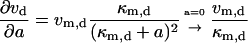 |
(21) |
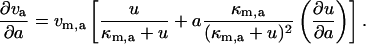 |
(22) |
The second term in Eq. 22 was neglected since a = 0. However, to evaluate the first term, we solved for u as a function of a. We solved for u as a function of a using the conservation of total protein (Eq. 4) and the steady-state expression for f:
 |
(23) |
which resulted in a quadratic expression for f as a function of a:
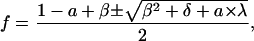 |
(24) |
where β = vm,f + κm,f, δ = 1 + 2(κm,f − vm,f), and λ = a − 2(κm,f − vm,f + 1). Combining Eq. 24 with Eq. 4,
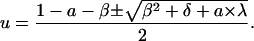 |
(25) |
The resulting expression for Eq. 22, at a = 0:
 |
(26) |
Therefore, the full Condition 2:
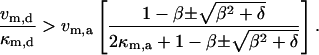 |
(27) |
References
- 1.Hartl, F. U. 1996. Molecular chaperones in cellular protein folding. Nature. 381:571–579. [DOI] [PubMed] [Google Scholar]
- 2.Volles, M. J., and P. T. Lansbury, Jr. 2002. Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 41:4595–4602. [DOI] [PubMed] [Google Scholar]
- 3.Lashuel, H. A., D. Hartley, B. M. Petre, T. Walz, and P. T. Lansbury, Jr. 2002. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 418:291. [DOI] [PubMed] [Google Scholar]
- 4.Soto, C. 2003. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4:49–60. [DOI] [PubMed] [Google Scholar]
- 5.Fink, A. L. 1999. Chaperone-mediated protein folding. Physiol. Rev. 79:425–449. [DOI] [PubMed] [Google Scholar]
- 6.Cyr, D. M., J. Hohfeld, and C. Patterson. 2002. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem. Sci. 27:368–375. [DOI] [PubMed] [Google Scholar]
- 7.Wickner, S., M. R. Maurizi, and S. Gottesman. 1999. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 286:1888–1893. [DOI] [PubMed] [Google Scholar]
- 8.Bagowski, C. P., J. Besser, C. R. Frey, and J. E. Ferrell, Jr. 2003. The JNK cascade as a biochemical switch in mammalian cells: ultrasensitive and all-or-none responses. Curr. Biol. 13:315–320. [DOI] [PubMed] [Google Scholar]
- 9.Bagowski, C. P., and J. E. Ferrell, Jr. 2001. Bistability in the JNK cascade. Curr. Biol. 11:1176–1182. [DOI] [PubMed] [Google Scholar]
- 10.Thron, C. D. 1997. Bistable biochemical switching and the control of the events of the cell cycle. Oncogene. 15:317–325. [DOI] [PubMed] [Google Scholar]
- 11.Pomerening, J. R., E. D. Sontag, and J. E. Ferrell, Jr. 2003. Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol. 5:346–351. [DOI] [PubMed] [Google Scholar]
- 12.Tian, T., and K. Burrage. 2004. Bistability and switching in the lysis/lysogeny genetic regulatory network of bacteriophage-λ. J. Theor. Biol. 227:229–237. [DOI] [PubMed] [Google Scholar]
- 13.Morley, J. F., H. R. Brignull, J. J. Weyers, and R. I. Morimoto. 2002. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 99:10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ben-Zvi, A. P., P. De Los Rios, G. Dietler, and P. Goloubinoff. 2004. Active solubilization and refolding of stable protein aggregates by cooperative unfolding action of individual HSP70 chaperones. J. Biol. Chem. 36:37298–37303. [DOI] [PubMed] [Google Scholar]
- 15.Shorter, J., and S. Lindquist. 2004. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 304:1793–1797. [DOI] [PubMed] [Google Scholar]
- 16.Conway, K. A., S. J. Lee, J. C. Rochet, T. T. Ding, R. E. Williamson, and P. T. Lansbury, Jr. 2000. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA. 97:571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agorogiannis, E. I., G. I. Agorogiannis, A. Papadimitriou, and G. M. Hadjigeorgiou. 2004. Protein misfolding in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 30:215–224. [DOI] [PubMed] [Google Scholar]
- 18.Harper, J. D., and P. T. Lansbury, Jr. 1997. Models of amyloid seeding in Alzheimer's disease and Scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66:385–407. [DOI] [PubMed] [Google Scholar]
- 19.Come, J. H., P. E. Fraser, and P. T. Lansbury, Jr. 1993. A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc. Natl. Acad. Sci. USA. 90:5959–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kacser, H., and J. R. Small. 1996. How many phenotypes from one genotype? The case of Prion diseases. J. Theor. Biol. 182:209–218. [DOI] [PubMed] [Google Scholar]
- 21.Kodaka, M. 2004. Interpretation of concentration-dependence in aggregation kinetics. Biophys. Chem. 109:325–332. [DOI] [PubMed] [Google Scholar]
- 22.Kodaka, M. 2004. Requirements for generating sigmoidal time-course aggregation in nucleation-dependent polymerization model. Biophys. Chem. 107:243–253. [DOI] [PubMed] [Google Scholar]
- 23.Pallitto, M. M., and R. M. Murphy. 2001. A mathematical model of the kinetics of β-amyloid fibril growth from the denatured state. Biophys. J. 81:1805–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson, A. S., and D. A. Lauffenburger. 1996. Model for ER chaperone dynamics and secretory protein interactions. AIChE J. 42:1443–1453. [Google Scholar]
- 25.Palsson, B. O., J. D. Keasling, and S. G. Emerson. 1990. The regulatory mechanisms of human immunodeficiency virus replication predict multiple expression rates. Proc. Natl. Acad. Sci. USA. 87:772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatzimanikatis, V., and J. E. Bailey. 1997. Studies on glycolysis. 1. Multiple steady states in bacterial glycolysis. Chem. Eng. Sci. 52:2579–2588. [Google Scholar]
- 27.Feinberg, M. 1987. Chemical-reaction network structure and the stability of complex isothermal reactors. 1. The Deficiency-Zero and Deficiency-One theorems. Chem. Eng. Sci. 42:2229–2268. [Google Scholar]
- 28.Feinberg, M. 1988. Chemical-reaction network structure and the stability of complex isothermal reactors. 2. Multiple steady states for networks of Deficiency One. Chem. Eng. Sci. 43:1–25. [Google Scholar]
- 29.Mayer, M. P., H. Schroder, S. Rudiger, K. Paal, T. Laufen, and B. Bukau. 2000. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 7:586–593. [DOI] [PubMed] [Google Scholar]
- 30.McAdams, H. H., and A. Arkin. 1997. Stochastic mechanisms in gene expression. Proc. Natl. Acad. Sci. USA. 94:814–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McAdams, H. H., and A. Arkin. 1999. It's a noisy business! Genetic regulation at the nanomolar scale. Trends Genet. 15:65–69. [DOI] [PubMed] [Google Scholar]
- 32.Thattai, M., and A. van Oudenaarden. 2001. Intrinsic noise in gene regulatory networks. Proc. Natl. Acad. Sci. USA. 98:8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lai, K., M. J. Robertson, and D. V. Schaffer. 2004. The Sonic-Hedgehog signaling system as a bistable genetic switch. Biophys. J. 86:2748–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson, M. A., and J. Bruck. 2000. Efficient exact stochastic simulation of chemical systems with many species and many channels. J. Phys. Chem. A. 104:1876–1889. [Google Scholar]
- 35.Gillespie, D. T. 1976. General method for numerically simulating stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 22:403–434. [Google Scholar]
- 36.Gillespie, D. T. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81:2340–2361. [Google Scholar]
- 37.Kaarniranta, K., N. Oksala, H. M. Karjalainen, T. Suuronen, L. Sistonen, H. J. Helminen, A. Salminen, and M. J. Lammi. 2002. Neuronal cells show regulatory differences in the Hsp70 gene response. Brain Res. Mol. Brain Res. 101:136–140. [DOI] [PubMed] [Google Scholar]
- 38.Marcuccilli, C. J., S. K. Mathur, R. I. Morimoto, and R. J. Miller. 1996. Regulatory differences in the stress response of hippocampal neurons and glial cells after heat shock. J. Neurosci. 16:478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kahlem, P., H. Green, and P. Djian. 1998. Transglutaminase action imitates Huntington's disease: selective polymerization of Huntingtin containing expanded polyglutamine. Mol. Cell. 1:595–601. [DOI] [PubMed] [Google Scholar]
- 40.Nollen, E. A., S. M. Garcia, G. van Haaften, S. Kim, A. Chavez, R. I. Morimoto, and R. H. Plasterk. 2004. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA. 101:6403–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmberg, C. I., K. E. Staniszewski, K. N. Mensah, A. Matouschek, and R. I. Morimoto. 2004. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 23:4307–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]