

Abstract
The voltage-dependent activity of prestin, the outer hair cell (OHC) motor protein essential for its electromotility, enhances the mammalian inner ear's auditory sensitivity. We investigated the effect of prestin's activity on the plasma membrane's (PM) susceptibility to electroporation (EP) via cell-attached patch-clamping. Guinea pig OHCs, TSA201 cells, and prestin-transfected TSA cells were subjected to incremental 50 μs and/or 50 ms voltage pulse trains, or ramps, at rates from 10 V/s to 1 kV/s, to a maximum transmembrane potential of ±1000 mV. EP was determined by an increase in capacitance to whole-cell levels. OHCs were probed at the prestin-rich lateral PM or prestin-devoid basal portion; TSA cells were patched at random points. OHCs were consistently electroporated with 50 ms pulses, with significant resistance to depolarizing pulses. Although EP rarely occurred with 50 μs pulses, prior stimulation with this protocol had a significant effect on the sensitivity to EP with 50 ms pulses, regardless of polarity or PM domain. Consistent with these results, resistance to EP with depolarizing 10-V/s ramps was also found. Our findings with TSA cells were comparable, showing resistance to EP with both depolarizing 50-ms pulses and 10 V/s ramps. We conclude prestin significantly affects susceptibility to EP, possibly via known biophysical influences on specific membrane capacitance and/or membrane stiffness.
INTRODUCTION
In the mammalian cochlea, outer hair cells (OHC) enhance hearing sensitivity and frequency selectivity (1). This enhancement, known as the “cochlear amplifier” (2), results from OHC mechanical activity, as this unique neuroepithelial cell can undergo electrically induced rapid length changes (3–6) at acoustic rates (7–9). Prestin, the OHC motor protein (10), is fundamental for OHC electromotility (11). Mechanical, electrical, and morphological data indicate that these motors are distributed preferentially within the cell's lateral plasma membrane (PM) (12–17).
A variety of models for electromotility have been proposed, based on electro-osmosis (4), electrophoresis (18), membrane bending (19), stiffness changes (20), and via surface area modulation, designated the “area-motor” model (21–26). The latter three remain serious candidates. We have proposed, based on modeling the voltage dependent capacitance of the OHC, that prestin's conformational change alters tension within the lateral membrane (27).
It is well known that cell membranes break down when subjected to mechanical or electrical stress, and that these stimuli can interact (28,29). The vulnerability to electroporation (EP) can provide insight into the interaction between PM and cortical cytoskeleton (29–31); for instance, negative pressure has been found to be more effective in producing membrane breakdown, presumably due to the greater interaction with the cytoskeleton with positive pressure (29). In addition, the integrity of the cytoskeleton is critical for resealing (32). The composition of the membrane is also highly influential; an increase in cholesterol, protein content, or certain surfactants can alter the susceptibility of the lipid bilayer to applied electrical tension (28,33). A variety of models have been proposed to explain the molecular mechanism of EP. It is generally accepted that a progression of membrane defects occurs as the energy threshold for their production is surpassed and maintained, resulting in increased permeability.
We have studied EP via cell-attached patch-clamping in OHCs to determine whether the electromechanical activity of prestin alters the biophysical properties of the PM. We also tested prestin-transfected TSA201 cells to divorce prestin's activity from modulation by the OHC's specialized cytoskeletal lattice. We find significant differences in susceptibility to EP between the lateral and basal membranes of OHCs, as well as between prestin-transfected and untransfected TSA cells, which we attribute to the residence of prestin.
MATERIALS AND METHODS
Cell preparation
Guinea pigs were overdosed with halothane, according to Yale University's Animal Use and Care Committee guidelines. Their temporal bones were dissected, isolating the top two turns of the organ of Corti. OHCs were dissociated with dispase in calcium-free medium for 10–15 min, followed by gentle pipetting. The cell-enriched solution was transferred to a 700-μL perfusion chamber and allowed to settle on the coverglass bottom for 10–15 min.
TSA201 cells, clones of human embryonic kidney (HEK) 293 cells that express the simian virus 40 large T-antigen in a stable manner, were cultured in Dulbecco's modified Eagle's medium (Sigma Chemical, St. Louis, MO) with 5% fetal bovine serum, 100 U/ml of penicillin and 100 μg/ml of streptomycin. Cells were plated for 24 h before transfection using effectene (Qiagen). The transfection reaction mixture consisted of 1 μg/ml of Pres cDNA plasmid added to 0.1 μg/ml of green fluorescent protein (GFP) plasmid, as a transfection marker in the 3 ml of Dulbecco's modified Eagle's medium the TSA cells were cultured in. Experiments were carried out 24–48 h posttransfection in the same culture dish without any dissociating procedures to avoid trauma to the cells.
Experimental setup
The guinea pig OHC and TSA cell PMs were probed via cell-attached patch-clamp recording. All experiments were performed at room temperature (∼23°C). A Nikon Diaphot (Melville, NY) inverted microscope with Hoffman optics (Aurora, IL) and a Sony VCR Model EV-C200 (Tokyo, Japan), were used to observe and tape record the experiments with OHCs. A Leica DM-IRB inverted microscope (McHenry, IL) with differential interference contrast optics was used for experiments with TSA cells.
OHC patches were clamped with an Axon Instruments Axopatch 200B amplifier (Foster City, CA) at a holding potential of −20 mV, the resting potential of dissociated OHCs (6), to zero the transmembrane voltage drop. Gigohm seals were obtained at the OHC lateral PM, where there is a high concentration of prestin, and at the basal area, which is devoid of these motors. TSA cells were also patch-clamped on random points of their PM that appeared smooth with an Axon Instruments Axopatch 200 amplifier holding them at their estimated average resting potential of −10 mV (E. G. Navarrete, unpublished observations). An ionic blocking solution was used in the bath and pipette, consisting of (in mM) 20 TEA, 20 CsCl, 2 CoCl2, 1.47 MgCl2, 10 Hepes, 99 NaCl, and 2 CaCl2. CsOH and glucose were used to adjust pH to 7.2 and osmolarity to 300 mOsm. Ion channel blockers were used to avoid increases in membrane conductance due to channel activity and to measure prestin's voltage-dependent capacitance. Pipettes had an average aperture of 1.43 ± 0.03 μm (mean ± SE; n = 78 measurements) and initial resistances ranged from 1.5 to 3 MΩ.
Pulse and ramp protocols
In all cells, EP was investigated using square voltage pulses and ramps. We used stimulus protocols based on Akinlaja and Sachs (31) in the case of the pulse trains, and O'Neill and Tung (36) in the case of the ramp stimuli. Initially, in OHCs two different pulse protocols were used, varying the duration of the voltage stimulus. The first protocol consisted of a series of 50-μs voltage pulses with sequential increments of 50 mV and an interpulse delay of 5 s to a transmembrane potential (TMP) of at least ±1000 mV (−1020 mV, +1030 mV). The second protocol consisted of 50 ms voltage pulses with sequential increments of 20 mV and an interpulse delay of 7 s, to a TMP of ±1000 mV. The 50-ms protocol was administered after the 50-μs pulse train in all cells that did not undergo EP and additionally to a set of cells in isolation to check for prior stimulation effects. Because OHCs were rarely permeabilized with 50-μs protocols, and to avoid loss of seal resistance or sensitization, TSA cells were only subjected to a 50-ms pulse train starting at 400 mV. During the interpulse interval, membrane capacitance (Cm) was measured every 120 ms, with transient analysis (14). EP was determined by a step increase in capacitance to whole-cell levels (Fig. 1). Occasionally, transient EP occurred (Fig. 2), which we defined as an increase in Cm to whole-cell levels for a minimum of two data points.
FIGURE 1.

Increase in membrane capacitance (Cm) to whole-cell levels after a voltage pulse indicates electroporation (EP). Voltage pulses are indicated by the thick lines on the x axis.
FIGURE 2.

Episodes of transient membrane breakdown followed by fast resealing are indicated by the spikes in Cm on this data trace reaching whole-cell levels. Voltage pulses are indicated by the thick lines on the x axis. Electroporation is noted at the end of the trace by the sustained increase in Cm after a voltage pulse.
Ramps at speeds from 10 V/s to 1 kV/s were additionally used to study both cell types, also to a maximum voltage of ±1000 mV. Membrane breakdown (MB) was defined, as it was previously (32), as a step increase in the conductance of the membrane patch indicated by a sudden and sharp rise in current (I). As with the pulse protocol, Cm was measured with transient analysis after administering the stimulus, occasionally revealing an intact, resealed patch. Thus, we defined the permanent increase in Cm to whole-cell levels as patch breakdown (PB).
Although membrane conductance will increase with EP, only the increase in charging time/capacitance irrefutably demonstrates EP; however, it is possible that the plasma membrane was electroporated and quickly resealed. This would be unusual since cell membranes have relatively long sealing times, on the order of minutes; for example, in their study, O'Neill and Tung (34) found that conductance changes persisted when their cardiac myocytes were restimulated within a minute. Since the increase in capacitance may have been missed due to our sampling resolution, we proceeded to analyze the membrane instability (MI) and MB data.
Data analysis
Statistical analyses were performed with Excel (Microsoft) and Analyse-it (Analyse-it Software, Leeds, UK). To avoid obtaining a potentially misleading asymmetric polarity-dependent TMP at the onset of EP, the resting potential was subtracted/added to the applied voltage (35,36). For OHCs, absolute TMP at onset of EP was calculated taking into account the average resting potential of −20 mV (see above). With TSA we accounted for an average resting potential of −10 mV. Results were evaluated using parametric (t-tests, analysis of variance (ANOVA), and linear regression) and nonparametric (chi-square) statistical analyses when appropriate. After the electrical stimulation and establishment of whole-cell configuration, TSA cell capacitance was measured as fully described elsewhere (27,37) to confirm prestin's presence and functionality, and to measure the motor protein density. The density of prestin was assessed via charge density, a normalization metric measuring maximum charge moved per linear capacitance (Qmax/Clin), since the linear capacitance will be proportional to cell surface area. To avoid sensitization, charge density in the cell-attached patch configuration was not measured before the EP protocols. It is assumed that the concentration of prestin would be very similar to the whole-cell value. We also assumed consistent patch geometry in TSA cells and OHCs based on images in the literature (13,31), although we did not control for this variable for each individual cell. A Windows-based software package, jClamp (SciSoft, Branford, CT), was used for acquisition and analysis. Igorpro (Wavemetrics; Lake Oswego, OR) was used to fit the capacitance data. All data are presented as mean ± SE.
RESULTS
Pulse protocols in outer hair cells
Susceptibility to EP with 50 μs pulse trains in our population of OHCs is very similar to that reported previously with HEK cells (29) and lipid bilayers (28). EP was rarely produced at voltages <1 V in the absence of extrinsic pressure (6.8%, n = 59). Nevertheless, the integrity of the membrane was undoubtedly compromised in the OHC by this prior stimulation. Sufficient current was injected through the patch to produce a change in the TMP and evoke the OHC's unique electromotile responses, as fast reversible movements in phase with the stimulus polarity were evident starting at an absolute average TMP of 765 mV. Furthermore, a consistent decrease in seal resistance was frequently revealed, which analyzed in isolation could have been mistaken for a loss of the seal were it not for our ability to monitor the electromotile responses and interpulse capacitance. Significant differences in the absolute average voltages at which mechanical responses occurred during 50 ms pulse trains were found depending on prior stimulation with the 50 μs protocol (71 ± 9 mV with prior stimulation versus 214 ± 12 mV with no prior stimulation, p < 0.001, n = 81, t-test). In addition, multiway analysis of variance demonstrated that prior stimulation with 50 μs pulses was a significant factor influencing the onset of EP in the OHC PM (p < 0.001, n = 85) during the subsequent 50 ms protocol.
EP occurred consistently between 240 and 580 mV, similar to the values reported previously for HEK cells (29). There was little difference in the average absolute TMP at onset of EP at the OHC basal area (see Table 1). At the lateral PM our results demonstrated a significant difference when polarity was varied (p < .05, t-test). Transient reversible EP with fast resealing (Fig. 2) on a timescale as short as 240 ms was observed in almost 20% (seven cells) of OHC patches subjected to depolarization (n = 40) at a TMP ranging between 360 and 460 mV, regardless of location. Interestingly, this phenomenon was not observed with hyperpolarizing pulses, although its occurrence could have been possible at a timescale beyond the detection of our sampling frequency. Because of our 2-data-point minimum as criteria for determining transient reversible EP, it may also have been dismissed as noise.
TABLE 1.
Pulse protocols in OHCs: comparison of absolute TMP at onset of EP
| Location | Stimulus polarity | Absolute TMP at onset of EP |
|---|---|---|
| Basal | Hyperpolarizing | 388 ± 21 (n = 10) |
| Depolarizing | 382 ± 14 (n = 10) | |
| Lateral | Hyperpolarizing | 388 ± 13 (n = 10) |
| Depolarizing | 446 ± 20 (n = 10) |
OHCs demonstrated a statistically significant difference in susceptibility to EP due to the effect of polarity at the lateral PM (p <.05, t-test). The incidence of EP was 100% in all categories. The charge density of the guinea pig OHCs lateral wall is estimated to be ∼150–200 fC/pF.
Pulse protocols in prestin-transfected and nontransfected TSA cells
TSA cells transfected with gPres demonstrate significant resistance (p < .02, chi-square analysis) to EP with our depolarizing 50 ms pulse protocol, failing to be permeabilized in >50% of our sample (see Table 2). Untransfected TSA cells underwent EP in 90% of the cells tested at absolute TMPs between 400 and 580 mV, consistent with our OHC data. Regarding the average absolute TMP at onset of EP, no statistically significant trends were evident, although it is interesting that the average threshold TMPs required for EP were slightly higher in transfected cells, in addition to showing greater variance. Virtually every cell resealed within seconds of EP, regardless of transfection or stimulus polarity.
TABLE 2.
Pulse protocols in gPres transfected and nontransfected TSA cells: incidence of EP, absolute TMP at onset of EP
| DNA | Stimulus polarity | EP incidence | Absolute TMP at onset of EP (mV) | Charge density |
|---|---|---|---|---|
| None | Hyperpolarizing | 90% (n = 10) | 464 ± 20 | N/A |
| Depolarizing | 90% (n = 10) | 495 ± 13 | N/A | |
| gPres | Hyperpolarizing | 100% (n = 9) | 549 ± 50 | 13.1 ± 3.1 fC/pF |
| Depolarizing | 44% (n = 9) | 600 ± 122 | 14.5 ± 10.6 fC/pF |
Transfected TSA cells demonstrate significantly reduced EP incidence with depolarizing 50 ms pulse trains (p < .02, chi-square analysis). No statistically significant differences are evident comparing absolute TMP at onset of EP.
Ramp protocols in outer hair cells
Almost 100% of the OHCs displayed a period of MI before a rapid step increase in conductance attributed to MB (Fig. 3), as described in the literature (33), regardless of portion probed, polarity, or ramp speed. After the ramp stimulus, and despite evidence for MB during the ramp, Cm increase to whole-cell levels is not consistently observed at 1 kV/s (see Table 3). This resistance to EP was clearly a rate-dependent phenomenon as it occurred in almost 100% of the sample at 10 V/s (see Table 4). Significantly, the lateral PM was more resistant to PB with depolarizing 10 V/s ramps (p < .02, chi-square analysis). Although we find interesting polarity-dependent trends in both MI and MB, it is impossible to distinguish whether conductance increases were due to seal loss or EP without monitoring capacitance. Although it is plausible that the plasma membrane was electroporated and quickly resealed, only the increase in capacitance indisputably demonstrates EP.
FIGURE 3.

Response to a depolarizing voltage ramp at 1 kV/s in an OHC. Membrane instability is noted by the deviation from the preceding linear trace. Membrane breakdown is indicated by the rapid step increase in conductance.
TABLE 3.
Ramp protocols at 1 kV/s in OHCs: absolute TMP at onset of MI and MB and incidence of PB
| Location | Polarity | MI (mV) | MB (mV) | PB |
|---|---|---|---|---|
| Basal | Hyperpolarizing | 299 ± 4 (n = 19) | 559 ± 9 | 16% |
| Depolarizing | 538 ± 12 (n = 19) | 606 ± 11 | 37% | |
| Lateral | Hyperpolarizing | 290 ± 10 (n = 18) | 590 ± 10 | 28% |
| Depolarizing | 527 ± 19 (n = 13) | 582 ± 16 | 15% |
PB did not occur consistently with this stimulus type. MI occurs at pronouncedly higher TMPs with depolarizing 1 kV/s ramps, demonstrating the significant effect of polarity (p < .0001, two-way ANOVA). MI and MB values are given as mean ± SE.
TABLE 4.
Ramp protocols at 10 V/s in OHCs: absolute TMP at onset of MI and MB and Incidence of PB
| Location | Polarity | MI (mV) | MB (mV) | PB |
|---|---|---|---|---|
| Basal | Hyperpolarizing | 314 ± 14 (n = 12) | 445 ± 30 | 92% |
| Depolarizing | 273 ± 16 (n = 13) | 374 ± 16 | 100% | |
| Lateral | Hyperpolarizing | 317 ± 12 (n = 11) | 454 ± 31 | 91% |
| Depolarizing | 284 ± 10 (n = 14) | 383 ± 15 | 57% |
The incidence of PB is >90% at the basal area with 10 V/s ramps. Significantly, the lateral PM was more resistant to PB with depolarizing ramps (p < .02, chi-square analysis), although MI and MB are polarity-dependent regardless of region (p < .05 and p < .01, respectively, two-way ANOVA). In contrast to 1 kV/s responses, with 10 V/s ramps there is more resistance to MI with hyperpolarizing stimuli.
Since the increase in capacitance may have been missed due to our sampling resolution, we present our findings and analysis of the MI and MB data. Location did not have a significant effect on either MI or MB TMPs with the 1-kV/s ramps (see Table 3). The effect of polarity on the onset of MI is obvious at both ramp speeds, but more significant with 1 kV/s ramps (p < .0001, two-way ANOVA); there is more resistance to MI with depolarizing ramps. Interestingly, we find the opposite effect with 10 V/s ramps, an increased resistance to MI with hyperpolarizing ramps (p < .05, two-way ANOVA). We performed additional experiments using 100 mV/s ramps and found no polarity dependence in MI or MB at either region of the OHC (data not shown). Although there are no polarity-dependent differences with respect to MB at the lateral wall with 1 kV/s ramps, we find resistance to depolarizing ramps at the base. Regarding MB with 10 V/s ramps, we find significant effects due to polarity and resistance to depolarizing ramps (p < .01, two-way ANOVA), regardless of location.
Ramp protocols in prestin-transfected and nontransfected TSA cells
Both transfected and untransfected cells undergo MI somewhat consistently (100% at 1 kV/s, 77–93% at 10 V/s). TMPs at onset of MI and MB were independent of transfection and polarity, although variance at 10 V/s in TSA cells was very large (see Tables 5 and 6).
TABLE 5.
Ramp protocols at 1 kV/s in gPres transfected and nontransfected TSA cells: absolute TMP at onset of MI and MB
| DNA | Polarity | MI (mV) | MB (mV) | Charge density |
|---|---|---|---|---|
| None | Hyperpolarizing | 551 ± 27 (n = 9) | 595 ± 35 | N/A |
| Depolarizing | 526 ± 27 (n = 10) | 601 ± 26 | N/A | |
| gPres | Hyperpolarizing | 541 ± 36 (n = 10) | 602 ± 36 | 30.3 ± 3.6 fC/pF |
| Depolarizing | 524 ± 20 (n = 10) | 617 ± 23 | 20 ± 2 fC/pF |
PB never occurred at this ramp speed.
TABLE 6.
Ramp protocols at 10 V/s in gPres transfected and nontransfected TSA cells: absolute TMP at onset of MI and MB and incidence of PB
| DNA | Polarity | MI (mV) | MB (mV) | PB | Charge density |
|---|---|---|---|---|---|
| None | Hyperpolarizing | 321 ± 44 mV (n = 9) | 514 ± 136 mV | 100% | |
| Depolarizing | 331 ± 118 mV (n = 9) | 579 ± 121 mV | 88% | ||
| gPres | Hyperpolarizing | 338 ± 45 mV (n = 10) | 592 ± 50 mV | 90% | 19.6 ± 4.2 fC/pF |
| Depolarizing | 364 ± 51 mV (n = 15) | 564 ± 66 mV | 53% | 16.4 ± 3.2 fC/pF |
Onset of MI and MB were independent of transfection and polarity. Increased resistance to PB with hyperpolarizing ramps (p < .05, chi-square analysis) in transfected TSA cells was consistent with the OHC lateral membrane results.
After the ramp stimulus, PB was never observed after the 1 kV/s ramps. Interestingly, the incidence of PB at 10 V/s in the transfected TSA cell was virtually identical to that of the OHC lateral membrane, showing increased resistance to PB with depolarizing ramps (p < .05, chi-square analysis; see Table 6).
DISCUSSION
We have found that the OHC lateral membrane, as well as the prestin-transfected TSA cell membrane, exhibit polarity-dependent sensitivity to EP. Prestin's influence is to significantly increase resistance to EP and PB with depolarizing 50 ms pulse trains and with depolarizing 10 V/s ramps. These features appear to result from prestin's residence and/or activity within the PM. There are, however, clear differences in responses between OHCs and TSA cells that likely do not depend on prestin expression. First, in the TSA cell there was, in general, a greater resistance to EP, most notably with 50 ms pulse trains and 1 kV/s ramps. Also, unlike the OHC, MI in TSA cells was not affected by polarity when 1 kV/s ramps were used (see Tables 3 and 5) and we find higher TMPs at onset of MB with 10 V/s ramps (see Tables 4 and 6). Finally, in TSA cells, transient EP with fast resealing occurs much more frequently with pulse stimuli. It is conceivable that some of the divergence we find between responses of OHCs and transfected TSA cells may relate to peculiarities of each cell type. Probably most significant is the structural complexity of the OHC. In OHCs, prestin is uniformly concentrated within the lateral PM, which is closely associated with a well developed submembranous cortical cytoskeleton and subsurface cisternae. Additionally, the OHC has a cytoplasmic turgor pressure of up to 1–2 kPa (38), which contributes to cell shape and the tensile forces the OHC PM experiences. The TSA cell lacks these features, and prestin may be heterogeneously distributed throughout the entire PM, possibly providing patch densities that vary significantly. This disparity in prestin density is highlighted by motor charge density estimates in OHCs (∼150–200 fC/pF (14,25)) and our TSA cells (13–30 fC/pF). Despite these cell-specific contributing factors, our data clearly indicate that the presence of prestin within a membrane will alter the membrane's susceptibility to electrical breakdown.
Mechanistic considerations
Based on the mechanistic model of Needham and Hochmuth (28), derived from their investigation of lipid bilayers, membrane breakdown will occur when the average energy density, T, exceeds a critical value. T arises from mechanical and/or electrical loads applied to the membrane,
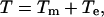 |
(1) |
where Tm is the mechanically induced tension and Te is the electrically induced tension. The addition of mechanical tension to the PM will lower the voltage that is required for EP.
Though we did not subject the patches to extrinsic mechanical tension, mechanical forces must be taken into account due to the voltage-dependent conformational changes of prestin. As mentioned previously, we have evidence based on modeling of capacitance measures that depolarization, which drives the OHC into a compact state, produces tension in the lateral membrane (27). Consequently, depolarization might have been expected to be a more effective stimulus for EP in the lateral PM, but we found the opposite. It is possible that our data were influenced by the time dependence of interactions between electrical and mechanical tension, since we proposed that the tension generated by prestin dissipates in a multiexponential fashion (27). Time-dependent interactions of electrical and mechanical tension have been observed previously. For example, though Akinlaja and Sachs (29) found that their results with HEK 293 cells probed with 50 μs pulse trains were consistent with the simple Needham and Hochmuth model (see above), mechanical tension had no effect with longer 50 ms voltage pulses. There are probably other prestin-induced mechanical changes in the OHC PM that may influence EP. For example, lipid lateral diffusion in the OHC PM is a sigmoid function of voltage, depolarization reducing membrane fluidity by half (39). Voltage-dependent membrane folding could also explain the biophysical effects on the diffusion constant as well as on EP. In addition, a number of experiments suggest that the PM is a major contributor to the OHC's axial stiffness (40–43) and that this stiffness is also voltage-dependent secondary to the activity of prestin (44). These investigators have found that stiffness increases with hyperpolarized potentials and decreases with depolarization. Thus, prestin's effects on membrane fluidity and stiffness, as well as on electrically induced membrane bending, may significantly influence our results.
A role for prestin's voltage-dependent specific capacitance
We have previously noted polarity-dependent biophysical properties that prestin imparts to the PM in OHCs while studying capacitance changes at extreme voltages (25,45). In the OHC, we found that the linear capacitance is actually voltage-dependent and is greater in the hyperpolarized region. The capacitance increase, which averages 3.3 pF at voltages <−200 mV, is too great to be explained solely by the area motor model, and we concluded that the membrane dielectric and/or thickness is altered (see Fig. 4). The electrical tension is described by Needham and Hochmuth (28) as
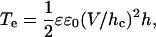 |
(2) |
or, as simplified by Akinlaja and Sachs (31),
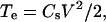 |
(3) |
where ɛ0 is the permittivity of free space, ɛ the dielectric constant of the membrane, V the transmembrane potential, h the membrane thickness, hc the capacitive thickness of the membrane, and Cs the specific membrane capacitance.
FIGURE 4.

OHC capacitance derives from the sum of the parallel capacitance proportional to membrane surface area (Csa) and the motor's voltage sensor-associated capacitance (Cv) to produce the total membrane capacitance (Cm).
It is clear that the effectiveness of a given voltage will depend directly on the specific capacitance of the membrane. If we assume that the specific capacitance of 0.5 μF/cm2 (48) characterizes the OHC PM at very positive voltage levels, then we estimate that the 3.3 pF increase at hyperpolarized extremes translates to an increased specific capacitance value of 0.61. Given this, we calculate for 50-ms pulse thresholds in the absence of prior stimulation:
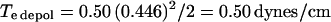 |
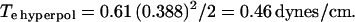 |
To some extent, then, accounting for the voltage dependence of the OHC's specific membrane capacitance reconciles differences in breakdown voltage thresholds, since the membrane would require greater voltages in the depolarizing direction to reach breakdown threshold. Our values for pulse-induced breakdown are in line with those obtained by Akinlaja and Sachs (29), who found a Te of 0.47 dynes/cm for hyperpolarizing pulses of 100 ms duration.
Time-dependent membrane breakdown and recovery
We find that the time integral of stimulation affects the membrane's susceptibility to EP; this is shown by the resistance to EP with 50 μs pulse trains compared to that with 50 ms pulse trains in the OHC. If the possibility of seal loss is eliminated, the short timescale of EP with 50 μs square voltage pulses would seem to have potential for gene transfer and drug delivery experiments in OHCs. Additionally, there is an increased susceptibility to PB with 10 V/s ramps, especially in TSA cells. After induced membrane instabilities, fast resealing was a common phenomenon in both cell types, although more frequent in TSA cells, probably due to prestin's reduced density, which likely results in faster lateral diffusion kinetics within the bilayer. Although fast resealing is commonly seen in pure lipid bilayers, such rapid resealing has never been documented in other biological membranes (35,47,48) despite the vast amount of work done on HEK cell lines (34,49,50). Fast resealing may be impeded by the presence of proteins that limit the bilayer's lipid lateral diffusion (51). In the lateral OHC membrane, besides the high-density prestin known to be present, other protein species are present; these include AE2 (52), aquaporins (53,54), glut 5 (55,56), stretch-activated channels (57,58), and a nonselective stretch-sensitive conductance with significant chloride permeability (59). Although we can expect membrane composition to play an important role in fast resealing, we must also remain cognizant that within patches protein residence may be dramatically altered. In addition, the OHC's highly organized cytoskeleton, with its intimate relationship to the plasma membrane via the pillars, most likely plays a role in resealing, as it does in other cell types (31,32,47).
We should make one final point on resealing after apparent membrane breakdown during ramp stimuli in the prestin-containing membrane. The membrane breakdown (defined according to O'Neill and Tung (34)) during depolarizing ramp stimulation probably represents a lower energy state before PB, since subsequent Cm measures frequently do not show an increase to whole-cell levels. According to EP theory (60–63), progression from reversible unstable electropores to permanent stable electropores requires breaching a higher energy threshold. MB as defined by O'Neill and Tung (34) and the transient EP with fast resealing we encountered are likely a lower energy state with a predominance of unstable reversible electropores. Apparently the presence of prestin increases the energy requirement to achieve PB with depolarizing stimuli and fosters patch recovery after the electrical stimulus in the absence of a transmembrane potential. The fact that the thresholds for those patches that did undergo PB and those that did not were not significantly different in OHCs or TSA cells supports this conclusion. The differences between prestin's effects during ramp stimulation and during pulse stimulation, where breakdown thresholds were clearly altered during depolarization, again points to stimulus-type and rate influences on prestin's interaction with the lipid bilayer.
Acknowledgments
We thank Linda Bartoshuk and Adam Mendizabal for sharing their expertise in statistics in some of the analyses, Peter Dallos for his general support of this project, and the reviewers for their constructive and insightful comments.
This work was supported by the National Institutes of Health National Institute on Deafness and Other Communication Disorders grant DC000273.
References
- 1.Dallos, P. 1992. The active cochlea. J. Neurosci. 12:4575–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis, H. 1983. An active process in cochlear mechanics. Hear. Res. 9:79–90. [DOI] [PubMed] [Google Scholar]
- 3.Brownell, W. E., C. R. Bader, D. Bertrand, and Y. de Ribaupierre. 1985. Evoked mechanical responses of isolated cochlear outer hair cells. Science. 227:194–196. [DOI] [PubMed] [Google Scholar]
- 4.Kachar, B., W. E. Brownell, R. Altschuler, and J. Fex. 1986. Electrokinetic shape changes of cochlear outer hair cells. Nature. 322:365–368. [DOI] [PubMed] [Google Scholar]
- 5.Ashmore, J. F. 1987. A fast motile response in guinea-pig outer hair cells: the cellular basis of the cochlear amplifier. J. Physiol. 388:323–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santos-Sacchi, J., and J. P. Dilger. 1988. Whole cell currents and mechanical responses of isolated outer hair cells. Hear. Res. 35:143–150. [DOI] [PubMed] [Google Scholar]
- 7.Santos-Sacchi, J. 1992. On the frequency limit and phase of outer hair cell motility: effects of the membrane filter. J. Neurosci. 12:1906–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dallos, P., and B. N. Evans. 1995. High-frequency motility of outer hair cells and the cochlear amplifier. Science. 267:2006–2009. [DOI] [PubMed] [Google Scholar]
- 9.Frank, G., W. Hemmert, and A. W. Gummer. 1999. Limiting dynamics of high-frequency electromechanical transduction of outer hair cells. Proc. Natl. Acad. Sci. USA. 96:4420–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng, J., W. Shen, D. Z. He, K. B. Long, L. D. Madison, and P. Dallos. 2000. Prestin is the motor protein of cochlear outer hair cells. Nature. 405:149–155. [DOI] [PubMed] [Google Scholar]
- 11.Liberman, M. C., J. Gao, D. Z. He, X. Wu, S. Jia, and J. Zuo. 2002. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature. 419:300–304. [DOI] [PubMed] [Google Scholar]
- 12.Dallos, P., B. N. Evans, and R. Hallworth. 1991. Nature of the motor element in electrokinetic shape changes of cochlear outer hair cells. Nature. 350:155–157. [DOI] [PubMed] [Google Scholar]
- 13.Kalinec, F., M. C. Holley, K. H. Iwasa, D. J. Lim, and B. Kachar. 1992. A membrane-based force generation mechanism in auditory sensory cells. Proc. Natl. Acad. Sci. USA. 89:8671–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang, G., and J. Santos-Sacchi. 1993. Mapping the distribution of the outer hair cell motility voltage sensor by electrical amputation. Biophys. J. 65:2228–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang, G., and J. Santos-Sacchi. 1994. Motility voltage sensor of the outer hair cell resides within the lateral plasma membrane. Proc. Natl. Acad. Sci. USA. 91:12268–12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi, S., and J. Santos-Sacchi. 2001. Non-uniform mapping of stress-induced, motility-related charge movement in the outer hair cell plasma membrane. Pflugers Arch. 441:506–513. [DOI] [PubMed] [Google Scholar]
- 17.Belyantseva, I., H. J. Adler, R. Curi, G. I. Frolenkov, and B. Kachar. 2000. Expression and localization of Prestin and the sugar transporter GLUT-5 during development of electromotility in cochlear outer hair cells. J. Neurosci. 20:RC116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jen, D. H., and C. R. Steele. 1987. Electrokinetic model of cochlear hair cell motility. J. Acoust. Soc. Am. 82:1667–1678. [DOI] [PubMed] [Google Scholar]
- 19.Raphael, R. M., A. S. Popel, and W. E. Brownell. 2000. A membrane bending model of outer hair cell electromotility. Biophys. J. 78:2844–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dallos, P., and D. Z. He. 2000. Two models of outer hair cell stiffness and motility. J. Assoc. Res. Otolaryngol. 1:283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dallos, P., R. Hallworth, and B. N. Evans. 1993. Theory of electrically driven shape changes of cochlear outer hair cells. J. Neurophysiol. 70:299–323. [DOI] [PubMed] [Google Scholar]
- 22.Iwasa, K. H. 1993. Effect of stress on the membrane capacitance of the auditory outer hair cell. Biophys. J. 65:492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santos-Sacchi, J. 1993. Harmonics of outer hair cell motility. Biophys. J. 65:2217–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwasa, K. H. 1994. A membrane motor model for the fast motility of the outer hair cell. J. Acoust. Soc. Am. 96:2216–2224. [DOI] [PubMed] [Google Scholar]
- 25.Santos-Sacchi, J., and E. Navarrete. 2002. Voltage-dependent changes in specific membrane capacitance caused by prestin, the outer hair cell lateral membrane motor. Pflugers Arch. 444:99–106. [DOI] [PubMed] [Google Scholar]
- 26.Dong, X. X., and K. H. Iwasa. 2004. Tension sensitivity of prestin: comparison with the membrane motor in outer hair cells. Biophys. J. 86:1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santos-Sacchi, J., S. Kakehata, and S. Takahashi. 1998. Effects of membrane potential on the voltage dependence of motility-related charge in outer hair cells of the guinea-pig. J. Physiol. 510:225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Needham, D., and R. M. Hochmuth. 1989. Electro-mechanical permeabilization of lipid vesicles. Role of membrane tension and compressibility. Biophys. J. 55:1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akinlaja, J., and F. Sachs. 1998. The breakdown of cell membranes by electrical and mechanical stress. Biophys. J. 75:247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ko, K. S., and C. A. McCulloch. 2000. Partners in protection: interdependence of cytoskeleton and plasma membrane in adaptations to applied forces. J. Membr. Biol. 174:85–95. [DOI] [PubMed] [Google Scholar]
- 31.Teissie, J., and M. P. Rols. 1994. Manipulation of cell cytoskeleton affects the lifetime of cell membrane electropermeabilization. Ann. N. Y. Acad. Sci. 720:98–110. [DOI] [PubMed] [Google Scholar]
- 32.Rols, M. P., and J. Teissie. 1992. Experimental evidence for the involvement of the cytoskeleton in mammalian cell electropermeabilization. Biochim. Biophys. Acta. 1111:45–50. [DOI] [PubMed] [Google Scholar]
- 33.Troiano, G. C., K. J. Stebe, R. M. Raphael, and L. Tung. 1999. The effects of gramicidin on electroporation of lipid bilayers. Biophys. J. 76:3150–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Neill, R. J., and L. Tung. 1991. Cell-attached patch clamp study of the electropermeabilization of amphibian cardiac cells. Biophys. J. 59:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teissie, J., M. Golzio, and M. P. Rols. 2005. Mechanisms of cell membrane electropermeabilization: A minireview of our present (lack of?) knowledge. Biochim. Biophys. Acta. 1724:270–280. [DOI] [PubMed] [Google Scholar]
- 36.Barrau, C., J. Teissie, and B. Gabriel. 2004. Osmotically induced membrane tension facilitates the triggering of living cell electropermeabilization. Bioelectrochemistry. 63:327–332. [DOI] [PubMed] [Google Scholar]
- 37.Santos-Sacchi, J. 2004. Determination of cell capacitance using the exact empirical solution of partial differential Y/partial differential Cm and its phase angle. Biophys. J. 87:714–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ratnanather, J. T., M. Zhi, W. E. Brownell, and A. S. Popel. 1996. Measurements and a model of the outer hair cell hydraulic conductivity. Hear. Res. 96:33–40. [DOI] [PubMed] [Google Scholar]
- 39.Oghalai, J. S., H. B. Zhao, J. W. Kutz, and W. E. Brownell. 2000. Voltage- and tension-dependent lipid mobility in the outer hair cell plasma membrane. Science. 287:658–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holley, M. C., and J. F. Ashmore. 1988. A cytoskeletal spring in cochlear outer hair cells. Nature. 335:635–637. [DOI] [PubMed] [Google Scholar]
- 41.Russell, I. J., and C. Schauz. 1996. Salicylate ototoxicity: effects on the stiffness and electromotility of outer hair cells isolated from the guinea pig cochlea. Audit. Neurosci. 1:309–320. [Google Scholar]
- 42.Tolomeo, J. A., C. R. Steele, and M. C. Holley. 1996. Mechanical properties of the lateral cortex of mammalian auditory outer hair cells. Biophys. J. 71:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He, D. Z. Z., S. P. Jia, and P. Dallos. 2004. Mechanoelectrical transduction of adult outer hair cells studied in a gerbil hemicochlea. Nature. 429:766–770. [DOI] [PubMed] [Google Scholar]
- 44.He, D. Z., and P. Dallos. 1999. Somatic stiffness of cochlear outer hair cells is voltage-dependent. Proc. Natl. Acad. Sci. USA. 96:8223–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navarrete, E., and J. Santos-Sacchi. 2001. Electropermeabilization and fast resealing in the cellular elements of the mammalian cochlea. Assoc Res Otolaryngol Abs No. 22058.
- 46.Solsona, C., B. Innocenti, and J. M. Fernandez. 1998. Regulation of exocytotic fusion by cell inflation. Biophys. J. 74:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abidor, I. G., V. B. Arakelyan, L. V. Chernomordik, Y. A. Chizmadzhev, V. F. Pastushenko, and M. R. Tarasevich. 1979. Electric breakdown of bilayer lipid-membranes. 1. Main experimental facts and their qualitative discussion. Bioelectrochem. Bioenerg. 6:37–52. [Google Scholar]
- 48.Gehl, J. 2003. Electroporation: theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 177:437–447. [DOI] [PubMed] [Google Scholar]
- 49.Kinosita, K., Jr., and T. Y. Tsong. 1977. Formation and resealing of pores of controlled sizes in human erythrocyte membrane. Nature. 268:438–441. [DOI] [PubMed] [Google Scholar]
- 50.Zimmermann, U., and G. A. Neil. 1996. Electromanipulation of Cells. CRC Press, Boca Raton, FL.
- 51.Golan, D. E., M. R. Alecio, W. R. Veatch, and R. R. Rando. 1984. Lateral mobility of phospholipid and cholesterol in the human-erythrocyte membrane: effects of protein-lipid interactions. Biochemistry. 23:332–339. [DOI] [PubMed] [Google Scholar]
- 52.Kalinec, F., G. Kalinec, C. Negrini, and B. Kachar. 1997. Immunolocalization of anion exchanger 2 alpha in auditory sensory hair cells. Hear. Res. 110:141–146. [DOI] [PubMed] [Google Scholar]
- 53.Belyantseva, I. A., G. I. Frolenkov, J. B. Wade, F. Mammano, and B. Kachar. 2000. Water permeability of cochlear outer hair cells: characterization and relationship to electromotility. J. Neurosci. 20:8996–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orem, A. R., and J. Zheng. 2004. Investigating a Novel Aquaporin Mrna Expression in Organ of Corti. Assoc Res Otolaryngol Abs No. 1493.
- 55.Nakazawa, K., S. S. Spicer, and B. A. Schulte. 1995. Postnatal expression of the facilitated glucose transporter, GLUT 5, in gerbil outer hair cells. Hear. Res. 82:93–99. [DOI] [PubMed] [Google Scholar]
- 56.Geleoc, G. S., S. O. Casalotti, A. Forge, and J. F. Ashmore. 1999. A sugar transporter as a candidate for the outer hair cell motor. Nat. Neurosci. 2:713–719. [DOI] [PubMed] [Google Scholar]
- 57.Ding, J. P., R. J. Salvi, and F. Sachs. 1991. Stretch-activated ion channels in guinea pig outer hair cells. Hear. Res. 56:19–28. [DOI] [PubMed] [Google Scholar]
- 58.Iwasa, K. H., M. X. Li, M. Jia, and B. Kachar. 1991. Stretch sensitivity of the lateral wall of the auditory outer hair cell from the guinea pig. Neurosci. Lett. 133:171–174. [DOI] [PubMed] [Google Scholar]
- 59.Rybalchenko, V., and J. Santos-Sacchi. 2003. Cl− flux through a non-selective, stretch-sensitive conductance influences the outer hair cell motor of the guinea-pig. J. Physiol. 547:873–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benz, R., F. Beckers, and U. Zimmermann. 1979. Reversible electrical breakdown of lipid bilayer membranes: charge-pulse relaxation study. J. Membr. Biol. 48:181–204. [DOI] [PubMed] [Google Scholar]
- 61.Weaver, J. C., and K. T. Powell. 1989. Theory of electroporation. In Electroporation and Electrofusion in Cell Biology. E. Neumann, A. E. Sowers, and C. A. Jordan, editors. Plenum Press, New York. 111–126.
- 62.Weaver, J. C., and A. Barnett. 1992. Progress toward a theoretical model for electroporation mechanism: membrane electrical behaviour and molecular transport. In Guide to Electroporation and Electrofusion. D. C. Chang, B. M. Chassy, J. A. Saunders, and A. E. Sowers, editors. Academic Press, San Diego. 91–118.
- 63.Ho, S. Y., and G. S. Mittal. 1996. Electroporation of cell membranes: a review. Crit. Rev. Biotechnol. 16:349–362. [DOI] [PubMed] [Google Scholar]