

Abstract
Selectively deuterated N-palmitoyl sphingomyelins were studied by deuterium nuclear magnetic resonance spectroscopy (2H-NMR) to elucidate the backbone conformation as well as the interaction of the sphingolipids with glycerophospholipids. Macroscopic alignment of the lipid bilayers provided good spectral resolution and permitted the convenient control of bilayer hydration. Selective deuteration at the acyl chain carbons C2 and C3 revealed that the N-acyl chain performs a bend, similar to the sn-2 chain of the phosphatidylcholines. Profiles of C-D bond order parameters were derived from the segmental quadrupolar splittings for sphingomyelin alone and for sphingomyelin-phosphatidycholine mixtures. In the liquid-crystalline state, the N-acyl chain of sphingomyelin alone revealed significantly more configurational order than the chains of homologous disaturated or monounsaturated phosphatidylcholines. The average chain order parameters and the relative width of the order parameter distribution were correlated over a range of bilayer compositions. The temperature dependence of the 2H-NMR spectra revealed phase separation in bilayers composed of sphingomyelin and monounsaturated phosphatidylcholine, in broad agreement with existing phase diagrams.
INTRODUCTION
The sphingomyelins represent a minor but essential phospholipid class of the eukaryotic cell membrane. Like phosphatidylcholine, sphingomyelin is mainly found in the outer leaflet of the cell membrane (1). The choline phospholipids together account for >50% of the phospholipid content in most tissues, whereas sphingomyelin is rather tissue specific (2,3). This sphingolipid is abundant in certain cell types, e.g., it constitutes >20% in erythrocytes and up to 18% in brain tissue (2). An even higher proportion (∼35%) was found in the apical renal brushborder membrane (4,5). In human brain tissue the sphingomyelin content increases with age, both in the cerebral cortex and in white matter (6). It has been shown that in a mammalian cell culture sphingomyelin, but not glucosylceramide, is able to rescue the cells from a defective sphingosine biosynthesis, which further underscores the fundamental role of this lipid class for cell proliferation and cell survival (7). Sphingomyelin is also intimately involved in cell signaling by means of its degradation products (3).
Sphingomyelin shares the same headgroup with phosphatidylcholine, which represents the major glycerophospholipid class. The interfacial segments and the hydrocarbon chains of the molecules are considerably different, however. The hydroxyl and amide residues of the sphingosine backbone render the sphingomyelin molecule capable of donating and accepting hydrogen bonds, whereas the phosphatidylcholine molecule is only a hydrogen bond acceptor by virtue of its carbonyl groups (8). An interfacial hydrogen bond network (9) may be the basis for the low permeability of SM vesicle membranes (10), for the tight interfacial cohesion, and for the favorable interaction with cholesterol (11). Moreover, the trans double bond between C4 and C5 of the sphingosine moiety may contribute to the tighter lipid packing in sphingomyelin versus phosphatidylcholine bilayers (12).
Deuterium nuclear magnetic resonance (2H-NMR) spectroscopy provides a convenient and conclusive means for an investigation of chain ordering (13), chain dynamics (14), molecular conformation (15), and phase behavior (16) in phospholipid membranes. Although the properties of glycero-phospholipids have been studied extensively, there are only a few reports on the properties of sphingolipids that make use of this versatile technique. This prompted us to perform an exploratory study using selectively deuterated N-palmitoyl sphingomyelin, alone and in the presence of a partially unsaturated phosphatidylcholine. 2H-NMR spectra were obtained from macroscopically aligned and hydrated multibilayers over a range of temperatures including the main phase transition (17). Our results reveal structural similarities of sphingomyelin and phosphatidylcholine with regard to the interfacial segments of the two phospholipid classes. However, the acyl chain of N-palmitoyl-d31-sphingomyelin (PSM-d31) has remarkably less configurational freedom than the chains of 1,2-diperdeuteropalmitoyl-sn-glycero-3-phosphocholine (DPPC-d62) according to the C-D bond order parameters derived from the 2H-NMR spectra.
MATERIALS AND METHODS
Chemicals
Synthetic phospholipids (DPPC, POPC) and deuterated phospholipids (DPPC-d62 and POPC-d31) were purchased from Avanti Polar Lipids (Alabaster, AL). Perdeuterated palmitic acid (d31-palmitic acid) and deuterium oxide were from Cambridge Isotopes (Promochem GmbH, Wesel, Germany). Palmitic acid-3,3-d2 was obtained from C/D/N Isotopes (Pointe-Claire, Quebec, Canada) and palmitic acid-2,2-d2 from Cambridge Isotopes.
N-palmitoyl-SM (PSM), as well as the labeled SMs (C2-d2-palmitoyl-, C3-d2-palmitoyl, and d31-palmitoyl-SM) were synthesized by N-acylation of D-erythro-sphingosylphosphocholine as described previously (18). Briefly, the p-nitrophenyl esters of palmitic acid and anhydrous potassium carbonate were added to lyso-SM (prepared from egg SM as described by Bittman and Verbicky (18)) in a mixture of anhydrous dimethylformamide and dichloromethane under nitrogen, and the mixture was stirred at room temperature for 1 day. The products were purified by silica gel column chromatography (elution with chloroform-methanol-water 65:35:5), and the suspended silica gel was removed by filtration through a Cameo filter.
Sample preparation
Lipid multibilayers were prepared and macroscopically aligned as described previously (17,19). Briefly, 30 mg of the lipid or lipid mixture were dissolved in 5 ml of methanol. The solutions were spread onto 50 ultrathin glass plates (8 × 18 × 0.08 mm; Marienfeld Lab. Glassware, Lauda-Königshofen, Germany) and dried for 20 min under a stream of warm air and then at room temperature for at least 18 h in vacuo (20–30 Pa). The glass plates were stacked on top of each other with gentle pressure and inserted, along with a pair of glass cylinder segments, into an open glass tube (inner diameter 9.8 mm). Two small paper strips were soaked in D2O and carefully dried to exchange labile hydrogen for deuterium. The strips were attached at the short sides of the glass stacks and a few microliters of D2O or of a D2O/H2O 10/90 v/v mixture were applied onto the paper surface. The tube was rapidly stoppered by two appropriately machined Teflon plugs with silicon O-rings. The membranes were annealed for 8 h at 46°C. The annealing process and the final hydration level were monitored by 2H-NMR spectroscopy (17).
NMR spectroscopy
A Varian VXR-400 spectrometer operating at 9.4 T (2H-frequency 61.4 MHz) was employed for the acquisition of 2H-NMR spectra. Spectra of macroscopically aligned samples were obtained using a 10 mm flat wire solenoid. A home built goniometer, driven by a stepper motor under software control, was used for accurate orientation in the magnetic field at θ = 0° where θ denotes the angle between the normal to the bilayer stack with respect to the field direction (17). Order parameters for individual carbon positions in the N-palmitoyl chain of PSM-d31 or in the sn-1 chain of POPC-d31 were obtained from the resolved quadrupolar splittings 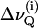 according to
according to
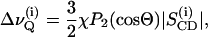 |
(1) |
where χ denotes the quadrupolar coupling constant (170 kHz for a C2H bond), P2(cos Θ) the second Legendre polynomial, and 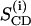 the order parameter of the i-th carbon-deuterium bond in the hydrocarbon chain. The mean square deviation from the mean of the distribution of order parameters was calculated for the deuterated positions 3–15 in the palmitic acid chains of the lipids :
the order parameter of the i-th carbon-deuterium bond in the hydrocarbon chain. The mean square deviation from the mean of the distribution of order parameters was calculated for the deuterated positions 3–15 in the palmitic acid chains of the lipids :
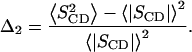 |
(2) |
The quadrupolar echo sequence (22) was applied for signal excitation using composite pulses with a 90° pulse width of 7 μs and a pulse spacing of 20 μs. The number of water molecules per lipid headgroup (nw) was obtained by calculating the integral ratio of the respective 2H signals from D2O and from the labeled lipids. The hydration level was adjusted to obtain full hydration of the respective bilayers (25 ≤ nw ≤ 35). The recycle delays were chosen so as to avoid saturation. The angle Θ between the normal to the glass plates and the magnetic field was varied until the quadrupolar splittings 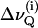 reached a maximum, indicating that Θ = 0 (see Eq. 1). The alignment procedure warrants good spectral resolution so that accurate quadrupolar splittings can be extracted without resorting to deconvolution techniques (23). Signal assignments for DPPC were obtained as described by Salmon et al. (24), based on selectively deuterated DPPC (25).
reached a maximum, indicating that Θ = 0 (see Eq. 1). The alignment procedure warrants good spectral resolution so that accurate quadrupolar splittings can be extracted without resorting to deconvolution techniques (23). Signal assignments for DPPC were obtained as described by Salmon et al. (24), based on selectively deuterated DPPC (25).
RESULTS
Signal assignments and backbone conformation
Liquid crystalline bilayers of palmitoyl sphingomyelin (PSM) and of the homologous glycerophospholipid dipalmitoyl phosphatidylcholine (DPPC) are remarkably different regarding the equilibrium alkyl chain ordering. This is shown in Fig. 1, which compares the 2H-NMR spectra of N-perdeuteropalmitoyl sphingomyelin (PSM-d31), of an equimolar mixture of PSM-d31 with 1-pamitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC), and of DPPC-d62. The 2H-NMR spectrum of PSM-d31 in the liquid crystalline state (52°C) displays unusually large quadrupolar splittings with amplitude maxima at ±32 kHz (Fig. 1 A). This may be contrasted with the 2H-NMR spectrum of DPPC-d62, which shows maxima at ±27 kHz (Fig. 1 C). In an equimolar mixture of PSM-d31 with POPC the splitting of the maxima is somewhat reduced (29 kHz versus 32 kHz; Fig. 1 B) but still significantly larger than in the DPPC-d62 bilayers (cf. Fig. 1 C), whereas individual 2H signals are narrower than those obtained for the sphingolipid alone. There are additional small signals at the outer edges in the spectrum of SM-d31 with splittings of ∼ ±33 kHz and ±35 kHz, respectively, the former appearing as unresolved shoulders. This feature is particularly well resolved in the PSM-d31/POPC mixture but is not detectable in the spectrum of DPPC-d62 (see below).
FIGURE 1.

2H-NMR spectra of macroscopically aligned membranes. (A) PSM-d31. (B) PSM-d31/POPC, 1:1 mol/mol. (C) DPPC-d62. Orientation of the bilayer normal, Θ = 0 (Eq. 1). Temperature, 52°C. nw ≈ 32 mol/mol.
The large amplitude outer edge signals in Fig. 1, A–C, can be assigned to the first 6–7 methylene segments, starting with the carbonyl groups of the PSM-d31 or DPPC-d62 acyl chains. Quadrupolar splittings are not resolved here or, equivalenty, there are nearly identical C-D bond order parameters (Eq. 1) for the corresponding chain segments (the order parameter “plateau”). The plateau order parameters 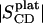 are 0.26 and 0.21 for PSM-d31 and DPPC-d62, respectively, which suggests tighter interfacial chain packing in the sphingolipid versus the glycerophospholipid. Tighter chain packing may also contribute to the significant broadening of individual signals in the PSM-d31 spectrum (Fig. 1 A). Other possibilities, e.g., inhomogeneous line broadening as a result of larger mosaic spread cannot be excluded, however. The chain melting temperatures Tm of both (nondeuterated) lipids are almost the same (Tm ≈ 41.5°C; (26)) indicating that the notion that the different widths of the 2H-NMR spectra are due to the unique packing properties of SM-d31 rather than to the different relative distances of the lipids from their respective phase transition temperatures.
are 0.26 and 0.21 for PSM-d31 and DPPC-d62, respectively, which suggests tighter interfacial chain packing in the sphingolipid versus the glycerophospholipid. Tighter chain packing may also contribute to the significant broadening of individual signals in the PSM-d31 spectrum (Fig. 1 A). Other possibilities, e.g., inhomogeneous line broadening as a result of larger mosaic spread cannot be excluded, however. The chain melting temperatures Tm of both (nondeuterated) lipids are almost the same (Tm ≈ 41.5°C; (26)) indicating that the notion that the different widths of the 2H-NMR spectra are due to the unique packing properties of SM-d31 rather than to the different relative distances of the lipids from their respective phase transition temperatures.
The temperature dependence of the 2H-NMR spectra of PSM-d31 and DPPC-d62 is shown in Fig. 2. The broadening of the spectra observed at ∼38°C is due to the main phase transition of the lipids. It may be noted that chain perdeuteration lowers the chain melting temperature of phospholipids by a few degrees, which causes the deviation from the transition temperature of the nondeuterated lipids (Tm ≈ 41.5°C). The splitting of the outer edges in the PSM-d31 spectrum reaches >120 kHz around 30°C. This is close to the quadrupolar splitting of 127.5 kHz that one expects for bilayers aligned at Θ = 0 (see Eq. 1) when the alkyl chains assume an all-trans configuration with the molecular axes being parallel to the bilayer normal. In contrast, the 2H spectra of DPPC-d62 show a wide distribution of resonance frequencies below 30°C with broad maxima, equivalent to quadrupolar splittings of <100 kHz (see Discussion).
FIGURE 2.

Liquid crystalline-gel state transition in aligned multibilayers of (A) PSM-d31 and (B) DPPC-d62. The transition for PSM-d31 will be complete at 42°C (spectrum not shown).
The assignment of individual 2H signals in a well-resolved spectrum is usually based on the assumption that the quadrupolar splittings decrease monotonically on going from the lipid-water interface toward the terminal methyl group. An obvious exception from this rule is the sn-2 chain of the glycerophospholipids that makes a bend as a result of the average orientation of the glycerol backbone (13). Thus, the deuterons on C2 of the sn-2 chain are magnetically nonequivalent as a result of the backbone conformation of the glycerophospholipids (27). This nonequivalence has been also shown by 2H-NMR spectroscopy for the C2-position of the acyl chain in N-galactosyl ceramide (28). Using 13C-NMR techniques, the plane of the amide linkage of this lipid was later found to be almost parallel to the membrane surface (29), which again suggests that the attached fatty acid performs a bend, similar to the glycerophospholipid sn-2 chain. To ascertain whether a similar backbone conformation applies to the sphingomyelins, PSM was selectively labeled at C2 and C3 of the palmitic acid chain (C2-2H-PSM and C3-2H-PSM). The pro-R and pro-S deuterons at C2 are indeed nonequivalent as shown by the two 2H doublets obtained from C2-2H-PSM (quadrupolar splittings 38.8 kHz and 57.8 kHz, respectively), suggesting that the C-D bonds of the respective alkyl chain segment assume different orientations with respect to the axis of motional averaging of the liquid crystalline lipid bilayer (the director axis), in agreement with the assumption of a bent N-acyl chain (Fig. 3 A). The nonequivalence of the C2 deuterons persisted in a 1:2 mol/mol mixture of C2-2H-PSM and POPC, whereas the splittings were reduced in the mixture, e.g., at 56°C from 57.8/38.8 kHz to 50.1/37.0 kHz, respectively. It may be further noted that the quadrupolar splittings in the mixed system at 26°C were almost identical with those of PSM-d31 alone at 56°C (spectra not shown).
FIGURE 3.

2H-NMR spectra of aligned multibilayers of (A) C2-2H-PSM and (B) C3-2H-PSM. Temperature, 56°C.
Selective 2H labeling of the C3 methylene group yielded a large quadrupolar splitting (74 kHz; Fig. 3 B). This seems to confirm the assignment in Fig. 1 B, although an additional splitting as in Fig. 1, A and B, was not observed for which no satisfactory explanation is available at present. The difference may be due to a slightly different average tilt of the C2H2 plane with respect to the director axis in the selectively versus the fully deuterated palmitic acid chain.
Chain order parameter profiles in the liquid crystalline state
An assignment of the 2H spectra of PSM-d31 can now be made, assuming that the quadrupolar splittings decrease from C3 toward the chain end. Order parameter profiles as calculated from the respective quadrupolar splittings (Eq. 1) are summarized in Fig. 4 A; comparison of the profiles for PSM-d31 alone and for PSM-d31-POPC mixtures is shown in Fig. 4 A, whereas Fig. 4 B presents the complementary information for PSM/POPC-d31 mixtures. The figure includes the order parameter profiles for DPPC-d62 and for a DPPC/POPC-d31 mixture at 1:2 molar ratio. All profiles shown were obtained at 48°C where the components are likely to be completely miscible and where the PSM-POPC system is in the liquid crystalline state (30,31).
FIGURE 4.

Order parameter profiles of pure lipids and of binary lipid mixtures. (A) PSM or DPPC with perdeuterated acyl chains. (B) POPC, perdeuterated in the SN1-acyl chain. Temperature, 48°C.
PSM-d31 alone exhibits unusually large C-D bond order parameter values for the entire alkyl chain as compared to the deuterated glycero-phospholipids, DPPC-d62 and POPC-d31. The order parameter “plateau”, 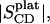 has been obtained here from Eq. 1 by taking the average of the quadrupolar splittings attributable to the chain segments from C4 to C6 (
has been obtained here from Eq. 1 by taking the average of the quadrupolar splittings attributable to the chain segments from C4 to C6 (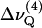 to
to 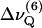 ). The plateau values are larger for PSM-d31 than for DPPC-d62, even in the mixtures with POPC (Table 1 and Fig. 4 A). Likewise, the presence of PSM results in a significant increase of the profiles in PSM/POPC-d31 mixtures as compared to POPC-d31 alone (Fig. 4 B). It is also worth noting that the effect of DPPC is almost negligible in a mixture of DPPC with POPC at a 1:2 molar ratio, whereas the presence of PSM at the same molar ratio increases the order parameters of the interfacial chain segments by >5% (Table 1).
). The plateau values are larger for PSM-d31 than for DPPC-d62, even in the mixtures with POPC (Table 1 and Fig. 4 A). Likewise, the presence of PSM results in a significant increase of the profiles in PSM/POPC-d31 mixtures as compared to POPC-d31 alone (Fig. 4 B). It is also worth noting that the effect of DPPC is almost negligible in a mixture of DPPC with POPC at a 1:2 molar ratio, whereas the presence of PSM at the same molar ratio increases the order parameters of the interfacial chain segments by >5% (Table 1).
TABLE 1.
Evaluation of average bilayer properties at 48°C from chain order parameters
| Sample composition |
 * *
|
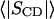 |
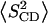 ×102 ×102
|
Δ2 |
|---|---|---|---|---|
| PSM-d31 | 0.258 | 0.221 | 5.111 | 0.047 |
| DPPC-d62 | 0.214 | 0.183 | 3.507 | 0.045† |
| POPC-d31 | 0.185 | 0.146 | 2.305 | 0.081 |
| PSM-d31/POPC, 2:1 | 0.238 | 0.200 | 4.216 | 0.054 |
| PSM-d31 - POPC, 1:1 | 0.235 | 0.196 | 4.089 | 0.064 |
| PSM-d31/POPC, 1:2 | 0.229 | 0.188 | 3.765 | 0.065 |
| DPPC-d62/POPC, 1:2 | 0.198 | 0.164 | 2.837 | 0.055† |
| PSM/POPC-d31, 2:1 | 0.206 | 0.166 | 2.972 | 0.079 |
| PSM/POPC-d31, 1:2 | 0.195 | 0.154 | 2.575 | 0.086 |
| DPPC/POPC-d31, 1:2 | 0.185 | 0.147 | 2.346 | 0.086 |
The average plateau and average chain order parameters were determined for carbon positions 4–6 and 3–15, respectively. See text for details.
Order parameters were obtained from spectra recorded at 48°C where the lipid components are completely miscible.
Only the order parameters for the sn-2 chain enter the calculation. Estimated errors of order parameter determination, ±2%.
These data were further evaluated in terms of the mean square deviation Δ2 (Eq. 2, see Methods), which represents the width of the order parameter distribution (Table 1). The values of Δ2 decrease with increasing average chain order 〈SCD〉. Notably, these parameters seem to be almost linearly related for mixtures composed of POPC and PSM, including bilayers containing these lipids alone. DPPC follows the same trend although it is characterized by lower Δ2 values (Fig. 5). An analogous observation has been made for POPC-d31, POPE-d31, and for POPC-d31/cholesterol mixtures when the chain order parameters were studied as a function of temperature, indicating that this behavior is a general feature of bilayer membranes (32).
FIGURE 5.

Correlation between average chain order parameters 〈SCD〉 (chain positions 3–15) and relative width of the order parameter distribution Δ2. Data from Table 1.
The above analysis provides some qualitative insight into the packing properties of sphingomyelin mixed bilayers, e.g., the Δ2-parameter indicates that the chain order falls off more rapidly for POPC because it is more disordered on average. A more elaborate statistical mechanical approach (24,33), based on a broader set of experimental data, including variations of composition and chain lengths will enable us to obtain reliable data on the bilayer thickness, on the area per lipid as a function of sphingomyelin concentration, and on thermal expansion coefficients of sphingomyelins alone and in mixed bilayers.
Domain formation in the presence of POPC
Resolved 2H-NMR subspectra from liquid crystalline and ordered bilayer domains may be observed when a multicomponent lipid mixture crosses a thermotropic phase boundary. As an example, Fig. 6 A reveals the phase separation in the 2:1 mol/mol PSM-d31/POPC system below 30°C. The onset of the transition can be easily recognized, considering the broad outer wings that appear at 26°C in the spectrum and the partially resolved splitting of the components of the methyl doublet signal (inset in Fig. 6 A). The drastic line broadening indicates that the sphingomyelin component of the binary mixture undergoes a liquid crystalline-gel state transition. The entire transition seems to occur over an approximate temperature range of 10°. The onset of the phase transition was observed analogously in 1:1 and in 1:2 mol/mol mixtures of these lipids between 26°C and 20°C and between 20°C and 16°C, respectively (not shown). It may be noted that a second methyl signal component was also found for the equimolar mixture, whereas line broadening rather than splitting was observed between 20°C and 16°C at 1:2 molar ratio. These observations are in good agreement with a published phase diagram for the binary system bovine brain sphingomyelin/egg lecithin obtained previously by calorimetry and small angle x-ray scattering (30).
FIGURE 6.

Domain separation in aligned PSM/POPC mixtures. (A) PSM-d31/POPC, 2:1 mol/mol. (B) PSM/POPC-d31, 2:1 mol/mol. (Insets) Expanded versions showing the 2H-methyl resonances.
A different result was obtained when POPC-d31 was the labeled component in the above PSM/POPC system (Fig. 6 B). The 2H spectrum indicates that POPC-d31 remains largely in the fluid state over the temperature range where PSM-d31 undergoes a phase transition. Likewise, there is no splitting of the methyl doublet signal. However, scaling the vertical amplitude of the spectra by a factor of 5.5 (Fig. 6 B) reveals small amplitude shoulders at ±50 kHz, which clearly shows that there is a minor fraction of POPC in the gel state domains where PSM forms the major lipid component. According to a phase diagram (30) a pure sphingomyelin gel phase rather than a solid mixture would coexist with a liquid crystalline mixed phase under the conditions shown in Fig. 6 (PSM/POPC molar ratio 2:1, temperature range 16°C–30°C). The observation of solid POPC in the 2H-NMR spectra therefore suggests that a small fraction of this lipid enters an ordered state when PSM undergoes the disorder-order phase transition.
DISCUSSION
PSM conformation and phase transition
This study investigates the backbone conformation and acyl chain order in macroscopically aligned multibilayers of N-perdeuteropalmitoyl sphingomyelin (PSM) by 2H-NMR spectroscopy. Sphingolipids have only rarely been analyzed using this technique although the properties of the phospholipid bilayer were successfully explored by 2H-NMR spectroscopy (15,34,35). PSM has nearly the same phase transition temperature as its natural homologs (12,36), suggesting that chain deuterated PSM reliably reproduces conformational and motional properties of the sphingomyelins encountered in biological membranes (31,36–38).
Some scattered information is available on the sphingolipid backbone in bilayer membranes, which consistently suggests that the amide linkage of the fatty acyl chain assumes a nearly parallel conformation with respect to the bilayer-water interface (8,19,29). A closely related structural detail, i.e., a bent conformation of the sn-2 acyl chain has been discovered for the phosphatidylcholines by 2H-NMR (25), in agreement with earlier x-ray data (39). The different quadrupolar splittings suggest that the diastereotopic C2 deuterons of the PSM N-acyl chain are motionally nonequivalent, in analogy to the corresponding deuterons of the phosphatidylcholine sn-2 chain (Fig. 3). The alternative assumption of two long-lived lipid conformations in the liquid crystalline bilayer (40) seems less likely, but a decision may be only possible after stereospecific replacement of just one of the C-2 protons by deuterium (27). In any case, it seems justified to assume that the average orientation of the normal to the plane spanned by the C2 methylene segment (2H-C2-2H) is not parallel to membrane normal.
The C3 segment, in contrast to C2, assumes an orientation with both C2H bonds nearly perpendicular to the bilayer normal as shown by the large 2H quadrupolar splittings obtained after selective deuteration of this chain position (Fig. 3). These deuterons seem to be slightly nonequivalent when the chain is fully deuterated (cf. Fig. 1), which is not evident in Fig. 3 where only a single quadrupolar splitting is resolved. Even a very small change in the average orientation of the 2H-C3-2H-plane may result in the coalescence of the doublets that appear at the outer edges of the spectra in Fig. 1, A and B, given the tentative assignment of the corresponding resonances in Fig. 1.
The drastic broadening of the 2H spectra of PSM-d31 and DPPC-d62 below 40°C clearly reflects the main phase transition (Fig. 2). The outer edges of the PSM-d31 spectra appear at ∼±60 kHz, which is close to the limiting value for rigid orientation of the carbon-deuteron bonds perpendicular with respect to the bilayer normal (127.5 kHz). The transition occurs around 38°C for both lipids, which is somewhat lower than the transition temperature determined calorimetrically (41.5°C; (26)). This temperature shift can be attributed to the deuteration of the acyl chains (41). In agreement with earlier reports on the phase transition of DPPC (42), the spectra also reveal narrow two phase regions for both lipids. The gel state 2H-NMR spectra of PSM-d31 and DPPC-d62 show notably different line shapes, regardless of the virtually identical transition temperatures of the nonlabeled lipids. PSM-d31 displays rather uniform maxima centered at ±55 kHz at 36°C and ±58 kHz at 16°C. In agreement with recent x-ray diffraction data of hydrated PSM obtained at 29°C (43), this line shape provides evidence for the presence of an Lβ (with perpendicular chains) rather than an Lβ′ (characterized by tilted hydrocarbon chains). In contrast, the inhomogeneous lines obtained from the aligned DPPC bilayers below Tm indicate that there is a broad distribution of orientational angles between the relevant methylene C2H bonds and the bilayer normal. This observation can be attributed qualitatively to the tilted hydrocarbon chains in the Pβ′ and in the Lβ′ phase structures of DPPC as revealed earlier by x-ray diffraction (44,45). For PSM there seems to be no such tilted chain packing, i.e., the sphingolipid structure below 30°C can be characterized as Lβ—rather than as Lβ′ or Pβ′. Whether there is a ripple or Pβ′ phase below Tm cannot be decided on the basis of the current NMR data alone. It may be further noted that no subtransition was detected in PSM (38), which underscores the difference between the sphingo- and glycerophospholipids.
The binary system PSM/POPC
Phospholipid mixtures containing sphingomyelin, phosphatidylcholine, and cholesterol are suitable model systems for the investigation of phase separation and domain formation in biological membranes (46). Here the focus was on the binary system PSM/POPC rather than on a comprehensive determination of phase boundaries in the more complicated ternary model systems. A number of partial phase diagrams have been published for sphingomyelin/phosphatidylcholine mixtures (30,36,38,47). A common denominator of these reports was the sensitivity of the mixing behavior of PSM on the chain length of the glycerophospholipid component, e.g., mixing of PSM with DMPC is nearly ideal (47), whereas a highly nonideal behavior was found for the mixture of PSM with DPPC (38).
A phase diagram for the binary mixture PSM/POPC has been recently determined using the anisotropy of 1,6-diphenyl-1,3,5-hexatriene fluorescence (31), which in addition to a liquid crystalline mixture identifies two different solids with different PSM/POPC ratios. The sensitivity of 2H-NMR for the motional anisotropy of the lipid alkyl chains allows for an assessment of the chain ordering of both components of the PSM/POPC system within the temperature range where the mixture undergoes phase separation. Our data (Fig. 6) are in broad agreement with the phase diagram by de Almeida et al. (31), although the transition into the two phase region as detected by 2H-NMR occurs at a somewhat lower temperature (∼6°C). One reason for this discrepancy may be the very different probe techniques used for the determination of phase boundaries. A somewhat lower transition temperature may also be due to the deuteration of the lipid acyl chains (16).
The data collected in Table 1 reflect the unique interfacial properties of the sphingomyelins. Comparing the plateau order parameter values of PSM-d31, DPPC-d62, and POPC-d31 at 48°C (i.e., above the main phase transition temperature of the lipids), one may conclude that sphingomyelin is characterized by a more rigid lipid-water interface as compared to the glycerophospholipids, presumably as a consequence of tight intermolecular H-bonding. The average order parameter as defined in Table 1 is also significantly higher for PSM versus DPPC, whereas the width of the order parameter distribution (Δ2) is similar for these lipids (Fig. 5). In contrast, the low average order parameters for the plateau region and for the carbon segments 3–15 indicate that the entire POPC molecule is more disordered and flexible than PSM or DPPC.
Considering the order parameter values obtained for the PSM-d31/POPC mixtures, one arrives at somewhat different conclusions. The width of the order parameter distribution, Δ2, is larger for PSM-d31 in the mixtures than either for PSM-d31 or for DPPC-d62 alone, although the average order and the plateau order parameters for deuterated sphingomyelin in the presence of POPC are still higher than those for DPPC-d62. The broader distribution of order parameters is a consequence of the abridged plateau in the order parameter gradient of the PSM-d31/POPC mixtures (Table 1 and Fig. 4). The correlation shown in Fig. 5 suggests that Δ2 reflects the flexibility of the deuterated chains, even in complex phospholipid mixtures. It may be noted that the parameter can be also derived from the first and second moments of the 2H-NMR spectra (20), i.e., it can be easily obtained without macroscopic bilayer orientation when the full assignment of the quadrupolar splittings may be difficult, providing an undistorted line shape and a high signal/noise can be achieved.
Intermolecular hydrogen bonding which leads to the high average chain order in bilayers of sphingomyelin alone may be partially disrupted by increasing the POPC concentration in the membrane (cf. Fig. 1). Conversely, the capability of establishing two intermolecular hydrogen bonds (12) may account for the fact that the order parameter plateau is higher in the binary mixtures of nondeuterated sphingomyelin and POPC-d31 than in POPC-d31 alone (Table 1). It may be concluded that in homogeneous lipid mixtures the sphingomyelins have a similar effect as cholesterol on the isomerization of the surrounding glycerophospholipid chains, most probably due to the formation of transient intermolecular hydrogen bonds.
References
- 1.Op den Kamp, J. A. F. 1979. Lipid asymmetry in membranes. Annu. Rev. Biochem. 48:47–71. [DOI] [PubMed] [Google Scholar]
- 2.Yorek, M. A. 1993. Biological distribution. In Phospholipids Handbook. G. Cevc, editor. Marcel Dekker, New York. 745–775.
- 3.Holthuis, J. C., T. Pomorski, R. J. Raggers, H. Sprong, and G. Van Meer. 2001. The organizing potential of sphingolipids in intracellular membrane transport. Physiol. Rev. 81:1689–1723. [DOI] [PubMed] [Google Scholar]
- 4.Molitoris, B. A., and F. R. Simon. 1985. Renal cortical brush-border and basolateral membranes: cholesterol and phospholipid composition and relative turnover. J. Membr. Biol. 83:207–215. [DOI] [PubMed] [Google Scholar]
- 5.Venien, C., and C. Le Grimellec. 1988. Phospholipid asymmetry in renal brush-border membranes. Biochim. Biophys. Acta. 942:159–168. [DOI] [PubMed] [Google Scholar]
- 6.Svennerholm, L. 1968. Distribution and fatty acid composition of phosphoglycerides in normal human brain. J. Lipid Res. 9:570–579. [PubMed] [Google Scholar]
- 7.Hanada, K., M. Nishijima, M. Kiso, A. Hasegawa, S. Fujita, T. Ogawa, and Y. Akamatsu. 1992. Sphingolipids are essential for the growth of Chinese hamster ovary cells. Restoration of the growth of a mutant defective in sphingoid base biosynthesis by exogenous sphingolipids. J. Biol. Chem. 267:23527–23533. [PubMed] [Google Scholar]
- 8.Pascher, I. 1976. Molecular arrangements in sphingolipids. Conformation and hydrogen bonding of ceramide and their implication on membrane stability and permeability. Biochim. Biophys. Acta. 455:433–451. [DOI] [PubMed] [Google Scholar]
- 9.Boggs, J. M. 1987. Lipid intermolecular hydrogen bonding: influence on structural organization and membrane function. Biochim. Biophys. Acta. 906:353–404. [DOI] [PubMed] [Google Scholar]
- 10.Hertz, R., and Y. Barenholz. 1975. Permeability and integrity properties of lecithin-sphingomyelin liposomes. Chem. Phys. Lipids. 15:138–156. [DOI] [PubMed] [Google Scholar]
- 11.Bittman, R., C. R. Kasireddy, P. Mattjus, and J. P. Slotte. 1994. Interaction of cholesterol with sphingomyelin in monolayers and vesicles. Biochemistry. 33:11776–11781. [DOI] [PubMed] [Google Scholar]
- 12.Barenholz, Y., and T. E. Thompson. 1999. Sphingomyelin: biophysical aspects. Chem. Phys. Lipids. 102:29–34. [DOI] [PubMed] [Google Scholar]
- 13.Seelig, J. 1977. Deuterium magnetic resonance: theory and application to lipid membranes. Q. Rev. Biophys. 10:353–418. [DOI] [PubMed] [Google Scholar]
- 14.Brown, M. F. 1996. Membrane structure and dynamics studied with NMR spectroscopy. In Biological Membranes: A Molecular Perspective from Computation and Experiment. K. M. Merz and B. Roux, editors. Birkhäuser, Boston. 175–252.
- 15.Seelig, J., and A. Seelig. 1980. Lipid conformation in model membranes and biological membranes. Q. Rev. Biophys. 13:19–61. [DOI] [PubMed] [Google Scholar]
- 16.Vist, M. R., and J. H. Davis. 1990. Phase equilibria of cholesterol/dipalmitoylphosphatidylcholine mixtures: 2H nuclear magnetic resonance and differential scanning calorimetry. Biochemistry. 29:453–464. [DOI] [PubMed] [Google Scholar]
- 17.Kurze, V., B. Steinbauer, T. Huber, and K. Beyer. 2000. A 2H NMR study of macroscopically aligned bilayer membranes containing interfacial hydroxyl residues. Biophys. J. 78:2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bittman, R., and C. A. Verbicky. 2000. Methanolysis of sphingomyelin. Toward an epimerization-free methodology for the preparation of D-erythro-sphingosylphosphocholine. J. Lipid Res. 41:2089–2093. [PubMed] [Google Scholar]
- 19.Steinbauer, B., T. Mehnert, and K. Beyer. 2003. Hydration and lateral organization in phospholipid bilayers containing sphingomyelin: a 2H-NMR study. Biophys. J. 85:1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis, J. H. 1979. Deuterium magnetic resonance study of the gel and liquid crystalline phases of dipalmitoyl phosphatidylcholine. Biophys. J. 27:339–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lafleur, M., P. R. Cullis, and M. Bloom. 1990. Modulation of the orientational order profile of the lipid acyl chain in the L alpha phase. Eur. Biophys. J. 19:55–62. [DOI] [PubMed] [Google Scholar]
- 22.Davis, J. H., K. R. Jeffrey, M. Bloom, and M. I. Valic. 1976. Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 42:390–394. [Google Scholar]
- 23.Bloom, M., J. H. Davis, and A. L. MacKay. 1981. Direct determination of the oriented sample NMR spectrum from the powder spectrum for systems with local axial symmetry. Chem. Phys. Lett. 80:198–202. [Google Scholar]
- 24.Salmon, A., S. W. Dodd, G. D. Williams, J. M. Beach, and M. F. Brown. 1987. Configurational statistics of acyl chains in polyunsaturated lipid bilayers from 2H NMR. J. Am. Chem. Soc. 109:2600–2609. [Google Scholar]
- 25.Seelig, A., and J. Seelig. 1975. Bilayers of dipalmitoyl-3-sn-phosphatidylcholine conformational differences between the fatty acyl chains. Biochim. Biophys. Acta. 406:1–5. [DOI] [PubMed] [Google Scholar]
- 26.Cevc, G., and D. Marsh. 1987. Phospholipid Bilayers, Physical Principles and Models. John Wiley & Sons, New York.
- 27.Engel, A. K., and D. Cowburn. 1981. The origin of multiple quadrupole couplings in the deuterium NMR spectra of the 2 chain of 1,2 dipalmitoyl-sn-glycero-3-phosphorylcholine. FEBS Lett. 126:169–171. [DOI] [PubMed] [Google Scholar]
- 28.Skarjune, R., and E. Oldfield. 1979. Physical studies of cell surface and cell membrane structure deuterium nuclear magnetic resonance investigation of deuterium-labelled N-hexadecanoylgalactosylceramides (cerebrosides). Biochim. Biophys. Acta. 556:208–218. [DOI] [PubMed] [Google Scholar]
- 29.Ruocco, M. J., D. J. Siminovitch, J. R. Long, S. K. Das Gupta, and R. G. Griffin. 1996. 2H and 13C nuclear magnetic resonance study of N-palmitoylgalactosylsphingosine (cerebroside)/cholesterol bilayers. Biophys. J. 71:1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Untracht, S. H., and G. G. Shipley. 1977. Molecular interactions between lecithin and sphingomyelin. Temperature- and composition-dependent phase separation. J. Biol. Chem. 252:4449–4457. [PubMed] [Google Scholar]
- 31.de Almeida, R. F., A. Fedorov, and M. Prieto. 2003. Sphingomyelin/phosphatidylcholine/cholesterol phase diagram: boundaries and composition of lipid rafts. Biophys. J. 85:2406–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lafleur, M., P. R. Cullis, and M. Bloom. 1990. Modulation of the orientational order profile of the lipid acyl chain in the L3 phase. Eur. Biophys. J. 19:53–62. [DOI] [PubMed] [Google Scholar]
- 33.Schindler, H., and J. Seelig. 1975. Deuterium order parameters in relation to thermodynamic properties of a phospholipid bilayer. Biochemistry. 14:2283–2287. [DOI] [PubMed] [Google Scholar]
- 34.Davis, J. H. 1983. The description of membrane lipid conformation, order and dynamics by 2H-NMR. Biochim. Biophys. Acta. 737:117–171. [DOI] [PubMed] [Google Scholar]
- 35.Brown, M. F. 1996. Membrane structure and dynamics studied with NMR spectroscopy. In Biological Membranes. K. Merz, J. Roux, and B. Roux, editors. Birkhäuser, Boston. 175–252.
- 36.Calhoun, W. I., and G. G. Shipley. 1979. Sphingomyelin–lecithin bilayers and their interaction with cholesterol. Biochemistry. 18:1717–1722. [DOI] [PubMed] [Google Scholar]
- 37.Filippov, A., G. Oradd, and G. Lindblom. 2003. The effect of cholesterol on the lateral diffusion of phospholipids in oriented bilayers. Biophys. J. 84:3079–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nyholm, T. K., M. Nylund, and J. P. Slotte. 2003. A calorimetric study of binary mixtures of dihydrosphingomyelin and sterols, sphingomyelin, or phosphatidylcholine. Biophys. J. 84:3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hauser, H., I. Pascher, R. H. Pearson, and S. Sundell. 1981. Preferred conformation and molecular packing of phosphatidylethanolamine and phosphatidylcholine. Biochim. Biophys. Acta. 650:21–51. [DOI] [PubMed] [Google Scholar]
- 40.Rance, M., K. R. Jeffrey, A. P. Tulloch, K. W. Butler, and I. C. Smith. 1980. Orientational order of unsaturated lipids in the membranes of Acholeplasma laidlawii as observed by 2H-NMR. Biochim. Biophys. Acta. 600:245–262. [DOI] [PubMed] [Google Scholar]
- 41.Davis, J. H. 1979. Deuterium magnetic resonance study of the gel and liquid crystalline phases of dipalmitoyl phosphatidylcholine. Biophys. J. 27:339–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.König, S., E. Sackmann, C. Carlile, and T. M. Bayerl. 1994. Molecular dynamics of water in oriented DPPC multilayers studied by quasielastic neutron scattering and deuterium-nuclear magnetic resonance relaxation. J. Chem. Phys. 100:3307–3316. [Google Scholar]
- 43.Maulik, P. R., and G. G. Shipley. 1996. N-palmitoyl sphingomyelin bilayers: structure and interactions with cholesterol and dipalmitoylphosphatidylcholine. Biochemistry. 35:8025–8034. [DOI] [PubMed] [Google Scholar]
- 44.Janiak, M. J., D. M. Small, and G. G. Shipley. 1979. Temperature and compositional dependence of structure of hydrated dimyristoyl lecithin. J. Biol. Chem. 254:6068–6078. [PubMed] [Google Scholar]
- 45.Hosemann, R., M. Hentschel, and W. Helfrich. 1980. Direct x-ray study of the molecular tilt in dipalmitoyllecithin bilayers. Z. Naturforschung. 35a:643–644. [Google Scholar]
- 46.Edidin, M. 2003. The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 32:257–283. [DOI] [PubMed] [Google Scholar]
- 47.Bar, L. K., Y. Barenholz, and T. E. Thompson. 1997. Effect of sphingomyelin composition on the phase structure of phosphatidylcholine-sphingomyelin bilayers. Biochemistry. 36:2507–2516. [DOI] [PubMed] [Google Scholar]