

Abstract
Using fluorescence resonance energy transfer spectroscopy we demonstrate that thymosin β4 (tβ4) binding induces spatial rearrangements within the small domain (subdomains 1 and 2) of actin monomers in solution. Tβ4 binding increases the distance between probes attached to Gln-41 and Cys-374 of actin by 2 Å and decreases the distance between the purine base of bound ATP (ɛATP) and Lys-61 by 1.9 Å, whereas the distance between Cys-374 and Lys-61 is minimally affected. Distance determinations are consistent with tβ4 binding being coupled to a rotation of subdomain 2. By differential scanning calorimetry, tβ4 binding increases the cooperativity of ATP-actin monomer denaturation, consistent with conformational rearrangements in the tβ4-actin complex. Changes in fluorescence resonance energy transfer are accompanied by marked reduction in solvent accessibility of the probe at Gln-41, suggesting it forms part of the binding interface. Tβ4 and cofilin compete for actin binding. Tβ4 concentrations that dissociate cofilin from actin do not dissociate the cofilin-DNase I-actin ternary complex, consistent with the DNase binding loop contributing to high-affinity tβ4-binding. Our results favor a model where thymosin binding changes the average orientation of actin subdomain 2. The tβ4-induced conformational change presumably accounts for the reduced rate of amide hydrogen exchange from actin monomers and may contribute to nucleotide-dependent, high affinity binding.
INTRODUCTION
The actin cytoskeleton plays a central role in the basic functions of eukaryotic cells such as maintenance of cell shape, cytoplasmic organization, cell movement, and cell division. This is achieved through a dynamic equilibrium between polymeric and monomeric actin. Many actin-binding proteins (ABP) that regulate the monomer-to-polymer transition by binding monomers, filaments or both, have been identified (1). Thymosin β4 (tβ4) is a small (Mr < 5 kDa) member of a highly conserved family of actin monomer-sequestering proteins present in actively motile cells, developing chick brain, and embryonic skeletal muscle (1,2). Tβ4 forms a 1:1 complex with monomeric actin and can bind to F-actin at high concentrations (3,4). β-thymosins are the principal in vivo regulators of unpolymerized actin, and are essential for maintaining the cytoplasmic pool of actin monomers required for rapid filament elongation (reviewed in (1)).
The exact tβ4 binding surfaces on the actin monomer are unknown and several models have been proposed (4–7). There is a general agreement that subdomains 1 and 3 (SD1 and SD3) of actin form part of the interface, similar to gelsolin and profilin, accounting for competitive binding (8,9). Binding to SD1 and SD3 is expected to restrict propeller-type opening and closing motion of the two major domains (10). However, there is disagreement over the role of SD2 of actin in binding tβ4. It has been proposed that the C-terminal region of tβ4 binds directly to the DNase I-binding loop (5) because it can be chemically crosslinked to residues of the DNase I-binding loop in SD2 (5,8). The nucleotide-dependence of SD2 conformation (11) could therefore account for the nucleotide-dependence of tβ4 binding affinity (12). Subtilisin cleavage of actin in the DNase I-binding loop causes a twofold decrease in the affinity of tβ4 (13), consistent with SD2 contributing to the stability and strength of the actin-tβ4 interaction. Image reconstruction of actin filaments chemically crosslinked to tβ4 (4) suggests tβ4 binds SD2 and SD4 as well as the gap between SD1 and SD2 (5). Binding of tβ4 to actin monomers is coupled to a change in heat capacity, dissociation of bound waters, a reduction in amide hydrogen exchange (5), and inhibition of nucleotide exchange (3). It was hypothesized that tβ4 folding upon binding could account for the change in heat capacity (5), and recent evidence favors this interpretation (13). It was also proposed that tβ4 binding inhibits the movement and separation of SD2 and SD4, closing the nucleotide-binding cleft and reducing the rates of nucleotide and amide hydrogen exchange (5). In this report, we used fluorescence spectroscopy to test the hypothesis that tβ4 changes the actin monomer conformation (5) and evaluate the contributions of actin SD2 to tβ4 binding. Our results are consistent with tβ4 binding to SD1, SD2 and SD3, causing changes in the spatial orientation of actin monomer subdomains.
MATERIALS AND METHODS
5-(((2-Iodoacetyl)amino)ethyl)aminonaphthalene-1-sulfonic acid (IAEDANS), 1,N6-ethenoadenosine-5′-triphosphate (ɛATP), dansyl cadaverine (DC), and fluorescein-5-isothiocyanate (FITC) were purchased from Molecular Probes (Eugene, OR). N-(4-(dimethylamino)-3,5-dinitrophenyl)-maleimide (DDPM) and N-ethylmaleimide (NEM) were from Sigma-Aldrich (St. Louis, MO). Rabbit skeletal muscle actin was prepared in G buffer (5 mM imidazole, pH 7.0, 0.2 mM ATP, 0.1 mM CaCl2, 0.5 mM DTT, and 1 mM NaN3). Tβ4 was isolated from bovine spleen (8). Bovine pancreatic DNase I (DPRF grade) was from Worthington Biochemicals (Lakewood, NJ). Cofilin was prepared as a recombinant protein using a cDNA sequence derived from chick embryo, kindly supplied by Dr. Takashi Obinata (Chiba, Japan). Concentrations of actin, DNase I, and cofilin were determined using extinction coefficients of 0.63 cm−1 (0.1%, 290 nm), 1.1 cm−1 (0.1%, 280 nm), and 0.98 cm−1 (0.1%, 280 nm), respectively. Actin was labeled with the following fluorescent probes as described: a), DC (14); b), DC-actin further modified with DDPM (14); c), IAEDANS (15); d), NEM or FITC (16); and e), ɛ-ATP (17). The following molar extinction coefficients were used for probes bound to actin: DC, 4640 M−1 cm−1 at 330 nm and DDPM, 3050 M−1 cm−1 at 440 nm (14); IAEDANS, 6100 M−1 cm−1 at 336 nm (15); and FITC, 74,500 M−1 cm−1 at 493 nm (18). The concentration of actin in labeled samples was determined by the DC Protein Assay (λ = 795 nm, Bio-Rad Laboratories, Hercules, CA) using unlabeled actin as the standard. The typical labeling ratios were: 0.63–0.9 for DC (n = 10); 0.5–0.85 for DDPM (n = 7); 0.8–0.98 for IAEDANS (n = 10); and 0.85–1.0 for FITC (n = 9). Bound ATP was exchanged with ɛATP as described earlier (17). In brief, nonlabeled or FITC-labeled G-actin (24–48 μM) was treated with 0.1 volumes of Dowex-1 on ice for 10 min to remove free ATP. Dowex-1 was removed by filtration and the solution was supplemented with 0.5 mM ɛATP, 0.1 mM CaCl2, and 1 mM Tris/HCl (pH 8.0) and equilibrated on ice overnight. This procedure was repeated again to ensure a complete exchange of bound ATP with ɛATP. Free nucleotide was removed with Dowex-1 immediately before fluorescence experiments. Because the actin concentrations used are much higher than the Kd for nucleotide binding (19), bound nucleotide will not dissociate to any appreciable extent during the time-course of the experiments.
Fluorescence measurements were carried out in G buffer at 10°C in a temperature-controlled cuvette using an SLM 48000 S Multiple Frequency Lifetime Spectrofluorometer (SLM Aminco, Foster City, CA) operating on a xenon arc lamp essentially as described (14). All experiments were repeated 5–10 times and results were presented as means ± SE. Fluorescence resonance energy transfer (FRET) efficiency (E) was determined from the intensities of the donor (D) in the presence (FDA) and absence (FD) of the acceptor (A) according to Eq. 1: E = (1 − FDA/FD)/α, where α is a degree of labeling with the acceptor in the double-labeled actin. Intensities were normalized to the D-concentration after background subtraction. Energy transfer efficiency is related to distance separating D- and A-probes according to Eq. 2: 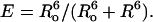 Ro is the Förster critical distance between the D and A (when E = 0.5) defined by Eq. 3:
Ro is the Förster critical distance between the D and A (when E = 0.5) defined by Eq. 3: 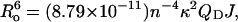 where n is the refractive index of the medium, κ2 is the orientation factor, QD is the quantum yield of the D on actin in the absence of the A, and J is the overlap integral given in M−1 cm−1 nm4. Ro distances of actin alone were taken as 49.6 Å for IAEDANS/FITC (20) and 47.4 Å for ɛATP/FITC (17). These values were corrected for changes in the donor quantum yield (21) due to tβ4 binding. The Ro for DC/DDPM was obtained experimentally using fluorescence emission spectrum of the DC-actin in the presence of nonfluorescent DDPM and the absorption spectrum of the DDPM-actin, taking κ2 = 2/3. FRET measurements between the following positions were done using the following donor-acceptor pairs as described: a), DC (Gln-41) and DDPM (Cys-374) (14); b), IAEDANS (Cys-374) and FITC (Lys-61) (17); and c), ɛATP (nucleotide-binding cleft) and FITC (Lys-61) (17). Intensities of the probe in the D-only and D/A samples were normalized to the fluorophore concentration by overnight tryptic digestion, as described (17). In brief, 0.2–0.4 mg ml−1 of trypsin in G-buffer was added into the D and D/A sample solutions after FRET measurements. After digestion, the amino-acid residues with D- and A-probes were separated, abolishing FRET. Thus, fluorescence intensities were directly proportional to the concentration of the D-probe in solution. The ratio of these fluorescence intensities was taken to be proportional to FDA × Ci/FD × C′i (Eq. 4), where Ci and C′i are the concentrations of D in D and D/A samples, respectively. Acrylamide quenching of DC-actin fluorescence (λex = 332 nm, λem = 512 nm) was performed at 22°C in a thermostated cuvette as described (14). The apparent Stern-Volmer constant (KSV) was obtained from the slope of a plot of Fo/F versus [acrylamide] by fitting to Eq. 5 as Fo/F = 1 + KSV [Q], where Q is the quencher, i.e., acrylamide.
where n is the refractive index of the medium, κ2 is the orientation factor, QD is the quantum yield of the D on actin in the absence of the A, and J is the overlap integral given in M−1 cm−1 nm4. Ro distances of actin alone were taken as 49.6 Å for IAEDANS/FITC (20) and 47.4 Å for ɛATP/FITC (17). These values were corrected for changes in the donor quantum yield (21) due to tβ4 binding. The Ro for DC/DDPM was obtained experimentally using fluorescence emission spectrum of the DC-actin in the presence of nonfluorescent DDPM and the absorption spectrum of the DDPM-actin, taking κ2 = 2/3. FRET measurements between the following positions were done using the following donor-acceptor pairs as described: a), DC (Gln-41) and DDPM (Cys-374) (14); b), IAEDANS (Cys-374) and FITC (Lys-61) (17); and c), ɛATP (nucleotide-binding cleft) and FITC (Lys-61) (17). Intensities of the probe in the D-only and D/A samples were normalized to the fluorophore concentration by overnight tryptic digestion, as described (17). In brief, 0.2–0.4 mg ml−1 of trypsin in G-buffer was added into the D and D/A sample solutions after FRET measurements. After digestion, the amino-acid residues with D- and A-probes were separated, abolishing FRET. Thus, fluorescence intensities were directly proportional to the concentration of the D-probe in solution. The ratio of these fluorescence intensities was taken to be proportional to FDA × Ci/FD × C′i (Eq. 4), where Ci and C′i are the concentrations of D in D and D/A samples, respectively. Acrylamide quenching of DC-actin fluorescence (λex = 332 nm, λem = 512 nm) was performed at 22°C in a thermostated cuvette as described (14). The apparent Stern-Volmer constant (KSV) was obtained from the slope of a plot of Fo/F versus [acrylamide] by fitting to Eq. 5 as Fo/F = 1 + KSV [Q], where Q is the quencher, i.e., acrylamide.
SDS PAGE and native 10% PAGE gels were done following standard protocols (22). After FRET measurements, labeled actin and its complexes were mixed with equal volumes of native sample buffer (62.5 mM Tris-Cl, pH 6.8, 10% glycerol, and 0.1% bromophenol blue) and separated by native PAGE gel electrophoresis at 70–110 V for 90–120 min in an ice bath (23). Differential scanning calorimetry (DSC) was performed on a DASM-4M differential scanning microcalorimeter (Institute for Biological Instrumentation, Pushchino, Russia) as described (24). The scanning rate was 1 K min−1. Transition temperatures (Tm) were determined from the peak of thermal transition, and the apparent enthalpies (ΔH°app) were calculated from the integrated areas of the denaturation peaks.
RESULTS
Effect of tβ4 on the distance between Gln-41 and Cys-374 of actin
A donor probe, DC, at Gln-41 and an acceptor, DDPM, at Cys-374 (Fig. 1), located on opposite sides of the small domain of actin were used to measure FRET along the SD1 and SD2 axis. Fig. 2 A shows corrected and averaged (n = 5) fluorescence emission spectra of DC-actin (curve 1) and DDPM-DC-actin (curve 2). A calculated value of 30.3 Å for R0 of the DC/DDPM pair in actin alone (J = 9.8 × 1015 M−1 cm−1 nm4) and a FRET efficiency of 0.268 ± 0.03 correspond to a distance of 35.8 ± 1.0 Å between these probes. Note that the emission maximum of DC is shifted by 15 nm to the longer wavelength in the presence of DDPM, suggestive of a possible allosteric effect caused by a modification of Cys-374 consistent with reports on the allosteric relationship between SD1 and SD2 (25).
FIGURE 1.

Position of the donor/acceptor probe pairs used for FRET measurements in actin. Gln-41, Lys-61, Cys-374, and ATP are shown as space-filled structures (26). The numerals refer to the four subdomains.
FIGURE 2.

Effect of tβ4 on the distance between Gln-41 and Cys-374 of actin. Corrected and averaged fluorescence spectra (λex = 332 nm and λem = 525 nm) of DC- and DC/DDPM-actin in the presence and absence of tβ4. (A) DC- and DC/DDPM-actin (2–5 μM) before (solid curves 1 and 2, respectively) and after addition of a 10-fold molar excess of tβ4 (dashed curves 1′ and 2′, respectively); and DC/NEM-actin in the presence and absence of tβ4 (dash-dot curves 3 and 3′, respectively). (B) (Left) A Coomassie-stained 12% native PAGE gel of DC/DDPM-actin (lane 1) and its binary complex with tβ4 (lane 2). (Right) The same gel viewed under UV light. (Symbols: A, monomeric actin; T, tβ4; and AT, actin/tβ4 complex.) (C) Stern-Volmer quenching plots: open squares, DC-actin monomers; solid squares, DC-actin filaments; circles, the actin monomer/tβ4 complex; and triangles, the DNase I/actin monomer complex. All samples were in G-buffer except DC-actin filaments that additionally contained 50 mM KCl and 2 mM MgCl2. The temperature was constant (22°C).
Bound tβ4 decreases the intensity of DC-actin by 12 ± 2% (dashed curve 1′, n = 5) in agreement with a previous report (8). However, in the presence of DDPM this decrease is abolished (dashed curve 2′). The resulting FRET efficiency calculated using R0 = 29.4 Å (corrected for changes in the QD) is 0.182 ± 0.03, which corresponds to a distance of 37.8 ± 1.2 Å between DC and DDPM in the actin-tβ4 complex. Thus, tβ4 increases the distance between the probes attached to Cys-374 and Gln-41 by 2 Å (p < 0.000). Since DDPM modification of Cys-374 induces an allosteric effect on the actin monomer conformation, we modified DC-actin with NEM at Cys-374, and repeated the FRET experiments using DC/NEM-actin as a donor-only sample. The NEM probe has no absorbance in the overlap region of the donor emission, and therefore, there is no FRET between these two probes. Fluorescence intensity of DC slightly increases in the presence of NEM (dash-dot curve 3, n = 5). However, it has no significant effect on the final distance. Using R0 = 31.05 Å (recalculated for changes in the QD of DC in the presence of NEM), and the efficiency of 0.307 ± 0.03 (n = 5), the calculated distance between the probes attached to Gln-41 and Cys-374 is 35.5 ± 0.9 Å. The addition of tβ4 to DC/NEM-actin (dash-dot curve 3′) results in a decrease in fluorescence intensity by 13 ± 4%, i.e., similar to NEM-unmodified DC-actin. The calculated efficiency of 0.191 ± 0.02 (n = 5) yields a distance of 37.4 ± 1.0 Å between DC and DDPM in the presence of tβ4. Thus, tβ4 increases the measured distance by ∼2 Å. Native PAGE gels (Fig. 2 B) demonstrate that concentrations of tβ4 used in FRET experiments are sufficient to saturate labeled actin as expected from the equilibrium binding affinities (5). The tβ4-induced increase in the Gln-41/Cys-374 distance is accompanied by a reduction in the solvent accessibility of Gln-41 (Fig. 2 C). Acrylamide quenches the fluorescence emission of DC in the G-actin/tβ4 complex (curve GT, KSV = 1.0 ± 0.3, n = 6) less efficiently than in G-actin alone (curve G, KSV = 2.4 ± 0.1, n = 6). A similar effect is observed in F-actin (curve F, KSV = 1.3 ± 0.1, n = 5) and in the actin-DNase I complex (curve GD, KSV = 1.2 ± 0.1, n = 6) where Gln-41 is protected from solvent because it forms part of the binding interface (26).
Effect of tβ4 on the distance between Lys-61 and Cys-374 of actin
Actin labeled with an IAEDANS (donor) at Cys-374 and a FITC (acceptor) at Lys-61 (Fig. 1) has a FRET efficiency of 0.65 ± 0.04 corresponding to a distance of 44.7 ± 1.1 Å, in agreement with a previous report (20). The fluorescence emission spectrum of IAEDANS-actin (Fig. 3 A, solid curve 1) demonstrates that tβ4 binding (dashed curve 1′) generates a 39 ± 3% (n = 10) decrease in fluorescence intensity at 475 nm and an 11-nm red shift of the emission maximum wavelength (5). However, in the presence of FITC (solid curve 2), tβ4 causes only a 27 ± 4% decrease (λ = 475 nm, n = 9) of the D-fluorescence intensity (dashed curve 2′). The difference demonstrates that there is a reduction in FRET efficiency between the probes at Cys-374 and Lys-61 when tβ4 is bound. The ratios of corrected fluorescence intensities (FDA to FD at λ = 475 nm) of the IAEDANS-actin/tβ4 (dashed curve 1′) and IAEDANS/FITC-actin/tβ4 complexes (dashed curve 2′) yields a FRET efficiency of 0.59 ± 0.04 (n = 9). QD of the D significantly decreases in the presence of tβ4 (0.48 in actin alone compared to ∼0.3 in the actin/tβ4 complex, n = 6). This reduces R0 from 49.6 Å (in actin alone) to 46.5 Å (in the actin/tβ4 complex), yielding a distance between IAEDANS and FITC in the actin/tβ4 complex of 43.8 ± 0.9 Å, a mean reduction of only 0.9 Å (p = 0.018).
FIGURE 3.

Effect of tβ4 on the distance between Lys-61 and Cys-374 of actin. (A) Corrected and averaged fluorescence spectra (λex = 365 nm and λem = 520 nm) of IAEDANS-actin and IAEDANS/FITC-actin (1.5–3 μM) in the presence (dashed curves 1′ and 2′) and absence (solid curves 1 and 2) of 10-fold molar excess of tβ4. Conditions: G-buffer, 22°C. Spectra were normalized to a 100% acceptor labeling ratio. (Inset) The emission spectrum of FITC-actin excited at 520 nm (solid curve) and at 360 nm (dashed curve). (B) Effect of DNase I on the corrected and averaged fluorescence spectra of IAEDANS- and IAEDANS/FITC-actin (1.5–3 μM; solid curves 1 and 2, respectively) in the presence (dashed curves 1 and 2) and absence (dotted curves 1 and 2) of a 10-fold molar excess of tβ4. Curve 3 shows an emission spectrum of G-buffer. (C) The reverse order of ABP addition, i.e., tβ4 (dashed curves) followed by the DNase I (dotted curves). (D) A 10% Native PAGE gel. (Lane 1) IAEDANS/FITC-actin (3 μM) alone; (lane 2) 3 μM actin + 20 μM tβ4; (lanes 3–5) 1.5, 3, and 6 μM DNase I was added to the actin/tβ4 complex; (lane 6) 3 μM actin + 6 μM DNase I; (lane 7) 20 μM tβ4 + actin/DNase I complex (shown in lane 6); and (lane 8) 20 μM tβ4 + 6 μM DNase I.
Therefore, although the binding of tβ4 significantly decreases the FRET efficiency, the effect on the distance between these probes is not substantial. Fig. 3 A (inset) shows the emission spectrum of actin labeled with acceptor-only (solid curve), excited at 520 nm, and the same sample excited at 360 nm (dashed curve). From these data it is clear there can be no contribution of FITC fluorescence at 475 nm where the FRET is determined. DNase I binding does not alter the emission spectra of IAEDANS- or IAEDANS/FITC-actin (Fig. 3 B) nor does it affect the FRET efficiency between these sites (27). DNase I inhibits the tβ4-induced spectroscopic changes (Fig. 3) because it prevents tβ4 from binding to actin as assayed by native gel electrophoresis (Fig. 3 D), consistent with common or partially overlapping binding sites (8).
Effect of tβ4 on the distance between bound ɛ-ATP and Lys-61 of actin
FRET efficiencies between ɛATP and FITC were calculated from comparison of normalized fluorescence intensities of ɛATP/FITC- and ɛATP-actin in the absence and presence of tβ4. The distance between ɛATP and FITC attached to Lys-61 of actin (in the absence of tβ4 or DNase I) is 38.4 ± 0.8 Å, the R0 is 47.4 Å, and the FRET efficiency is 0.78 ± 0.02 (17). Binding of tβ4 results in a 14 ± 3% (n = 6) increase in the ɛATP-actin fluorescence (Fig. 4), which is almost completely eliminated when tβ4 binds to ɛATP/FITC-actin, indicating that tβ4 binding increases the FRET efficiency between bound ɛATP and Lys-61. The precise value of the change in FRET efficiency was obtained by incubating samples with 0.4 mg/ml of trypsin in the presence of 2 mM ATP, which results in separation of the D- and A-probes and allows normalization of the reference fluorescence with respect to the ɛATP concentration (17). Unlabeled ATP readily competes with ɛATP bound to actin. The comparison of fluorescence intensities of digested ɛ-ATP/FITC- and ɛATP-actin is used for final normalization of FRET efficiency (see Methods). In the presence of tβ4, the normalized ratio of the fluorescence intensities of ɛATP/FITC-actin to that of ɛATP-actin is 0.16 ± 0.02, i.e., a FRET efficiency of 0.84 ± 0.02. Using an R0 value of 48.1 Å (corrected for the tβ4-induced increased QD), the distance between the probes in the actin/tβ4 complex is 36.5 ± 1.0 Å. Thus, tβ4 closes the gap between the FITC probe on Lys-61 and ɛATP by 1.9 Å (p < 0.000).
FIGURE 4.

Effect of tβ4 on the distance between bound ɛATP and Lys-61 of actin. Corrected and averaged fluorescence spectra (λex = 315 nm and λem = 415 nm) of ɛATP- and ɛATP/FITC-actin in the absence (solid curves) and presence of 10-fold molar excess tβ4 (dashed curves). The actin concentration was 1.5 μM in G-buffer containing 1 mM Tris/HCl pH 8.0 and 0.1 mM CaCl2. The temperature was 10°C. Spectra were normalized to a labeling ratio of FITC to actin equal to one.
Effect of tβ4 on the thermal stability of G-actin
Binding of tβ4 to G-actin results in thermal stabilization of actin monomers (Fig. 5). The thermal transition of actin monomers has a single maximum at 59.9°C, and an apparent calorimetric enthalpy (ΔH°app) of 550 ± 20 kJ/mol (n = 5). Tβ4 alone has no thermal transition (i.e., it is undistinguishable from the buffer baseline, data not shown), consistent with a disordered solution structure in the absence of actin (13). The thermal transition of actin in the presence of tβ4 occurs at 61.7°C, and the ΔH°app increases to 620 ± 25 kJ/mol (n = 5). The width at the half-height of the thermal denaturation peak, ΔT0.5, is equal to 6.5°C for the actin/tβ4 complex but it is 7.5°C for actin alone, suggesting a more cooperative thermal transition with tβ4 binding. The thermal denaturation of actin in the presence and absence of tβ4 was irreversible (data not shown). It is important to clarify that for calorimetric enthalpies of thermal transitions to be thermodynamically significant, denaturation must be reversible. Since actin denaturation is an irreversible process, the measured calorimetric enthalpies are best considered to be apparent thermodynamic parameters reflecting the nonequilibrium denaturation of actin (24).
FIGURE 5.

Effect of tβ4 on the thermal unfolding of actin. Representative DSC scans of G-actin (A) and actin-tβ4 (AT). The trace of tβ4 alone is not shown because it superimposes the buffer baseline. Conditions: 24 μM actin and 30 μM tβ4 in G-buffer. Heating rate is 1 K min−1.
Cofilin and tβ4 compete for binding to actin
Cofilin, tβ4, and their binary complexes with actin can be successfully resolved by native polyacrylamide gel electrophoresis (22,23), which has been employed previously to show an increased affinity of cofilin for actin in the presence of DNase I (23). The actin/tβ4 complex migrates as a distinct band ahead of the actin monomers with free tβ4 appearing below the actin/tβ4 band (Fig. 6 A). Cofilin addition (molar ratios of A/T/C = 1:5:2) results in the appearance of a new band corresponding to the actin/cofilin complex. Excess cofilin (A/T/C molar ratios of 1:5:7) competes and displaces tβ4 from the actin/tβ4 complex. Cofilin and tβ4 do not bind each other (lane 1, Fig. 6 A). The higher affinity of cofilin for actin monomers (1) accounts for the concentration-dependence of the competition. As expected for a reversible equilibrium reaction, cofilin and tβ4 competitions are independent of the order of addition (Fig. 6 B). However, tβ4 does not compete with cofilin for actin binding in the presence of DNase I (Fig. 6 C), and the cofilin-DNase I-actin ternary complex (23) is stable even after the addition of a 15-fold molar excess tβ4 (Fig. 6 C). However, the addition of the same excess tβ4 to the actin/cofilin complex in the absence of DNase I leads to the dissociation of the actin/cofilin complex (Fig. 6 D). Importantly, under experimental conditions, binding of tβ4 to actin is completely abolished in the presence of DNase I, irrespective of the presence of cofilin (Fig. 3, D and E). We interpret these results to mean that accessibility to the DNase I binding loop is necessary for tβ4 to compete with cofilin for actin binding, presumably because SD2 forms part of the tβ4 binding site on actin and contributes to the high affinity.
FIGURE 6.

Effect of tβ4 on cofilin-actin and cofilin-actin-DNase I complexes. (Panel A) A representative 10% Native PAGE gel shows the effects of progressive addition of cofilin (C) to the preformed actin-tβ4 complex (AT). (Lane 1) A mixture of 14 μM C and 10 μM tβ4 (T); (Lane 2) 2 μM actin alone (A); (Lanes 3–5) a mixture of 2 μM A and 4, 6, and 10 μM T, respectively; (Lanes 6–9) titration of the preformed AT complex (2 μM A and 10 μM T) with 2, 6, 10, and 14 μM C, respectively; and (Lane 10) a mixture of 2 μM A and 14 μM C. (Panel B) A representative 10% Native PAGE gel shows the reverse order of ABP addition, i.e., progressive addition of T to the preformed AC complex. (Lane 1) A alone (3 μM); (Lane 2) C alone (9 μM); (Lane 3) a mixture of 3 μM A and 9 μM C; (Lanes 4 and 5) addition of 30 and 45 μM T, respectively, to the preformed AC complex (3 μM A and 9 μM C); and (Lane 6) a mixture of 3 μM A and 45 μM T. (Panel C) A representative 10% Native PAGE gel shows C (12 μM), A (3 μM), DNase I (D, 5 μM), and T (45 μM) alone (lanes 1, 2, 3, and 10, respectively). The binary complexes of A with C, D, and T are shown (lanes 4, 5, and 9, respectively). (Lane 6) The ternary complex formed by A, C, and D (A/C/D = 1:1.6:4) in the absence of T. (Lane 7) The addition of a 15-fold molar excess T to the preformed ternary ACD complex (shown in lane 6). (Lane 8) The addition of D to preformed AT (1:15) complex in the presence of a fourfold unbound C (this sample before addition of D is shown in lane 12, Gel D). (Panel D) A representative 10% Native PAGE gel shows the AC (1:4) complex (lane 11), and the addition of a 15-fold molar excess T to it (lane 12). (Panel E) This 10% Native PAGE gel shows the AT (1:15) complex (lane 13); the addition of a 1.6-fold molar excess D to it (lane 14); and the control AD (1:1.6) binary complex (lane 15). Gels were run simultaneously.
DISCUSSION
This investigation was initiated to determine if tβ4 binding induces a conformational change in actin and to evaluate models of tβ4 binding to G-actin. Tβ4 binding induces significant changes in the average conformation of actin monomers as measured by FRET. The distance between probes along one side of the monomer axis (Cys-374 to Gln-41) is increased, whereas the distance down the center (Cys-374 to Lys-61) decreases slightly. There is a significant reduction in the accessibility of the probe attached at Gln-41 when tβ4 binds, suggesting it forms part of the actin-tβ4 binding interface (5). The observation that tβ4 competes with cofilin and dissociates the cofilin-actin complex but not the cofilin-actin-DNase I ternary complex (Fig. 6) favors a role for SD2 in high affinity tβ4 binding. We conclude that tβ4 binds SD2 and rotates the DNase I-binding loop toward the nucleotide-binding site and away from SD1 (Fig. 1), providing a plausible structural mechanism for how tβ4 enhances the fluorescence of bound ɛATP (Fig. 4), changes the hydration (5), and reduces amide hydrogen exchange from actin (5). The nucleotide-dependence of SD2 conformation likely contributes to the nucleotide-dependence of the tβ4 binding affinity (5,28). When ATP bound to actin is exchanged for ADP, the DNase I binding loop adopts a folded α-helical conformation (11,12,29). Tβ4 has a much higher affinity for ATP-actin than ADP-actin monomers (5,28), suggesting that the conformation of SD2 dictates how tightly tβ4 binds actin (12) and that the higher affinity of tβ4 for ATP-actin results from stabilizing contacts made with SD2 (5,8).
The detailed three-dimensional structure of the actin-tβ4 complex has not been determined experimentally. However, two alternatives have been proposed for tβ4 binding to actin monomers, based on structures of actin bound to tβ4 homologs (6,7). Hertzog et al. (6) reported the structure of actin bound to domain 1 of ciboulot, which shares 30% sequence identity with tβ4, while Irobi et al. (7) reported the structure of actin bound to a construct consisting of tβ4 residues 21–43 fused to the C-terminus of gelsolin segment 1 (gelsolin residues 27–152). Both Hertzog's and Irobi's structures indicate that the N-terminal helix of tβ4 binds SD1 and SD3 of actin, and show the central region of tβ4 (residues 15–29) in an extended conformation overlying the nucleotide binding cleft. In both models, the C-terminus of tβ4 contacts the pointed end of the actin monomer, although the two models differ in the site of contact. Hertzog et al. (6) favor the C-terminus of tβ4 binding directly to SD2 as we proposed (5,8), while Irobi et al. (7) showed residues 30–40 of tβ4 in an α-helix positioned between actin SD2 and SD4 on the pointed end; the three C-terminal residues of tβ4 are unresolved in this structure. This is consistent with our crosslinking studies (5,8), which showed that the N-terminus of tβ4 contacts the barbed end of actin, while its C-terminus contacts the pointed end. However, some features of the crystallographic structures, and the models for actin-tβ4 based on them, are likely to differ from the actual structure of actin-tβ4.
Ciboulot and tβ4 have opposite effects on actin tryptophan fluorescence and on the fluorescence of AEDANS-labeled actin. In addition, while tβ4 acts as a pure sequestering protein, ciboulot promotes monomer addition to barbed ends (31). Ciboulot and tβ4 show substantial sequence homology. However, nonhomologous substitutions occur at several residues, which have been shown by NMR to form close contacts with actin (13). In particular, polar residues T20 and N26 in tβ4 are replaced by hydrophobic residues in ciboulot, and Domanski et al. (13) suggest that these substitutions result in differences between the binding sites of the C-terminal segments of ciboulot and tβ4. Such differences in the binding of the C-terminal segment may contribute to the observed differences in activity. The design of the gelsolin-tβ4 construct was similarly based on limited sequence homology, in this case between tβ4 residues 17–23 (LKKTETQ) and gelsolin residues 149–155 (FKHVVPN) (7). Considering that gelsolin segment 1 binds actin with ∼100-fold higher affinity than even full-length tβ4 (32), the gelsolin domain must provide most of the binding energy for the gelsolin-tβ4 construct. Thus the position of the gelsolin binding site on actin may constrain the binding of the tβ4 segment of the construct, so as to favor a binding mode that is less favorable for full-length tβ4.
The conformation of actin SD2 and its contact with tβ4 in the complex remain to be determined, since it was not fully resolved in either structure. Our results, particularly the reduced solvent exposure of Gln-41, strongly favor direct interaction of tβ4 with SD2 of actin, and therefore support this feature of the Hertzog structure, in contrast to that of Irobi et al. The fact that limited proteolysis of actin on SD2 did not affect the binding of tβ4 in an earlier study (13), rather than indicating a lack of contact, may indicate that tβ4 binding favors a protease-susceptible conformation of SD2 consistent with conformational rearrangement of this region of actin. The position of tβ4 residues 30–40, which Irobi et al. (7) observe in their crystal structure, may be influenced by several factors. Our original NMR and CD study of the actin-tβ4 complex (8) showed that although tβ4 becomes more structured when bound to actin, it retains substantial segmental mobility. The high mobility in the bound state suggests that tβ4 may bind actin in multiple modes with comparable energies that exist in dynamic equilibrium as demonstrated for tropomyosin and actin filaments (30,33). The equilibrium between these modes would likely be dependent on experimental conditions, and thus be influenced by crystal packing forces, ionic composition, and osmolarity of the crystallization solvent. In addition, the structure of the gelsolin-tβ4 fusion protein and the position of the gelsolin binding site on actin may favor a binding mode that is less favorable for full-length tβ4. A dynamic tβ4 C-terminus could account for the apparently conflicting biochemical solution studies demonstrating that tβ4 can be chemically crosslinked to SDs 1, 2, and 3 (5,8) as well as the actin-bound nucleotide (34).
The principal biological effect of tβ4 is to maintain a reservoir of unpolymerized ATP-actin monomers. Spontaneous polymerization is inhibited by blocking sites involved in intersubunit interactions (22). The FRET data indicate that stable binding to ATP-actin monomers is coupled to reorganization of actin SD2, which is likely to account for the specificity for ATP-actin over ADP-actin monomers. In this respect tβ4 differs from cofilin, which also inhibits nucleotide exchange but binds preferably to ADP-actin monomers (1). In addition, the competition between cofilin and tβ4 for binding actin may contribute to the balance between F- and G-actin in living cells by modulating dynamics of filament assembly/disassembly at the barbed and pointed ends (1). The ability to modulate the conformation of SD2 may account for the different affinities of tβ4 for ATP- and ADP-actin, and allows tβ4 to maintain a large reservoir of unpolymerized ATP-actin monomers.
Acknowledgments
We thank Dr. D. I. Levitsky (Bach Research Institute, Moscow, Russia) and Dr. D. Chhabra (University of Sydney) for advice.
I.V.D. was supported by grants from the Australian Research Council and the National Health and Medical Research Council of Australia, The Young Australian Researcher 2000 Award from The Australian Academy of Science, and The James Kentley Memorial Scholarship from the University of Sydney, Australia. E.M.D.L.C. was supported by a Hellman Family Fellowship and grants from the American Heart Association (No. 0235203N) and the National Science Foundation (No. MCB-0216834).
References
- 1.dos Remedios, C. G., D. Chhabra, M. Kekic, I. V. Dedova, M. Tsubakihara, D. A. Berry, and N. J. Nosworthy. 2003. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol. Rev. 83:433–473. [DOI] [PubMed] [Google Scholar]
- 2.Devineni, N., L. S. Minamide, M. Niu, D. Safer, R. Verma, J. R. Bamburg, and V. T. Nachmias. 1999. A quantitative analysis of G-actin binding proteins and the G-actin pool in developing chick brain. Brain Res. 823:129–140. [DOI] [PubMed] [Google Scholar]
- 3.Goldschmidt-Clermont, P. J., M. I. Furman, D. Wachsstock, D. Safer, V. T. Nachmias, and T. D. Pollard. 1992. The control of actin nucleotide exchange by thymosin β4 and profilin. A potential regulatory mechanism for actin polymerization in cells. Mol. Biol. Cell. 3:1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballweber, E., E. Hannappel, T. Huff, H. Stephan, M. Haener, N. Taschner, D. Stoffler, U. Aebi, and H. G. Mannherz. 2002. Polymerization of chemically cross-linked actin·thymosin β4 complex to filamentous actin: alteration in helical parameters and visualization of thymosin β4 binding on F-actin. J. Mol. Biol. 315:613–625. [DOI] [PubMed] [Google Scholar]
- 5.De La Cruz, E. M., E. M. Ostap, R. A. Brundage, K. S. Reddy, H. L. Sweeney, and D. Safer. 2000. Thymosin-β4 changes the conformation and dynamics of actin monomers. Biophys. J. 78:2516–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hertzog, M., C. van Heijenoort, D. Didry, M. Gaudier, J. Coutant, B. Gigant, G. Didelot, T. Preat, M. Knossow, E. Guittet, and M. F. Carlier. 2004. The β-thymosin/WH2 domain; structural basis for the switch from inhibition to promotion of actin assembly. Cell. 117:611–623. [DOI] [PubMed] [Google Scholar]
- 7.Irobi, E., A. H. Aguda, M. Larsson, C. Guerin, H. L. Yin, L. D. Burtnick, L. Blanchoin, and R. C. Robinson. 2004. Structural basis of actin sequestration by thymosin-β4: implications for WH2 proteins. EMBO J. 23:3599–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Safer, D., T. R. Sosnick, and M. Elzinga. 1997. Thymosin β4 binds actin in an extended conformation and contacts both the barbed and pointed ends. Biochemistry. 36:5806–5816. [DOI] [PubMed] [Google Scholar]
- 9.Ballweber, E., K. Giehl, E. Hannappel, T. Huff, B. M. Jockusch, and H. G. Mannherz. 1998. Plant profilin induces actin polymerization from actin: β-thymosin complexes and competes directly with β-thymosins and with negative co-operativity with DNase I for binding to actin. FEBS Lett. 425:251–255. [DOI] [PubMed] [Google Scholar]
- 10.Tirion, M. M., and D. ben-Avraham. 1993. Normal mode analysis of G-actin. J. Mol. Biol. 230:186–195. [DOI] [PubMed] [Google Scholar]
- 11.Otterbein, L. R., P. Graceffa, and R. Dominguez. 2001. The crystal structure of uncomplexed actin in the ADP state. Science. 293:708–711. [DOI] [PubMed] [Google Scholar]
- 12.De La Cruz, E. M., and T. D. Pollard. 2001. Structural biology. Actin' up. Science. 293:616–618. [DOI] [PubMed] [Google Scholar]
- 13.Domanski, M., M. Hertzog, J. Coutant, I. Gutsche-Perelroizen, F. Bontems, M. F. Carlier, E. Guittet, and C. van Heijenoort. 2004. Coupling of folding and binding of thymosin β4 upon interaction with monomeric actin monitored by nuclear magnetic resonance. J. Biol. Chem. 279:23637–23645. [DOI] [PubMed] [Google Scholar]
- 14.Dedova, I. V., V. N. Dedov, N. J. Nosworthy, B. D. Hambly, and C. G. dos Remedios. 2002. Cofilin and DNase I affect the conformation of the small domain of actin. Biophys. J. 82:3134–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao, T., and J. Cho. 1979. Fluorescence lifetime quenching studies on the accessibilities of actin sulfhydryl sites. Biochemistry. 18:2759–2765. [DOI] [PubMed] [Google Scholar]
- 16.Burtnick, L. D. 1984. Modification of actin with fluorescein isothiocyanate. Biochim. Biophys. Acta. 791:57–62. [DOI] [PubMed] [Google Scholar]
- 17.Miki, M., C. G. dos Remedios, and J. A. Barden. 1987. Spatial relationship between the nucleotide-binding site, Lys-61 and Cys-374 in actin and a conformational change induced by myosin subfragment-1 binding. Eur. J. Biochem. 168:339–345. [DOI] [PubMed] [Google Scholar]
- 18.Bernhardt, R., N. T. Ngoc Dao, H. Stiel, W. Schwarze, J. Friedrich, G. R. Janig, and K. Ruckpaul. 1983. Modification of cytochrome P-450 with fluorescein isothiocyanate. Biochim. Biophys. Acta. 745:140–148. [DOI] [PubMed] [Google Scholar]
- 19.De La Cruz, E. M., and T. D. Pollard. 1995. Nucleotide-free actin: stabilization by sucrose and nucleotide binding kinetics. Biochemistry. 34:5452–5461. [DOI] [PubMed] [Google Scholar]
- 20.Nyitrai, M., G. Hild, Z. Lakos, and B. Somogyi. 1998. Effect of Ca2+-Mg2+ exchange on the flexibility and/or conformation of the small domain in monomeric actin. Biophys. J. 74:2474–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Der Meer, B. W. 1994. The donor quantum yield. In Resonance Energy Transfer. B.W. Van Der Meer, G. Coker, and S.-Y. Chen, editors. VCH Publishers, New York. 20–23.
- 22.Safer, D., R. Golla, and V. T. Nachmias. 1990. Isolation of a 5-kiloDalton actin-sequestering peptide from human blood platelets. Proc. Natl. Acad. Sci. USA. 87:2536–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kekic, M., N. J. Nosworthy, I. Dedova, C. A. Collyer, and C. G. dos Remedios. 2001. Regulation of the cytoskeleton assembly: a role for a ternary complex of actin with two actin-binding proteins. In Molecular Interactions of Actin. Actin Structure and Actin-Binding Proteins. C.G. dos Remedios and D.D. Thomas, editors. Springer-Verlag, Berlin. 165–181. [DOI] [PubMed]
- 24.Nikolaeva, O. P., I. V. Dedova, I. S. Khvorova, and D. I. Levitsky. 1994. Interaction of F-actin with phosphate analogues studied by differential scanning calorimetry. FEBS Lett. 351:15–18. [DOI] [PubMed] [Google Scholar]
- 25.Kim, E., and E. Reisler. 1996. Intermolecular coupling between loop 38–52 and the C-terminus in actin filaments. Biophys. J. 71:1914–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabsch, W., H. G. Mannherz, D. Suck, E. F. Pai, and K. C. Holmes. 1990. Atomic structure of the actin·DNase I complex. Nature. 347:37–44. [DOI] [PubMed] [Google Scholar]
- 27.dos Remedios, C. G., P. C. Kiessling, and B. D. Hambly. 1994. DNase I binding induces a conformational change in the actin monomer. In Synchrotron Radiation in the Bioscience. B. Chance, editor. Oxford University Press, New York. 418–425.
- 28.Carlier, M. F., C. Jean, K. J. Rieger, M. Lenfant, and D. Pantaloni. 1993. Modulation of the interaction between G-actin and thymosin β4 by the ATP/ADP ratio: possible implication in the regulation of actin dynamics. Proc. Natl. Acad. Sci. USA. 90:5034–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graceffa, P., and R. Dominguez. 2003. Crystal structure of monomeric actin in the ATP state. Structural basis of nucleotide-dependent actin dynamics. J. Biol. Chem. 278:34172–34180. [DOI] [PubMed] [Google Scholar]
- 30.Xu, C., R. Craig, L. Tobacman, R. Horowitz, and W. Lehman. 1999. Tropomyosin positions in regulated thin filaments revealed by cryoelectron microscopy. Biophys. J. 77:985–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hertzog, M., E. G. Yarmola, D. Didry, M. R. Bubb, and M.-F. Carlier. 2002. Control of actin dynamics by proteins made of β-thymosin repeats. J. Biol. Chem. 277:14786–14792. [DOI] [PubMed] [Google Scholar]
- 32.Bryan, J. 1988. Gelsolin has three actin-binding sites. J. Cell Biol. 106:1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bacchiocchi, C., and S. S. Lehrer. 2002. Ca2+-induced movement of tropomyosin in skeletal muscle thin filaments observed by multi-site FRET. Biophys. J. 82:1524–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reichert, A., D. Heintz, H. Echner, W. Voelter, and H. Faulstich. 1996. Identification of contact sites in the actin-thymosin β4 complex by distance-dependent thiol cross-linking. J. Biol. Chem. 271:1301–1308. [DOI] [PubMed] [Google Scholar]