

Abstract
We explore the means by which immobilization of a substrate on a surface can increase the rate of a diffusion-controlled enzymatic reaction. A quasichemical approach is developed and compared with Brownian dynamics simulations. We use these methods to show that restricting only the orientation of the enzyme by long-range interactions with the surface is sufficient for enhancing catalysis.
INTRODUCTION
Enormous interest in surface-based assays for biological interactions and activities is driving significant advances in associated technologies. Surfaces that are both robust and inert to nonspecific adsorption can be made readily by controlled means (1), and chips with a diverse range of chemical properties are now available commercially (2). Used in combination with sensitive in situ methods for detection of binding and reaction, in particular surface plasmon resonance spectroscopy (2) (but see also (3–9)), solid substrates with molecules immobilized in well-defined ways are enabling quantitative measurement of equilibrium and kinetic parameters.
For a diffusion-controlled enzymatic reaction, different catalytic rates are observed for substrate molecules free in solution and ones tethered on surfaces. The relative diffusion constant, the rotational freedom of substrate molecules, and the solid angle available for collision are all reduced in going from the former to the latter case; these changes tend to limit association. Mass transport effects (2,10), aggregation, and crowding (11–14) can further influence the kinetics of reaction. It is important to obtain quantitative corrections for the various factors to make meaningful connection between experimental measurements and natural situations. Here, we restrict our attention to the simplest case, systems with well-separated substrate molecules in the absence of flow.
Because in general the effects listed above decrease apparent rates, it is natural to ask whether there are circumstances under which restriction of a diffusion-controlled reaction to an interface can enhance the kinetics. One well-known way that a surface can facilitate interaction of molecules is that it can guide the translational diffusion of the mobile species ((15–18); A. Nag and R. S. Berry, unpublished). In other words, the search is broken into two steps: association with the surface followed by a random walk in two, rather than three, dimensions. The reduction in the available space drastically accelerates the enzymatic throughput.
In this article, we investigate the rotational analog of the mechanism described above. Namely, restriction of orientation in the presence of a surface is shown to be sufficient to allow an enzyme to convert immobilized substrate species more rapidly than like-molecules in solution. To this end, we extend the quasichemical scheme that Šolc and Stockmayer (19) introduced for diffusion-controlled reactions in solution. The idea is then made more explicit with Brownian dynamics simulations of a simple representation of a neutral enzyme with a dipole that interacts with a substrate molecule immobilized to a surface with a uniformly distributed charge. The relation to electrostatic steering (20–23) is discussed.
THEORY
Michaelis-Menten kinetics
In this section, we develop an approximate analytic theory to treat the kinetics of enzymatic reactions with substrate molecules in solution and on surfaces consistently. Our starting point is the standard Michaelis-Menten scheme (24):
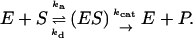 |
(1) |
Here, E is the enzyme, S is the substrate molecule, (ES) is a bound complex, and P is the product; ka, kd, and kcat are the rate constants for association, dissociation, and catalysis, respectively. The rate of product formation under the assumption that d(ES)/dt = 0 is
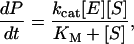 |
(2) |
where KM = (kcat + kd)/ka and [E] is the total enzyme concentration, which is the sum of the concentrations of both free and bound forms. Our specific goal is to relate ka and kd to molecular properties for the cases of mobile and immobile substrate species. To this end, we adapt a quasichemical scheme (19) and various expressions for its parameters (17,25) (reviewed below). The relations obtained are then used to show explicitly that tethering a substrate to a passive surface decreases kcat/KM and this effect can be overcome by allowing the surface to enhance enzyme reorientation.
It is important to note that the Michaelis-Menten expression for the rate assumes that the reversible formation of the enzyme-substrate complex (ES) by diffusional encounter and the irreversible conversion of the substrate to product P are both described by ordinary differential equations. In principle, diffusion introduces a time dependence to ka in Eq. 1, which can lead to deviation from Eq. 2. However, Zhou (26) showed that, for orientationally restricted sites typical of enzymes, ka very quickly approaches its infinite time value, so that Michaelis-Menten kinetics will be adequate in general.
Model geometry
We model the enzyme and substrate molecules as spheres with axially symmetric reactive patches. In the case of the surface-tethered substrate, only half the surface area of that species is available for collision and its reactive patch is centered on a vector orthogonal to the surface (Fig. 1). For simplicity, we assume here that there are no long-range forces between the molecules.
FIGURE 1.

Model system of spheres with axially symmetric reactive patches. θE is the angle associated with the reactive patch on the enzyme; θS (not indicated) is the corresponding angle for the substrate.
The case of two mobile spheres with angularly restricted reactive sites has been studied extensively (17,19,25,27–30). In contrast, there are relatively few studies that treat immobilized species as three- rather than two-dimensional objects (typically, circular disks (14,15,31)). Schmitz and Schurr (32,33) considered the case of a uniformly reactive hemispherical substrate interacting with a spherical enzyme with an axially symmetric reactive patch. However, the orientation constraint in Schmitz and Schurr (32) and Schurr and Schmitz (33) differed from that in studies of two mobile spheres and the present work in that the angle was measured relative to a fixed vector orthogonal to the surface rather than one along a line connecting the centers of the molecules (see (28) for a discussion).
Quasichemical approximation
Because our primary purpose is to gain qualitative insight into how tethering substrate molecules influences different aspects of association and dissociation, we explore the physically transparent but approximate quasichemical approach of Šolc and Stockmayer (19). The key simplification is that collision (due to translation) and reorientation to align the reactive patches (due to translation or rotation) can be treated separately (Fig. 2). Each species (C ∈ {E, S}) can be either oriented toward (C+) or away from (C−) the other, so there are four possible unbound but paired states: E+S+, E+S−, E−S+, and E−S−.
FIGURE 2.

Quasichemical scheme. Superscripts on enzyme-substrate pairs indicate whether the species are in reactive orientations: minus symbol (−) indicates a nonreactive orientation, and plus symbol (+) indicates a reactive orientation. Essentially, the enzyme and substrate can collide and separate in all possible orientations, but only E+S+, in which both species are correctly oriented, can form the ES complex (ES).
The E+S+ state forms a bound complex with rate constant kx; the corresponding parameter for the reverse process is k−x. The remaining elementary steps are described by the rate constants indicated in Fig. 2. Molecules come together with rate constant kt and partition into one of the four possible unbound but paired states with a rate proportional to the reactive fractional surface areas (φC for C+ and 1 – φC for C−). Paired species separate with rate constant k−t, which we take to be same for all orientations. In other words, there are no interactions associated with their nonreactive surfaces. Molecules are assumed to reorient one at a time; C− → C+ (C+ → C−) with rate constant kC (k−C).
Taking each of the four possible unbound but paired states to be at steady state, it can be shown by algebraic manipulation that (19)
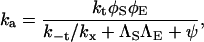 |
(3) |
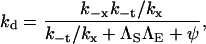 |
(4) |
with reorientation parameters
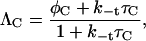 |
(5) |
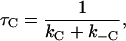 |
(6) |
and
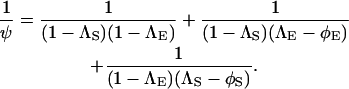 |
(7) |
The quasichemical approach has the advantage that its parameters can be varied directly without appeal to a specific microscopic picture, and we use this feature in Surface Enhancement of Reorientation Rates, below, to motivate the introduction of long-range interactions between the surface and the enzyme.
Relation of rate constants to molecular properties
We now relate the rate constants in Fig. 2 to molecular properties to make clear how substrate surface immobilization influences ka and kd. The spherical and hemispherical species in Fig. 1 can be described fully by their radii (RC) and the angles defining their reactive patches (θC).
Fraction of reactive surface area
The surface area of each axially symmetric reactive patch is
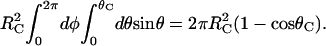 |
(8) |
Molecules in solution and on the surface differ with respect to their total areas available for collision: 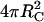 and
and 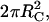 respectively. Using the half-angle trigonometric formulas, the reactive fractions are
respectively. Using the half-angle trigonometric formulas, the reactive fractions are
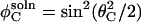 |
(9) |
and
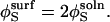 |
(10) |
Collision and separation
The separated species come together with the Smoluchowski diffusion-limited rate constant (kt) (34), and the corresponding parameter for the reverse process (k−t) can be derived by the same means (35). When both the spheres are mobile, kt and k−t are
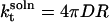 |
(11) |
and
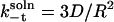 |
(12) |
with R = RE + RS. The parameter D is the relative translational diffusion constant, D = DE + DS. We estimate the diffusion constant of each molecule from the Stokes-Einstein relation DC = kBT/6πηRC, where kB is the Boltzmann constant, T is temperature, and η is the viscosity of the solution.
Tethering the substrate on the surface makes its translational diffusion negligible (DS = 0), and D reduces to DE. Also, the factor of 4π in Eq. 11 is decreased by a factor of 2 to account for the change in substrate solid angle available for collision. There is no corresponding modification of the expression for k−t because both the volume of the encounter complex and the surface area available for flux are reduced to the same extent. In other words, 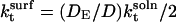 and
and 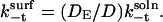
Reorientation
The ΛC parameters (Eq. 5) account for reorientation of the molecules in unbound but paired states. An approximate expression for ΛC in the solution case was obtained by Shoup et al. (28) and Zhou (25),
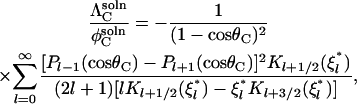 |
(13) |
where 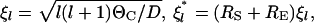 and
and 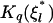 is the modified Bessel function of order q, Pl(cos θC) is the Legendre polynomial of order l, and ΘC is the rotational diffusion constant. We obtain the last of these from the Stokes-Einstein relation
is the modified Bessel function of order q, Pl(cos θC) is the Legendre polynomial of order l, and ΘC is the rotational diffusion constant. We obtain the last of these from the Stokes-Einstein relation 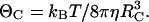 Equation 13 can also be used for
Equation 13 can also be used for 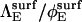 by reducing D to DE as discussed in Collision and Separation, above.
by reducing D to DE as discussed in Collision and Separation, above.
It is important to stress that, even when the substrate is immobilized, its reorientation parameter is nontrivial because this variable contains contributions from translation of the enzyme (see Appendix B of Shoup et al. (28)). An approximate expression for 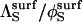 can be obtained by assuming that the diffusion-limited rate constant for association of a totally reactive enzyme sphere with an immobile hemispherical substrate bearing an axially symmetric reactive patch (
can be obtained by assuming that the diffusion-limited rate constant for association of a totally reactive enzyme sphere with an immobile hemispherical substrate bearing an axially symmetric reactive patch (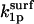 ) is half of the rate constant for association of the same reactive enzyme sphere with a spherical substrate, bearing two axially symmetric diametrically opposed reactive patches of equal size (
) is half of the rate constant for association of the same reactive enzyme sphere with a spherical substrate, bearing two axially symmetric diametrically opposed reactive patches of equal size (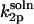 ). The latter system can be analyzed by the means introduced by Shoup et al. (28) and yields (Appendix)
). The latter system can be analyzed by the means introduced by Shoup et al. (28) and yields (Appendix)
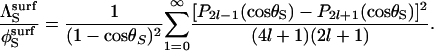 |
(14) |
Although Eqs. 13 and 14 are straightforward to evaluate numerically, Berg (17) introduced an approximate expression for 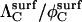 that does not require summation,
that does not require summation,
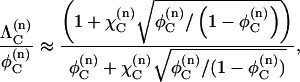 |
(15) |
where the general expression for χC is given by
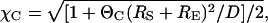 |
(16) |
and n ∈ {surf, soln}. The above approximation shows good agreement (within 10%) with Eq. 13. Somewhat larger errors (up to 20%) are observed when Eq. 15 is used to approximate Eq. 14.
Effects of substrate immobilization
As described above, restrictions associated with the surface decrease both ka and kd relative to solution in the absence of interactions that promote nonreactive surface sticking. We now use the quasichemical approximation to show that, on balance, these changes decrease the catalytic efficiency of the enzyme. Specifically, we argue that the ratio
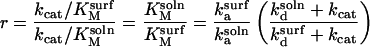 |
(17) |
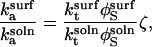 |
(18) |
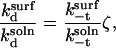 |
(19) |
where ζ is given by
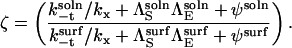 |
(20) |
Substituting the values of kt and k−t for the surface and solution cases into Eqs. 18 and 19,
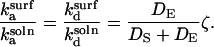 |
(21) |
Substituting into Eq. 17 and rearranging,
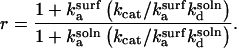 |
(22) |
Thus, r > 1 if and only if 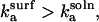 and simple physical considerations suggest this inequality is never satisfied.
and simple physical considerations suggest this inequality is never satisfied.
This argument can be made more precise in the following way. The minimum value for 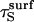 is
is 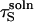 because limiting substrate mobility always decreases the rate of reorientation. In this case, ζ < 1, based on the separation rate constants in the surface and solution cases (Eqs. 12 and 20). Defining
because limiting substrate mobility always decreases the rate of reorientation. In this case, ζ < 1, based on the separation rate constants in the surface and solution cases (Eqs. 12 and 20). Defining 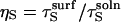 and
and 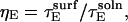 we can express the denominator of Eq. 20 in terms of
we can express the denominator of Eq. 20 in terms of 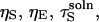 and
and 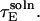 By writing out the derivative of the denominator with respect to ηS and grouping like-terms of the form
By writing out the derivative of the denominator with respect to ηS and grouping like-terms of the form 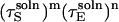 (for m, n ∈ {1, 2, 3}), it can be shown that the denominator increases monotonically with ηS; because the numerator is independent of ηS, ζ decreases monotonically (as ηS → ∞, ζ → 0). Thus DEζ can be taken to be bounded by DE + DS for the entire range of
(for m, n ∈ {1, 2, 3}), it can be shown that the denominator increases monotonically with ηS; because the numerator is independent of ηS, ζ decreases monotonically (as ηS → ∞, ζ → 0). Thus DEζ can be taken to be bounded by DE + DS for the entire range of 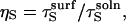 so that
so that 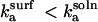 and r < 1.
and r < 1.
Surface enhancement of reorientation rates
How can surface immobilization of the substrate increase enzymatic throughput? It is well known that a long-range potential that leads to a bias in translations of the molecules relative to each other can enhance rates. Here, we show that a similar effect can arise from factors that limit enzyme orientations. In the presence of a long-ranged potential associated with the surface, the ratio of the forward and backward enzyme reorientation rates can be expressed as
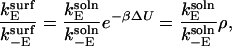 |
(23) |
where ΔU controls the extent to which one orientation is favored, and is defined by Eq. 23. For the case 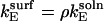 and
and 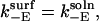 a plot of
a plot of 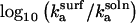 as a function of ρ (Fig. 3) shows that values of ρ ≈ 310 and higher lead to faster kinetics in the surface case. This ad hoc means of increasing the association rate motivates the simulations below in which we introduce an electrostatic interaction that serves to orient the enzyme in the surface case. At room temperature, the value of ρ at the crossover corresponds, for example, to an enzyme with a dipole moment of 100 Debye interacting with a surface with a uniform charge density of 0.3 e/nm2; these values are well within observed limits for natural systems (36,37).
as a function of ρ (Fig. 3) shows that values of ρ ≈ 310 and higher lead to faster kinetics in the surface case. This ad hoc means of increasing the association rate motivates the simulations below in which we introduce an electrostatic interaction that serves to orient the enzyme in the surface case. At room temperature, the value of ρ at the crossover corresponds, for example, to an enzyme with a dipole moment of 100 Debye interacting with a surface with a uniform charge density of 0.3 e/nm2; these values are well within observed limits for natural systems (36,37).
FIGURE 3.

Enhancing catalysis by restricting enzyme orientation. A positive value of the logarithm corresponds to faster kinetics in the surface case than in the solution case. In this example, 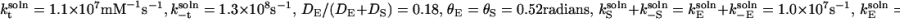 and
and 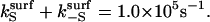 Calculations are in the diffusion-controlled limit.
Calculations are in the diffusion-controlled limit.
BROWNIAN DYNAMICS
In this section, we use Brownian dynamics simulations to show that reasonable electrostatic interactions between the surface and the enzyme can boost the catalytic efficiency of the enzyme through changes in reorientation rates. The rate constants obtained for a mobile enzyme sphere and an immobile substrate hemisphere are compared with the rate constants for the case when both the enzyme and substrate are mobile spheres in solution and no surface is present. The comparison is done for a range of values of the reactive patch widths on the molecular species and for a range of surface charge densities.
Simulation details
We model the enzyme as a neutral sphere with a point dipole at its center, and the surface as a homogeneously charged plane at z = 0. The sign of the charge is such that the reactive patch of the enzyme tends to point toward the surface. There is no electrostatic interaction between the enzyme and the substrate. The latter is fixed in space with its reactive patch orthogonal to the surface in the outward direction, as indicated in Fig. 1.
In the radial direction, the one-dimensional Smoluchowski diffusion equation was solved exactly for reflecting boundary conditions with the Lamm-Schulten algorithm (38) as described in Northrup et al. (39,40), but with a fixed time step. In the tangent directions, the Ermak-McCammon algorithm (41) in the absence of hydrodynamic interactions was used to integrate the equations of motion. When the latter yielded a position for the enzyme below the surface (z < 0), the z coordinate of the enzyme was set to its absolute value, which corresponds to reflection by the surface. The rotational degrees of freedom were varied independently using the scheme in Scherer (42). During the simulations, the escape probabilities for the specified reactive patch sizes and a finite simulation volume are accumulated. Rate constants for the full space were obtained from the calculated escape probabilities as in Northrup et al. (39,40), except that the Smoluchowski rate was adjusted to reflect the limited solid angle in the surface case, as described in Collision and Separation, above.
RESULTS AND DISCUSSION
Typical dipole moments of small globular proteins are 102 to 103 Debye (36) and surface charge densities of phospholipid bilayer membranes are of the order of magnitude 2.6 e/nm2 (37). Based on these data, we assign a dipole moment value of 800 D to the enzyme dipole and vary the charge density of the surface from 0 e/nm2 to 10 e/nm2.
The bimolecular association constants for the solution and for the charged and uncharged surface cases are plotted as a function of the enzyme reactive patch size in Fig. 4. As mentioned above, the bimolecular association rate in solution always exceeds that in the neutral surface case. For the substrate reactive patch size considered (20°), a crossover from faster association kinetics for the free substrate to faster association for the fixed one occurs at a surface charge density of ∼1 e/nm2 (Fig. 5). The enhancement is more pronounced for higher charge densities and smaller enzyme reactive patch sizes.
FIGURE 4.

Association rate constant as a function of enzyme reactive patch size; quasichemical theory evaluated using Eq. 15 (lines) and Brownian dynamics simulations (symbols) for the solution (+), uncharged (×), 2 e/nm2 (*), and 5 e/nm2 (⊡) surface cases. The radius of the enzyme was 15 Å and that of the substrate was 5 Å; θS = 20°. For computing the diffusion constants using the Stokes-Einstein relation, the temperature was taken to be 293 K and the viscosity of water was taken to be 1.002 × 10−3 Kg m−1 s−1. For the Brownian dynamics simulations, the starting radius was 25 Å and the terminating radius was 500 Å; 104 simulations were performed for each set of boundary conditions. In the case of the charged surface, the dielectric constant was taken to be 78.2. Calculations are in the diffusion-controlled limit.
FIGURE 5.

Association rate constant as a function of surface charge density; charged surface (solid line) and solution reference case (dashed line). Here, θE = 10° and the remaining parameters are as in Fig. 4.
The increased enzyme-substrate binding can be attributed to the effective reduction of dimensionality in rotational space of both the enzyme and the substrate. This can be viewed as the orientational analog of enzyme molecules sticking to and diffusing on a surface, which favors binding by reducing the dimensionality of the translational motion of the enzyme (15,18,31). It is also reminiscent of electrostatic steering (20–23). However in the case of electrostatic steering, specific receptor-ligand interactions lead to a bias in relative translational motion. Here, nonspecific interactions influence only the orientational degrees of freedom.
Acknowledgments
The authors thank R. Stephen Berry and Tong Zhao for critical readings of the manuscript.
The work was supported by the National Science Foundation Materials Research Science and Engineering Center at The University of Chicago (grant No. NSF-DMR-0213745).
APPENDIX
Here, we derive Eq. 14 for the substrate reorientation parameter in the surface case 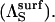 To this end, we consider the association of an enzyme with a mobile substrate in solution with two equal sized and diametrically oppositeed reactive patches
To this end, we consider the association of an enzyme with a mobile substrate in solution with two equal sized and diametrically oppositeed reactive patches 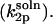 The rate constant
The rate constant 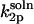 can be obtained along the lines of Shoup et al. (28), but with their Eqs. 3 and 5–8 modified to allow reaction over the ranges 0 ≤ θ ≤ θS and (π – θS) ≤ θ ≤ π with
can be obtained along the lines of Shoup et al. (28), but with their Eqs. 3 and 5–8 modified to allow reaction over the ranges 0 ≤ θ ≤ θS and (π – θS) ≤ θ ≤ π with 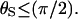
Shoup et al. (28) consider diffusion-controlled reactions between a molecule with an axially symmetric reactive patch and one that is uniformly reactive, so that the concentration (c) of the latter around the former can be expressed in terms of the vector separating their centers (r) and its orientation (θ). The key insight in Shoup et al. (28) is that the boundary condition
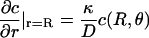 |
(24) |
can be well approximated by
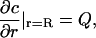 |
(25) |
where κ denotes reactivity, R = RE + RS, and Q is a constant such that Eq. 24 is satisfied on average over the reactive region. For the case of a sphere with two diametrically opposed patches, we find using the approach in Shoup et al. (28),
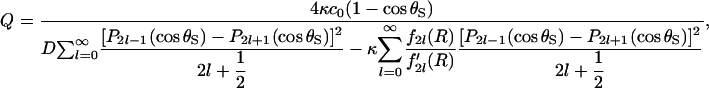 |
(26) |
where c0 is the bulk concentration of the enzyme at r = ∞. The function fl(r) is the solution of the radial part of the diffusion equation, which in our case is (28)
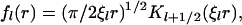 |
(27) |
with ξl defined as in the main text. The derivative
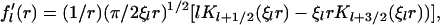 |
(28) |
follows directly from Eq. 27 and the identity
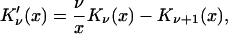 |
(29) |
which can be obtained by combining Eqs. 11.115 and 11.116 in Arfken and Weber (43). Equations 3 and 20 of Shoup et al. (28), together with
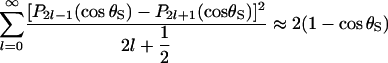 |
(30) |
yields
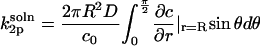 |
(31) |
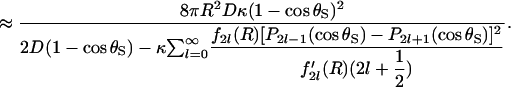 |
(32) |
To recover the immobilized hemisphere with one reactive patch, we let DS and ΘS go to zero. As DS → 0, ξl → 0; based on the identity (43)
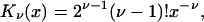 |
(33) |
we find that, in this limit,
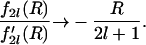 |
(34) |
Thus one obtains
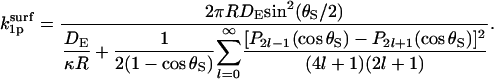 |
(35) |
In the diffusion-controlled limit, the reactivity κ → ∞. Thus,
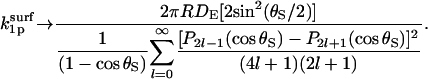 |
(36) |
The factor 2 sin2(θS/2) in the numerator is 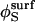 and the denominator is
and the denominator is 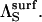 Inspection thus yields Eq. 14.
Inspection thus yields Eq. 14.
References
- 1.Mrksich, M. 2000. A surface chemistry approach to studying cell adhesion. Chem. Soc. Rev. 29:267–273. [Google Scholar]
- 2.Rich, R. L., and D. G. Myszka. 2000. Advances in surface plasmon resonance biosensor analysis. Curr. Opin. Biotechnol. 11:54–61. [DOI] [PubMed] [Google Scholar]
- 3.Yeo, W.-S., and M. Mrksich. 2003. Self-assembled monolayers that transduce enzymatic activities to electrical signals. Angew. Chem. Int. Ed. Engl. 42:3124–3131. [DOI] [PubMed] [Google Scholar]
- 4.Okano, T., N. Yamata, J. Sakai, and Y. Sakurai. 1995. Mechanism of cell detachment from temperature-modulated, hydrophilic-hydrophobic polymer surfaces. Biomaterials. 16:297–303. [DOI] [PubMed] [Google Scholar]
- 5.Collier, T. O., J. M. Anderson, A. Kikuchi, and T. Okano. 2002. Adhesion behavior of monocytes, macrophages, and foreign body giant cells on poly (n-isopropylacrylamide) temperature-responsive surfaces. J. Biomed. Mater. Res. 59:136–143. [DOI] [PubMed] [Google Scholar]
- 6.Blonder, R., E. Katz, I. Willner, V. Wray, and A. F. Bückmann. 1997. Application of a nitrospiropyran-FAD-reconstituted glucose oxidase and charged electron mediators as optobioelectronic assemblies for the amperometric transduction of recorded optical signals: control of the “on”-“off” direction of the photoswitch. J. Am. Chem. Soc. 119:11747–11757. [Google Scholar]
- 7.Blonder, R., S. Levi, G. Tao, I. Ben-Dov, and I. Willner. 1997. Development of amperometric and microgravimetric immunosensors using antigen and photoisomerizable antigen monolayer electrodes. J. Am. Chem. Soc. 119:10467–10478. [Google Scholar]
- 8.Hodneland, C. D., and M. Mrksich. 2000. Biomolecular surfaces that release ligands under electrochemical control. J. Am. Chem. Soc. 122:4235–4236. [Google Scholar]
- 9.Yeo, W.-S., C. D. Hodneland, and M. Mrksich. 2001. Electroactive monolayer substrates that selectively release adherent cells. Chem. Bio. Chem. 2:590–593. [DOI] [PubMed] [Google Scholar]
- 10.Wofsy, C., and B. Goldstein. 2002. Effective rate models for receptors distributed in a layer above a surface: applications to cells and Biacore. Biophys. J. 82:1743–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schurr, J. M. 1970. The role of diffusion in enzyme kinetics. Biophys. J. 10:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Northrup, S. H. 1988. Diffusion-controlled ligand binding to multiple competing cell-bound receptors. J. Phys. Chem. 92:5847–5850. [Google Scholar]
- 13.Goldstein, B. 1989. Diffusion limited effects of receptor clustering. Commun. Theor. Biol. 1:109–127. [Google Scholar]
- 14.Zwanzig, R. 1990. Diffusion-controlled ligand binding to spheres partially covered by receptors: an effective medium treatment. Proc. Natl. Acad. Sci. USA. 87:5856–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adam, G., and M. Delbrück. 1968. Reduction of dimensionality in biological diffusion processes. In Structural Chemistry and Molecular Biology. A. Rich, and N. Davidson, editors. W. H. Freeman and Co., San Francisco, CA. 198–215.
- 16.Bloomfield, V. A., and S. Prager. 1979. Diffusion-controlled reactions on spherical surfaces: application to bacteriophage tail fiber attachment. Biophys. J. 27:447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berg, O. G. 1985. Orientation constraints in diffusion-limited macromolecular association: the role of surface diffusion as a rate-enhancing mechanism. Biophys. J. 47:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berg, O. G., and P. H. von Hippel. 1985. Diffusion-controlled macromolecular interactions. Annu. Rev. Biophys. Biophys. Chem. 14:131–160. [DOI] [PubMed] [Google Scholar]
- 19.Šolc, K., and W. H. Stockmayer. 1973. Kinetics of diffusion-controlled reaction between chemically asymmetric molecules: II. Approximate steady-state solution. Int. J. Chem. Kinet. 5:733–752. [Google Scholar]
- 20.Davis, M. E., J. D. Madura, J. Sines, B. A. Luty, S. A. Allison, and J. A. McCammon. 1991. Diffusion-controlled enzymatic reactions. Methods Enzymol. 202:473–497. [DOI] [PubMed] [Google Scholar]
- 21.Tan, R. C., T. N. Truong, J. A. McCammon, and J. L. Sussman. 1993. Acetylcholinesterase: electrostatic steering increases the rate of ligand binding. Biochemistry. 32:401–403. [DOI] [PubMed] [Google Scholar]
- 22.Wade, R. C. 1996. Brownian dynamics simulations of enzyme-substrate encounter. Biochem. Soc. Trans. 24:254–259. [DOI] [PubMed] [Google Scholar]
- 23.Wade, R. C., R. R. Gabdoulline, S. K. Lüdemann, and V. Lounnas. 1998. Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations. Proc. Natl. Acad. Sci. USA. 95:5942–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stryer, L. 1995. Biochemistry, 4th Edition. W. H. Freeman and Company, New York.
- 25.Zhou, H.-X. 1993. Brownian dynamics study of the influences of electrostatic interaction and diffusion on protein-protein association kinetics. Biophys. J. 64:1711–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou, H.-X. 1997. Theory and simulation of the influence of diffusion in enzyme-catalyzed reactions. J. Phys. Chem. B. 101:6642–6651. [Google Scholar]
- 27.Šolc, K., and W. H. Stockmayer. 1971. Kinetics of diffusion-controlled reaction between chemically asymmetric molecules: I. General theory. J. Chem. Phys. 54:2981–2988. [Google Scholar]
- 28.Shoup, D., G. Lipari, and A. Szabo. 1981. Diffusion-controlled bimolecular reaction rates: the effect of rotational diffusion and orientation constraints. Biophys. J. 36:697–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlosshauer, M., and D. Baker. 2002. A general expression for bimolecular association rates with orientational constraints. J. Phys. Chem. B. 106:12079–12083. [Google Scholar]
- 30.Temkin, S. I. 1984. Diffusion-controlled reactions of chemically anisotropic molecules. J. Phys. Chem. 88:2679–2682. [Google Scholar]
- 31.Berg, H. C., and E. M. Purcell. 1977. Physics of chemoreception. Biophys. J. 20:193–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitz, K. S., and J. M. Schurr. 1972. The role of orientation constraints and rotational diffusion in bimolecular solution kinetics. J. Phys. Chem. 76:534–545. [Google Scholar]
- 33.Schurr, J. M., and K. S. Schmitz. 1976. Orientation constraints and rotational diffusion in bimolecular solution kinetics: a simplification. J. Phys. Chem. 80:1934–1936. [Google Scholar]
- 34.Smoluchowski, M. V. 1917. Attempt of a mathematical theory for the kinetics of coagulation of colloid solutions. Z. Phys. Chem. 92:129–168. [Google Scholar]
- 35.Steinfeld, J. I., J. S. Francisco, and W. L. Hase. 1989. Chemical Kinetics and Dynamics. Prentice Hall, Englewood Cliffs, NJ.
- 36.Takashima, S., and K. Asami. 1993. Calculation and measurement of the dipole moment of small proteins: use of protein data base. Biopolymers. 33:59–68. [DOI] [PubMed] [Google Scholar]
- 37.McLaughlin, S., G. Szabo, and G. Eisenman. 1971. Divalent ions and the surface potential of charged phospholipid membranes. J. Gen. Physiol. 58:667–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamm, G., and K. Schulten. 1983. Extended Brownian dynamics. II. Reactive, nonlinear diffusion. J. Chem. Phys. 78:2713–2734. [Google Scholar]
- 39.Northrup, S. H., S. A. Allison, and J. A. McCammon. 1984. Brownian dynamics simulation of diffusion-influenced bimolecular reactions. J. Chem. Phys. 80:1517–1524. [Google Scholar]
- 40.Northrup, S. H., M. S. Curvin, S. A. Allison, and J. A. McCammon. 1986. Optimization of Brownian dynamics methods for diffusion-influenced rate constant calculations. J. Chem. Phys. 84:2196–2203. [Google Scholar]
- 41.Ermak, D. L., and J. A. McCammon. 1978. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 69:1352–1360. [Google Scholar]
- 42.Scherer, C. 2004. Stochastic molecular dynamics of colloidal particles. Brazil. J. Phys. [E]. 34:442–447. [Google Scholar]
- 43.Arfken, G. B., and H. J. Weber. 1995. Mathematical Methods for Physicists, 4th Ed. Academic Press, San Diego, CA.