

Abstract
Hepatitis C virus (HCV) is an important human pathogen associated with chronic liver disease. Recently, based on a genotype 2a isolate, tissue culture systems supporting complete replication and infectious virus production have been developed. In this study, we used cell culture-produced infectious HCV to analyze the viral entry pathway into Huh-7.5 cells. Bafilomycin A1 and concanamycin A, inhibitors of vacuolar ATPases, prevented HCV entry when they were present prior to infection and had minimal effect on downstream replication events. HCV entry therefore appears to be pH dependent, requiring an acidified intracellular compartment. For many other enveloped viruses, acidic pH triggers an irreversible conformational change, which promotes virion-endosomal membrane fusion. Such viruses are often inactivated by low pH. In the case of HCV, exposure of virions to acidic pH followed by return to neutral pH did not affect their infectivity. This parallels the observation made for the related pestivirus bovine viral diarrhea virus. Low pH could activate the entry of cell surface-bound HCV but only after prolonged incubation at 37°C. This suggests that there are rate-limiting, postbinding events that are needed to render HCV competent for low-pH-triggered entry. Such events may involve interaction with a cellular coreceptor or other factors but do not require cathepsins B and L, late endosomal proteases that activate Ebola virus and reovirus for entry.
Hepatitis C virus (HCV) is the sole member of the Hepacivirus genus within the family Flaviviridae. HCV can cause a persistent infection in humans that is associated with chronic liver disease and hepatocellular carcinoma (27). Flaviviridae are enveloped viruses with a single-stranded RNA genome of positive polarity (reviewed in reference 24). Within the virion, HCV genomic RNA is complexed with multiple copies of capsid protein (C). The viral envelope bears on its surface two type I integral membrane envelope glycoproteins, E1 and E2, which form heterodimers (30). Both E1 and E2 have been shown to accumulate in the endoplasmic reticulum, where particles are thought to assemble (30).
Infection with an enveloped virus requires a fusion event between the viral membrane and a cellular membrane. This event can occur at the cell surface, as demonstrated by human immunodeficiency virus (HIV) and herpes simplex virus (HSV), where binding to one or more receptors induces conformational changes in the envelope glycoprotein, allowing membrane fusion at neutral pH. Alternatively, the fusion event can occur within an endosomal compartment in the presence of low pH (reviewed in reference 39), as has been previously described for classical flaviviruses and pestiviruses as well as alphaviruses (8, 12, 14, 15, 20).
Previous studies of HCV entry have been based on retroviral pseudotypes bearing HCV E1 and E2 glycoproteins (HCVpp) (3, 17). Such pseudotypes were shown to undergo pH-dependent entry into Huh-7 cells in a CD81-dependent manner and could be neutralized by certain anti-E2 monoclonal antibodies (17, 49). Additional receptors, including low-density lipoprotein receptor and scavenger receptor BI, have also been suggested to have a role in HCV entry (1, 4, 22, 36, 44).
By using the JFH genotype 2a strain of HCV, it has recently become possible to propagate infectious HCV particles in cell culture (HCVcc), allowing the study of the complete viral life cycle (23, 45, 50), including virus entry and fusion events. In this report, we demonstrate that HCVcc entry into Huh-7.5 cells is pH dependent although HCV virions are acid resistant, suggesting that HCV may have a mechanism of entry similar to that of the pestivirus bovine viral diarrhea virus (BVDV). The trigger(s) by which an acid-resistant virus becomes pH sensitive during entry was investigated. This work provides the first description of the mechanism of HCV entry into tissue culture cells.
MATERIALS AND METHODS
Cells, virus stocks, and chemicals.
Huh-7.5 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum and 1× nonessential amino acids. Media and reagents were purchased from GIBCO-BRL, Life Technologies Ltd. Stocks of J6/JFH and FL-J6/JFH-5′C19Rluc2AUbi were generated by transfection of in vitro-transcribed RNA into Huh-7.5 cells. Stocks of Toto1101/Luc (5), a Sindbis virus (SIN) expressing firefly luciferase, were generated by electroporation of in vitro-transcribed RNA into BHK-J cells (25). Stocks of KOS/Dlux/oriL (41), HSV-1 expressing firefly luciferase, were generated by infection of Vero cells at a multiplicity of infection (MOI) of 0.01 and harvesting of cell supernatants. Bafilomycin A1, concanamycin A, and CA-074 were purchased from Sigma (St. Louis, Mo.). FYdmk [Z-Phe-Tyr-(tBu)-diazomethylketone] was purchased from Calbiochem (San Diego, Calif.).
Construction of FL-J6/JFH-5′C19Rluc2AUbi.
FL-J6/JFH-5′C19Rluc2AUbi is a monocistronic, full-length HCV genome that expresses Renilla luciferase (Rluc) and was derived from the previously described infectious genotype 2a HCV genome J6/JFH1 (23). For the construction of FL-J6/JFH-5′C19Rluc2AUbi, an MluI site was introduced into the HCV C protein coding sequence of J6/JFH and was used to fuse Rluc, the gene encoding Renilla luciferase, to the first 19 residues of HCV C protein. Rluc was amplified from pRL-CMV (Promega) by PCR using primers that incorporated MluI and BglII sites. To allow removal of Rluc from the HCV polyprotein, a 17-residue fragment of the self-cleaving foot-and-mouth disease virus 2A protein (2A) and the ubiquitin monomer (Ubi) were fused to the C terminus of Rluc as follows. A DNA cassette encoding 2A protein and Ubi in tandem (2AUbi) was amplified from 213_HCVrep GFP-2A-Ubi-Neo (A. Kolykhalov and C. M. Rice, unpublished data) by PCR. 2AUbi was fused to the C terminus of Rluc using an engineered BglII site while the C terminus of Ubi was fused to the start of the HCV polyprotein using overlapping PCR.
RNA transcription.
FL-J6/JFH-5′C19Rluc2AUbi RNA for electroporation was generated essentially as described in reference 23 by in vitro transcription of XbaI-linearized DNA templates using the T7 MEGAscript kit (Ambion), followed by purification using the RNeasy Mini kit (QIAGEN) with on-column DNase treatment.
Electroporation and generation of FL-J6/JFH-5′C19Rluc2AUbi virus stocks.
Subconfluent Huh-7.5 cells were trypsinized, harvested by centrifugation (500 × g for 5 min), washed twice, and resuspended in ice-cold phosphate-buffered saline (PBS; Accugene) at 1.5 × 107 cells/ml. FL-J6/JFH-5′C19Rluc2AUbi RNA (2 μg) was mixed with 0.4 ml of cells in a 0.2-cm gap cuvette and immediately pulsed using a BTX ElectroSquarePorator essentially as described previously (23). Electroporated cells were allowed to recover for 10 min at room temperature prior to the addition of complete medium and were then plated in a 150-mm diameter cell culture dish. After 72 h, electroporated cells were trypsinized and replated in complete medium. Virus stocks containing supernatant from replated cells were then harvested after an additional 3 to 4 days of incubation. Virus stocks were stored at −80°C until use.
Luciferase assay.
To harvest samples for luciferase assays, cells were washed once with Dulbecco's PBS (DPBS), and then 100 μl of Renilla lysis buffer (HCV samples) (Promega) or passive lysis buffer (SIN or HSV-1 samples) (Promega) was added to each well of a 24-well plate. Lysates were harvested and frozen at −80°C. Frozen samples were thawed and vortexed, and 50 μl of sample was mixed with luciferase assay substrate (Promega). Luciferase activity was typically measured for 10 seconds, using a luminometer.
Growth kinetics of FL-J6/JFH-5′C19Rluc2AUbi.
Huh-7.5 cells, seeded in a 24-well plate, were chilled on ice for 5 min and incubated with 200 μl FL-J6/JFH-5′C19Rluc2AUbi (MOI, ∼0.01) for 2 h at 4°C. After incubation, the cells were washed twice with cold DPBS, and warm medium was added. Cells were incubated at 37°C for the appropriate time until being harvested for luciferase assays.
Chemical inhibitor experiments.
Huh-7.5 cells (typically 5 × 104/well) were seeded in a 24-well plate. Cells were pretreated with drug (bafilomycin A1, concanamycin A, or NH4Cl) for 1 h at 37°C. The cells were then chilled on ice for 5 min and infected with HCV (FL-J6/JFH-5′C19Rluc2AUbi), SIN (Toto1101/Luc), or HSV-1 (KOS/Dlux/oriL) for 2 h at 4°C. Once added to a particular well, the drug was present for the duration of the experiment. After binding, the cells were washed twice with cold DPBS, and then warm medium with or without drug was added. For postinfection drug addition, the drug was added directly to the medium in the well. SIN and HSV-1 samples were harvested at 12 hours postinfection (h p.i.). HCV samples were harvested at 24 h p.i.
Low-pH treatment.
HCV or SIN stocks, in medium containing 20 mM HEPES, were diluted 1:5 in a citric acid buffer (15 mM citric acid, 150 mM NaCl) at either pH 4.10 or 7.00. The final pHs of the solutions were pH 5 and 7, respectively. For experiments with reducing agent, 10 mM dithiothreitol (DTT) was included in the citric acid buffer. The buffers containing the diluted virus were incubated in a 37°C water bath for 10 min. After incubation, to neutralize the pH, a one-tenth volume of Dulbecco's modified Eagle's medium containing 150 mM HEPES was added. Each sample was then diluted 1:10 in medium containing 20 mM HEPES.
HCV and SIN entry at the plasma membrane.
Huh-7.5 cells were treated with bafilomycin A1 (25 nM) for 1 h at 37°C and then infected with HCV or SIN in the presence of bafilomycin for 2 h at 4°C. After 2 h, cells were sequentially washed with cold DPBS, warm citric acid buffer (pH 7 or 5), and medium. Medium containing bafilomycin was replaced, and cells were incubated at 37°C. Samples were harvested at 24 h p.i. For experiments with reducing agent, 10 mM DTT was included in the citric acid buffer. For HCV-infected cells that were shifted to 37°C prior to the low-pH wash, after 1 h at 37°C, cells were removed from the incubator and washed as indicated above with DPBS, citric acid buffer (pH 7 or 5), and medium. Medium containing bafilomycin was replaced, and cells were incubated at 37°C. Samples were harvested at 24 h p.i.
Cathepsin B/L inhibitor experiments.
Huh-7.5 cells, seeded in a 24-well plate, were pretreated with CA-074 (100 μM) or FYdmk (0.1 μM) for 3.5 h at 37°C. Cells were infected for 2 h at 37°C with HCV, HIV expressing firefly luciferase pseudotyped with vesicular stomatitis virus envelope glycoprotein (VSV-G) (HIV-VSV-G), or VSV expressing green fluorescent protein pseudotyped with Ebola virus glycoprotein (VSV-EboV GP) (7) (a generous gift from James Cunningham). Cells were washed and incubated for 24 h at 37°C. Drug was present in treated wells for the duration of the experiment. At 24 h p.i., cells were harvested for luciferase assays (HCV, HIV-VSV-G) or flow cytometry (VSV-EboV GP). Flow cytometry was performed on a FACSCalibur (Becton Dickinson), counting 104 cells.
RESULTS
HCV entry is pH dependent.
To examine the HCV entry pathway into Huh-7.5 cells, we utilized an HCV reporter virus termed FL-J6/JFH-5′C19Rluc2AUbi (Fig. 1A), which was derived from the previously reported infectious genotype 2a J6/JFH chimeric virus (23). FL-J6/JFH-5′C19Rluc2AUbi encodes the first 19 residues of the HCV C protein fused to the N terminus of Renilla luciferase. The C terminus of Rluc is fused to a 17-residue fragment from the self-cleaving foot-and-mouth disease virus 2A protein, a Ubi sequence, and finally the complete HCV polyprotein. Together, 2A and Ubi mediate removal of Rluc from the HCV polyprotein and generate the native N terminus of the HCV C protein. The expression of Rluc from FL-J6/JFH-5′C19Rluc2AUbi serves as highly sensitive assay for viral RNA replication after infection of permissive cells. To examine the replication kinetics of FL-J6/JFH-5′C19Rluc2AUbi in Huh-7.5 cells, virus was bound to cells at 4°C, and then the cells were shifted to 37°C for a 48-h period of infection. After a low-level infection (MOI, ∼0.01), a small increase in signal was observed after 8 to 12 h, followed by a logarithmic rise in the luciferase activity between 12 and 24 h p.i. (Fig. 1B).
FIG.1.

Characterization of FL-J6/JFH-5′C19Rluc2AUbi. (A) Schematic of HCV and FL-J6/JFH-5′C19Rluc2AUbi genomes. The 5′ nontranslated region contains an internal ribosome entry site (IRES), which allows translation of the HCV polyprotein (shaded boxes) containing structural (C, E1, E2) and nonstructural proteins (p7, NS2, NS4A, NS4B, NS5A, and NS5B). FL-J6/JFH-5′C19Rluc2AUbi is a full-length HCV genome encoding Rluc, a foot-and-mouth disease virus 2A sequence, and a Ubi sequence. The N terminus of Rluc is fused to the first 19 residues of the HCV C protein (see Materials and Methods). (B) Huh-7.5 cells were infected with FL-J6/JFH-5′C19Rluc2AUbi for 0, 4, 6, 8, 10, 12, 20, 24, 30, 36, or 48 h. At each time point, cells were harvested and luciferase activity was determined. Each point is the average value of triplicate wells; error bars show standard deviations. The dashed line indicates the background level of the assay from naive Huh-7.5 cells. RLU, relative light units.
To determine whether HCV entry is dependent on a low-pH step, we utilized bafilomycin A1, concanamycin A, and NH4Cl, agents that prevent the acidification of endosomal compartments. As controls for these experiments, we infected Huh-7.5 cells in parallel with SIN or HSV-1, which are well-characterized pH-dependent and -independent viruses, respectively. SIN undergoes receptor-mediated endocytosis, followed by fusion in the acidic environment of the endosome, while HSV-1 fuses at the plasma membrane at a neutral pH (39).
Huh-7.5 cells were pretreated with various concentrations of bafilomycin A1, an inhibitor of vacuolar type H+-ATPase, and were then infected with HCV, SIN, or HSV-1. At all concentrations tested, a 3-h pretreatment with bafilomycin reduced HCV-expressed luciferase activity to nearly background levels (Fig. 2A) while having no effect on HSV-1 infection (Fig. 2B). SIN infection was inhibited in a manner similar to that of HCV by pretreatment of cells with bafilomycin (data not shown). To confirm that the drug was acting at viral entry and not at subsequent replication steps, bafilomycin was added at 3 h p.i. with HCV, SIN, or HSV-1, with minimal effect on HCV or SIN luciferase activity (Fig. 2A and data not shown). As expected, HSV-1 was unaffected by the presence of bafilomycin at the 3-h p.i. time point (Fig. 2B).
FIG. 2.

HCV entry is sensitive to bafilomycin A1 (Baf). (A) Huh-7.5 cells were incubated with bafilomycin A1 (0, 25, 50, or 100 nM) either prior to infection (squares) or at 3 h p.i. (triangles) with HCV. Samples were harvested for luciferase assays at 24 h p.i. Each point is the average value of triplicate wells; error bars show standard deviations. The dashed line indicates the background level of the assay from naive Huh-7.5 cells. (B) Huh-7.5 cells were incubated with bafilomycin A1 (0, 25, 50, or 100 nM) either prior to infection (squares) or at 3 h p.i. (triangles) with HSV-1. Samples were harvested for luciferase assays at 12 h p.i. Each point is the average value of triplicate wells; error bars show standard deviations. (C) Huh-7.5 cells were untreated or incubated with bafilomycin A1 prior to infection (−3 h), at the time of infection (−2 h), or at 0, 1, 2, 3, 4, 5, or 6 h p.i. with HCV. Samples were harvested for luciferase assays at 24 h p.i. Values are expressed as percentages of untreated RLU and are the combined data from two independent experiments done in triplicate; error bars represent standard errors of the means.
To determine the time point postinfection at which HCV loses sensitivity to bafilomycin treatment, we performed a time course of bafilomycin addition during HCV infection. Cells were treated with bafilomycin prior to HCV infection (−3 h), at the time of infection at 4°C (−2 h), after inoculum was removed and cells were shifted to 37°C (0 h), or at 1, 2, 3, 4, 5, or 6 h p.i. (Fig. 2C). Drug addition at 1 h p.i. or after had little effect on HCV luciferase activity, but if bafilomycin was present at −3, −2, or 0 h, the luciferase signal was nearly eliminated (Fig. 2C). These data suggest that soon after the temperature shift to 37°C, bound HCV virions are able to enter Huh-7.5 cells, thereby becoming insensitive to bafilomycin treatment.
To further confirm that HCV requires a pH-dependent entry step, we tested another inhibitor of vacuolar type H+-ATPase, concanamycin A. Concanamycin A inhibited HCV and SIN entry into Huh-7.5 cells but had minimal effect on downstream replication events (Fig. 3A and data not shown). Pre- or postinfection treatment with concanamycin A had no effect on HSV-1 infection (Fig. 3B). Infection in the presence of NH4Cl, a lysosomotropic agent, was also tested for its ability to inhibit HCV entry. Pretreatment with 25 mM NH4Cl inhibited HCV (Fig. 3C), but similar inhibition was also observed when the drug was added at 3 h p.i. (data not shown). This observation was unlike that for SIN, which was inhibited in a dose-dependent manner by pretreatment with NH4Cl, but the inhibition was much decreased when the drug was present from only 3 h p.i. (data not shown). HSV-1 infection was unaffected by the presence of 25 mM NH4Cl, indicating that the decrease in HCV luciferase activity for the 3-h posttreatment was not likely due to nonspecific or cytotoxic effects of NH4Cl on the Huh-7.5 cells (Fig. 3C). These data suggest that HCV may require an additional low-pH step for downstream replication events that is inhibited by NH4Cl. Taken together, HCV sensitivity to bafilomycin A1, concanamycin A, and NH4Cl demonstrates that HCV entry requires a low-pH step.
FIG. 3.

HCV entry is sensitive to concanamycin A (Con A) and NH4Cl. (A) Huh-7.5 cells were incubated with concanamycin A (0, 12.5, 25, or 50 nM) prior to infection (squares) or at 3 h p.i. (triangles) with HCV. Samples were harvested for luciferase assays at 24 h p.i. The dashed line indicates the background level of the assay from naive Huh-7.5 cells. (B) Huh-7.5 cells were incubated with concanamycin A (0, 12.5, 25, or 50 nM) prior to infection (squares) or at 3 h p.i. (triangles) with HSV-1. Samples were harvested for luciferase assays at 12 h p.i. (C) Huh-7.5 cells were untreated (0 nM) or incubated with 25 mM NH4Cl prior to infection with HCV (black bars) or HSV-1 (white bars). Samples were harvested for luciferase assays at 24 h p.i. In panels A, B, and C, each point or bar is the average value of triplicate wells; error bars show standard deviations.
HCV infectivity is unaffected by low-pH treatment.
Enveloped viruses that are internalized by endocytosis are, in general, very sensitive to low-pH treatment. Exposure to acidic pH induces a conformational change in the envelope glycoproteins that prematurely exposes the fusion peptide and thereby diminishes the viral infectivity. To determine whether HCV infectivity is sensitive to acidic pH, we treated virions with pH 5 or 7 buffers. After neutralization, the virus was used to infect Huh-7.5 cells. SIN was again used as a control, being a well-characterized acid-sensitive virus that is readily inactivated by low-pH treatment. Treatment at pH 5 for 10 min at 37°C had minimal effect on HCV infectivity but reduced SIN luciferase activity to less than 1% of the pH 7 value (Fig. 4A). These data suggest that unlike SIN virions, which are primed to undergo fusion upon exposure to low pH, HCV requires an additional trigger to become acid sensitive. However, we cannot exclude the possibility that low pH induces a conformational change that is reversible upon neutralization, similar to what has been observed for VSV and rabies virus (10, 11). The observed insensitivity of HCV to low-pH treatment is reminiscent of the related pestivirus BVDV (20).
FIG. 4.
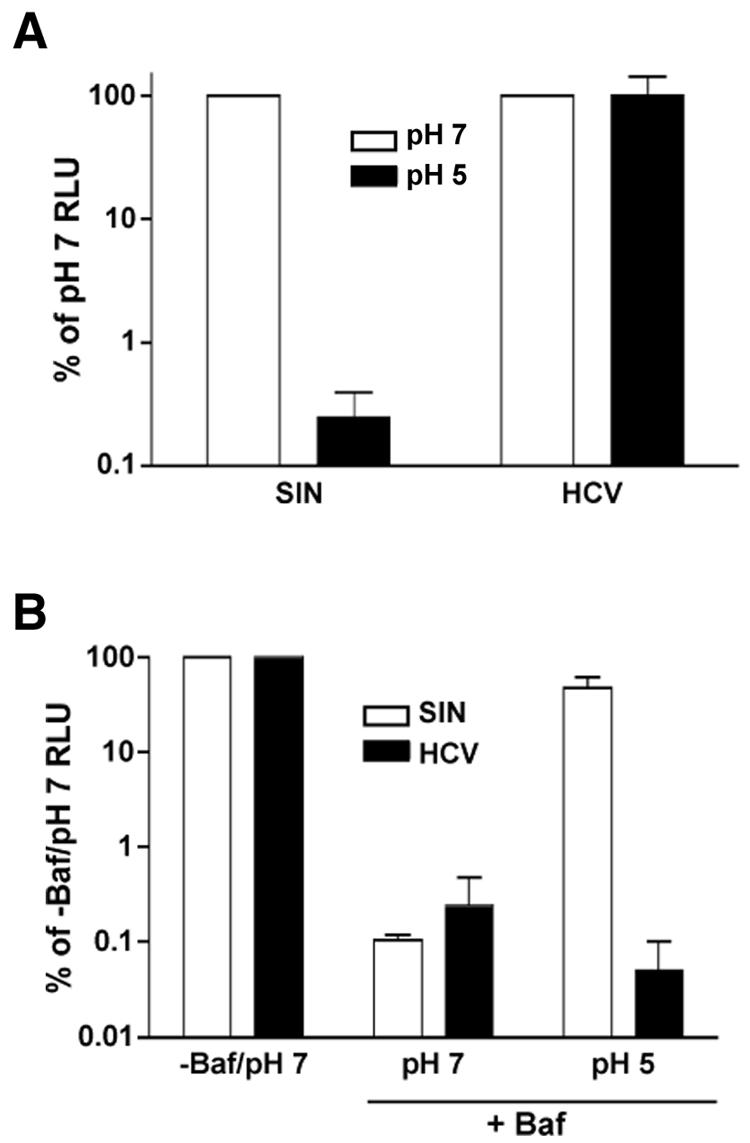
HCV infectivity is resistant to acidic pH. (A) HCV or SIN was diluted in citric acid buffer (15 mM citric acid, 150 mM NaCl) at pH 7 (white bars) or pH 5 (black bars) for 10 min at 37°C. Samples were then neutralized and used to infect Huh-7.5 cells. Samples were harvested at 24 h (SIN) or 48 h (HCV) for luciferase assays. Values, expressed as percentages of pH 7 RLU, are the combined data from two independent experiments done in triplicate; error bars represent standard errors of the means. (B) Huh-7.5 cells were treated with bafilomycin A1 (+Baf) (25 nM) and then were infected with HCV (black bars) or SIN (white bars) at 4°C for 2 h. Cells were washed to remove unbound virus and then washed with citric acid buffer at pH 7 or 5. Cells were incubated in the presence of bafilomycin A1 for 24 h when they were harvested for luciferase assays. Luciferase activity from untreated cells washed with the pH 7 buffer (−Baf/pH 7) was expressed as 100 percent. Values are the combined data from two independent experiments done in triplicate; error bars represent standard errors of the means.
HCV fails to enter bafilomycin-treated cells when exposed to low pH.
Many viruses that utilize the acidic environment of the endosome to trigger glycoprotein-mediated fusion can be induced to fuse at the plasma membrane with a brief low-pH treatment. Whereas free virus is inactivated, cell-bound virus can enter and initiate infection. Using SIN as a control, we tested whether HCV bound at the plasma membrane could enter when exposed to low pH. Huh-7.5 cells were pretreated with bafilomycin to prevent productive entry via endosomal acidification. Cells were then infected with HCV or SIN at 4°C. After the cells were washed to remove unbound virus, they were rinsed with prewarmed buffer at pH 5 or 7 and then incubated in the presence of bafilomycin at 37°C. While there was no increase in HCV luciferase activity from samples washed with the pH 5 buffer, SIN luciferase activity increased to nearly 50% of the untreated values in samples that had been washed with the pH 5 buffer (Fig. 4B). Therefore, cell surface-bound HCV is unable to enter at the plasma membrane under the same conditions as those of SIN. These results suggest that cell-bound HCV remains acid resistant and requires an additional trigger for low-pH-induced infection.
In the case of BVDV, the presence of reducing agent in addition to low pH has been reported to allow some, albeit inefficient, entry and replication (20). Therefore, we reproduced this experiment with HCV by including 10 mM DTT in the pH 5 buffer. No enhancement of the luciferase signal was observed (data not shown). However, this experiment is difficult to interpret since treatment of HCV with DTT, either at pH 5 or pH 7, abrogated detectable HCV infection (data not shown). It is unclear whether DTT is mediating this effect by destabilizing the HCV glycoproteins or cell surface receptors such as CD81, which contains a disulfide bond in its large extracellular loop that is required for E2 binding (16, 32).
HCV entry is not dependent on cathepsin B or L.
As described above for HCV, EboV entry is sensitive to inhibitors of endosomal acidification (42, 47), but acid pH cannot induce glycoprotein-dependent membrane fusion (18). A recent study demonstrated that infection with VSV virions pseudotyped with EboV GP depends on the action of pH-dependent endosomal proteases cathepsin B (CatB) and cathepsin L (CatL) (7). We therefore tested whether endosomal proteolysis by CatB or CatL is required for HCV entry.
Huh-7.5 cells pretreated with CA-074, an inhibitor of CatB, or with FYdmk, an inhibitor of both CatB and CatL, were infected with either HCV, HIV-VSV-G as a negative control, or VSV-EboV GP (7). Both HCV and HIV-VSV-G infections were unaffected by CA-074 or FYdmk, although either inhibitor reduced the efficiency of VSV-EboV GP infection by approximately 80% compared to untreated cells (Fig. 5). This experiment demonstrates that HCV infection is not sensitive to inhibitors of CatB and CatL, suggesting that in contrast to EboV, these endosomal proteases do not have a significant role in HCV entry.
FIG. 5.

HCV is unaffected by inhibitors of CatB and CatL. Huh-7.5 cells were pretreated with CA-074 (100 μM), a CatB selective inhibitor, or FYdmk (0.1 μM), a CatB/CatL inhibitor, and were then infected with HCV (black bars), HIV-VSV-G (gray bars), or VSV-EboV GP (white bars) for 2 h at 37°C. Cells were incubated in the presence of inhibitor for 24 h. At 24 h p.i., cells were harvested for luciferase assays (HCV, HIV-VSV-G) or fluorescence-activated cell sorter analysis (VSV-EboV GP). The luciferase activity or number of green fluorescent protein (GFP)-positive cells from untreated cells is expressed as 100 percent. Each bar is the average value of triplicate wells; error bars show standard deviations.
Evidence that additional postbinding steps are required to activate HCV for entry.
The inability of bound HCV to enter at the plasma membrane after exposure to low pH (Fig. 4B) suggested that an additional step(s) was required to activate the virus for pH-dependent entry. For some viruses, such as ALV (29), and Semliki Forest virus and VSV in certain cell types (28), fusion in the presence of inhibited endosomal acidification requires incubation at 37°C prior to acid treatment. We examined whether low pH could override the bafilomycin block to HCV infection if the cells were shifted to 37°C prior to the pH 5 wash. After 1 h at 37°C, the HCV-infected cells were washed with buffer at pH 5 or 7. In contrast to what we observed when the cells were washed immediately with low pH (Fig. 6, 0 h p.i.), luciferase activity from cells that had been incubated at 37°C prior to the low-pH wash increased to approximately 15% of the untreated value (Fig. 6, 1 h p.i.). Again, no increase in luciferase activity was observed from cells that had been washed with the pH 7 buffer (Fig. 6, 1 h p.i.). These results suggest that HCV requires an event that occurs within 1 h p.i., possibly at the cell surface or within a forming endosome, for low pH to trigger entry in the presence of bafilomycin.
FIG. 6.

Incubation at 37°C allows HCV to enter bafilomycin-treated cells. Huh-7.5 cells were treated with bafilomycin A1 (+Baf) (25 nM) and then infected with HCV at 4°C for 2 h. Cells were washed to remove unbound virus and then washed with citric acid buffer at pH 7 (white bars) or 5 (black bars) immediately (0 h p.i.) or after 1 h at 37°C (1 h p.i.). Cells were incubated in the presence of bafilomycin A1 for 24 h when they were harvested for luciferase assays. Luciferase activity from untreated cells washed with pH 7 buffer (−Baf) was expressed as 100 percent. Values are the combined data from two independent experiments done in triplicate; error bars represent standard errors of the means.
DISCUSSION
The sensitivity of HCV to agents that inhibit endosomal acidification strongly suggests that HCV requires a low-pH step to enter Huh-7.5 cells. The loss of sensitivity to bafilomycin and concanamycin, within 3 h p.i., demonstrates that these agents inhibit an early event in HCV infection and not downstream replication events. Such a pH-dependent route of entry is a common feature among viruses of the Flaviviridae. The observation that HCV virions are acid resistant is similar to that of members of the Pestivirus genus. The inability of HCV to infect a bafilomycin-treated cell after exposure to low pH under the same conditions that could induce fusion of the alphavirus SIN suggests that a postbinding maturation step is required to render the virus competent for low-pH-triggered entry.
The HCVpp system has provided clues to the mechanism of HCV entry into cells, which can now be tested directly using HCVcc. Many of the observations made with HCVpp, such as receptor usage and pH-dependent entry, have now been confirmed in the HCVcc system. For example, the role of CD81 in HCV infection, previously demonstrated using HCVpp (49), is also important for entry of HCVcc (23, 45, 50). As we have found for HCVcc entry, HCVpp entry is also pH dependent and sensitive to concanamycin A and NH4Cl (17). Interestingly, in contrast to the data presented here, Hsu et al. reported that HCVpp (genotype 1a, strain H77) were moderately susceptible to inactivation by low pH (17). The basis for this difference is unknown but could reflect differences in the HCV strains used in the two studies or the experimental systems themselves. If the incorporation of HCV E1E2 heterodimers into HCVpp is similar to that of gp120/gp41 into HIV particles (48), then each HCVpp may harbor fewer than 10 copies of functional HCV E1E2 heterodimers. In contrast, if HCVcc particles resemble those of the classical flaviviruses, which consist of 90 envelope protein dimers (21), the HCVcc surface may consist of tightly packed glycoprotein oligomers. One could imagine that such a lattice, stabilized by lateral interactions, might be more acid resistant. Thus, while entry studies using HCVpp and HCVcc have yielded remarkably convergent results, some features may differ. It will be important to continue to compare these two systems and attempt to extend entry studies to in vivo-produced viruses and other cell types, including primary hepatocytes.
Low pH induces conformational changes to the envelope glycoproteins of many viruses that enter cells by endocytosis, allowing fusion between viral and cellular membranes. Two classes of viral fusion proteins, class I and class II, that mediate entry of enveloped viruses have been defined (13). Influenza hemagglutinin (HA) is a prototype class I fusion protein (38). HA is primed for activation during assembly of influenza virions by proteolytic cleavage, which liberates an N-terminal fusion peptide. When exposed to low pH, HA undergoes a dramatic conformational change, which exposes the fusion peptide promoting interaction with membranes for fusion. Class II fusion proteins, found in alphaviruses and classical flaviviruses, are distinct from class I fusion proteins in their structure, mechanism of action, and apparent evolutionary origin. Alphavirus and flavivirus glycoprotein precursors PE2 and prM, respectively, serve dual roles in virus maturation. As precursors, PE2 and prM act as chaperones to protect the viral fusogens E1 and E from exposure to the low-pH environment of the trans-Golgi network, which would otherwise trigger exposure of the fusion peptide and premature fusion. During virion maturation, PE2 and prM are cleaved by furin, releasing their N-terminal fragments and rendering the viral fusogens competent for pH-triggered fusion during entry (13, 14, 26). These fusion proteins have a well-defined dimer-to-trimer transition upon exposure to low pH (2, 19, 40). With the exception of the G proteins of the rhabdoviruses, VSV (10) and rabies virus (11), the transition of class I or class II proteins to the fusogenic state is irreversible.
The observation that HCV infectivity is resistant to acidic pH is surprising given its route of transmission through direct blood-to-blood exposure. In contrast, pestiviruses have long been known to be acid resistant, which fits with their need for protection against low pH as they travel through the ruminant digestive system. This shared characteristic of acid resistance could be the result of a common evolutionary origin and may have been retained by HCV. Alternatively, production of acid-resistant virions may simply reflect a common strategy in the HCV and pestivirus life cycles. As mentioned earlier, the prM accessory protein of the classical flaviviruses provides protection during egress from the low-pH environment of the cellular secretory pathway upon virion maturation; cleavage of this protein prior to release renders the virus competent for acid-catalyzed fusion and activates the virions for fusion. For HCV and the pestiviruses, there is no evidence of such a proteolytic maturation event during assembly or release (31, 33, 43), suggesting that activation for subsequent low-pH-requiring steps must occur during entry. Activation could theoretically occur at any of the multiple steps involved in the viral entry pathway, e.g., binding to a receptor at the cell surface, sequential binding to a receptor and coreceptor, or assembly of multiple factors at the cell surface, or within a forming or mature endosome.
In the case of BVDV, evidence exists for activation via disulfide bond reshuffling. The three pestiviral glycoproteins are stabilized by a combination of intra- and intermolecular disulfide bonds. Treatment with DTT decreased BVDV's resistance to acidic pH and enhanced its ability to enter at the plasma membrane in the presence of low pH (20). It has been suggested that DTT might act by mimicking the action of protein disulfide isomerases (PDIs) at the cell surface or within the endocytic pathway that are required to destabilize the BVDV glycoproteins and prime them for fusion upon exposure to low pH. The role of PDIs in the entry of other viruses, such as HIV and Moloney murine leukemia virus, has been demonstrated (34, 35, 46). In our experiments, the possible contribution of disulfide bond reshuffling to HCV entry could not be assessed since DTT, at either pH 5 or 7, eliminated detectable HCVcc infection. The use of selective inhibitors of PDIs, as opposed to the relatively nonspecific action of DTT, may provide an alternative method for investigating the possible importance of disulfide bond reorganization in activation of HCV for productive entry.
Another possibility we explored is proteolytic activation of the HCV glycoproteins by endosomal proteases. Similar to our observations for HCV, Ebola virus enters via a pH-dependent pathway, but these particles cannot fuse at the plasma membrane upon low-pH exposure (18, 42, 47). Infection with VSV particles pseudotyped with EboV GP is inhibited when the activities of the CatB and CatL endosomal proteases are blocked by chemical inhibitors (7). This suggests that EboV entry requires a proteolytic cleavage, which triggers fusion in the endosome. Severe acute respiratory syndrome coronavirus infection was also shown to be dependent on CatL to cleave its spike glycoprotein within the endosome (37). Additionally, the nonenveloped reoviruses require CatB and CatL for efficient virus disassembly (6, 9). Although inhibitors of CatB/CatL had no effect on HCV infection, it is still possible that different endosomal proteases or other endosomal determinants are required to prime the HCV glycoproteins for fusion.
Alternatively, HCV may be primed for low-pH-induced entry by receptor binding or subsequent steps prior to the formation of mature endosomes. Simple binding of HCVcc to cells at 4°C was not sufficient to activate the virus for low-pH-induced triggered entry at the plasma membrane. One explanation is that HCV entry at the plasma membrane is nonproductive for downstream events in the viral life cycle, as previously observed for Semliki Forest virus and VSV in CHO cells (28) and avian leukosis virus (29). However, the fact that low pH could override the bafilomycin block to HCV infection after incubation at 37°C, shown in Fig. 6, suggests that HCV can undergo time- and temperature-dependent activation at the cell surface or in forming endosomes. Although the effects of the elevated temperature and incubation time warrant further study, several scenarios can be envisioned based on previous work with other enveloped viruses. In the case of avian leukosis virus and perhaps also for HCV, productive entry could require not only receptor binding but also a forming endosome, encountered only after incubation at 37°C (29). However, even before receptor-mediated endocytosis and formation of mature endosomes, HCV may require interactions that do not occur during binding at 4°C. For example, HCV attached to the cell surface via a primary binding determinant such as CD81 may require interaction with a coreceptor to stabilize binding and activate the virus for low-pH-induced entry. This interaction might require lateral diffusion in the plasma membrane or changes at the plasma membrane that occur during endosome formation, either of which could be compromised by low temperature and decreased membrane fluidity. Future experiments will be aimed at gaining a better understanding of the steps leading to productive HCV entry, paying particular attention to interaction between the HCV glycoproteins and specific molecules, such as CD81, scavenger receptor BI, low-density lipoprotein receptor, or as-yet-unidentified molecules required for entry.
Acknowledgments
We thank Margaret MacDonald and John William Carroll for stocks of Toto1101/Luc, Thomas von Hahn for stocks of HIV-VSV-G, James Cunningham for stocks of VSV-EboV GP, and David Leib for stocks of KOS/Dlux/oriL. We are also grateful to many colleagues for helpful discussions during the course of this work and especially to Margaret MacDonald for critical reading of the manuscript.
This work was supported by PHS grants CA57973 and AI40034 and the Greenberg Medical Research Institute. D.M.T. was supported in part through a Women and Science Fellowship.
REFERENCES
- 1.Agnello, V., G. Abel, M. Elfahal, G. B. Knight, and Q.-X. Zhang. 1999. Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 96:12766-12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison, S. L., J. Schalich, K. Stiasny, C. W. Mandl, C. Kunz, and F. X. Heinz. 1995. Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acidic pH. J. Virol. 69:695-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartosch, B., J. Dubuisson, and F. L. Cosset. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartosch, B., A. Vitelli, C. Granier, C. Goujon, J. Dubuisson, S. Pascale, E. Scarselli, R. Cortese, A. Nicosia, and F. L. Cosset. 2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 278:41624-41630. [DOI] [PubMed] [Google Scholar]
- 5.Bick, M. J., J.-W. N. Carroll, G. Gao, S. P. Goff, C. M. Rice, and M. R. MacDonald. 2003. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 77:11555-11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandran, K., and M. L. Nibert. 2003. Animal cell invasion by a large nonenveloped virus: reovirus delivers the goods. Trends Microbiol. 11:374-382. [DOI] [PubMed] [Google Scholar]
- 7.Chandran, K., N. J. Sullivan, U. Felbor, S. P. Whelan, and J. M. Cunningham. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeTulleo, L., and T. Kirchhausen. 1998. The clathrin endocytic pathway in viral infection. EMBO J. 17:4585-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebert, D. H., J. Deussing, C. Peters, and T. S. Dermody. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 277:24609-24617. [DOI] [PubMed] [Google Scholar]
- 10.Gaudin, Y. 2000. Reversibility in the fusion protein conformational changes: the intriguing case of rhabdovirus-induced membrane fusion. Subcell. Biochem. 34:379-408. [DOI] [PubMed] [Google Scholar]
- 11.Gaudin, Y., C. Tuffereau, D. Segretain, M. Knossow, and A. Flamand. 1991. Reversible conformational changes and fusion activity of rabies virus glycoprotein. J. Virol. 65:4853-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grummer, B., S. Grotha, and I. Greiser-Wilke. 2004. Bovine viral diarrhoea virus is internalized by clathrin-dependent receptor-mediated endocytosis. J. Vet. Med. B Infect. Dis. Vet. Public Health 51:427-432. [DOI] [PubMed] [Google Scholar]
- 13.Heinz, F. X., and S. L. Allison. 2001. The machinery for flavivirus fusion with host cell membranes. Curr. Opin. Microbiol. 4:450-455. [DOI] [PubMed] [Google Scholar]
- 14.Heinz, F. X., and S. L. Allison. 2000. Structures and mechanisms in flavivirus fusion. Adv. Virus Res. 55:231-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helenius, A., J. Kartenbeck, K. Simons, and E. Fries. 1980. On the entry of Semliki Forest virus into BHK-21 cells. J. Cell Biol. 84:404-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higginbottom, A., E. R. Quinn, C. C. Kuo, M. Flint, L. H. Wilson, E. Bianchi, A. Nicosia, P. N. Monk, J. A. McKeating, and S. Levy. 2000. Identification of amino acid residues in CD81 critical for interaction with hepatitis C virus envelope glycoprotein E2. J. Virol. 74:3642-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu, M., J. Zhang, M. Flint, C. Logvinoff, C. Cheng-Mayer, C. M. Rice, and J. A. McKeating. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 100:7271-7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito, H., K. A. Watanabe, A. Sanchez, M. A. Whitt, and Y. Kawaoka. 1999. Mutational analysis of the putative fusion domain of Ebola virus glycoprotein. J. Virol. 73:8907-8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klimjack, M. R., S. Jeffrey, and M. Kielian. 1994. Membrane and protein interactions of a soluble form of the Semliki Forest virus fusion protein. J. Virol. 68:6940-6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krey, T., H. J. Thiel, and T. Rumenapf. 2005. Acid-resistant bovine pestivirus requires activation for pH-triggered fusion during entry. J. Virol. 79:4191-4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn, R. J., W. Zhang, M. G. Rossmann, S. V. Pletnev, J. Corver, E. Lenches, C. T. Jones, S. Mukhopadhyay, P. R. Chipman, E. G. Strauss, T. S. Baker, and J. H. Strauss. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavillette, D., A. W. Tarr, C. Voisset, P. Donot, B. Bartosch, C. Bain, A. H. Patel, J. Dubuisson, J. K. Ball, and F. L. Cosset. 2005. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 41:265-274. [DOI] [PubMed] [Google Scholar]
- 23.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 24.Lindenbach, B. D., and C. M. Rice. 2001. Flaviviridae: the viruses and their replication, p. 991-1041. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 1. Lippincott-Raven Publishers, Philadelphia, Pa. [Google Scholar]
- 25.Lindenbach, B. D., and C. M. Rice. 1997. trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J. Virol. 71:9608-9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lobigs, M., and H. Garoff. 1990. Fusion function of the Semliki Forest virus spike is activated by proteolytic cleavage of the envelope glycoprotein precursor p62. J. Virol. 64:1233-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Major, M. E., B. Rehermann, and S. M. Feinstone. 2001. Hepatitis C viruses, p. 1127-1161. In D. M. Knipe, P. M. Howley, R. M. Chanock, T. P. Monath, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 28.Marsh, M., and R. Bron. 1997. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 110:95-103. [DOI] [PubMed] [Google Scholar]
- 29.Mothes, W., A. L. Boerger, S. Narayan, J. M. Cunningham, and J. A. T. Young. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103:679-689. [DOI] [PubMed] [Google Scholar]
- 30.Op de Beeck, A., L. Cocquerel, and J. Dubuisson. 2001. Biogenesis of hepatitis C envelope glycoproteins. J. Gen. Virol. 82:2589-2595. [DOI] [PubMed] [Google Scholar]
- 31.Op de Beeck, A., C. Voisset, B. Bartosch, Y. Ciczora, L. Cocquerel, Z. Keck, S. Foung, F. L. Cosset, and J. Dubuisson. 2004. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 78:2994-3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petracca, R., F. Falugi, G. Galli, N. Norais, D. Rosa, S. Campagnoli, V. Burgio, E. Di Stasio, B. Giardina, M. Houghton, S. Abrignani, and G. Grandi. 2000. Structure-function analysis of hepatitis C virus envelope-CD81 binding. J. Virol. 74:4824-4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rümenapf, T., G. Unger, J. H. Strauss, and H.-J. Thiel. 1993. Processing of the envelope glycoproteins of pestiviruses. J. Virol. 67:3288-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryser, H. J., E. M. Levy, R. Mandel, and G. J. DiSciullo. 1994. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc. Natl. Acad. Sci. USA 91:4559-4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanders, D. A. 2000. Sulfhydryl involvement in fusion mechansims. Subcell. Biochem. 34:483-514. [DOI] [PubMed] [Google Scholar]
- 36.Scarselli, E., H. Ansuini, R. Cerino, R. M. Roccasecca, S. Acali, G. Filocamo, C. Traboni, A. Nicosia, R. Cortese, and A. Vitelli. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017-5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simmons, G., D. N. Gosalia, A. J. Rennekamp, J. D. Reeves, S. L. Diamond, and P. Bates. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 102:11876-11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531-569. [DOI] [PubMed] [Google Scholar]
- 39.Smith, A. E., and A. Helenius. 2004. How viruses enter animal cells. Science 304:237-242. [DOI] [PubMed] [Google Scholar]
- 40.Stiasny, K., S. L. Allison, J. Schalich, and F. X. Heinz. 2002. Membrane interactions of the tick-borne encephalitis virus fusion protein E at low pH. J. Virol. 76:3784-3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Summers, B. C., and D. A. Leib. 2002. Herpes simplex virus type 1 origins of DNA replication play no role in the regulation of flanking promoters. J. Virol. 76:7020-7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takada, A., C. S. Robison, H. Goto, A. Sanchez, K. G. Murti, M. A. Whitt, and Y. Kawaoka. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. USA 94:14764-14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thiel, H.-J., R. Stark, E. Weiland, T. Rümenapf, and G. Meyers. 1991. Hog cholera virus: molecular composition of virions from a pestivirus. J. Virol. 65:4705-4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voisset, C., N. Callens, E. Blandchard, A. Op de Beeck, J. Dubuisson, and N. Vu-Dac. 2005. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 280:7793-7799. [DOI] [PubMed] [Google Scholar]
- 45.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wallin, M., M. Ekstrom, and H. Garoff. 2004. Isomerization of the intersubunit disulphide-bond in Env controls retrovirus fusion. EMBO J. 23:54-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wool-Lewis, R. J., and P. Bates. 1999. Endoproteolytic processing of the Ebola virus envelope glycoprotein: cleavage is not required for function. J. Virol. 73:1419-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang, X., S. Kurteva, X. Ren, S. Lee, and J. Sodroski. 2005. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J. Virol. 79:12132-12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang, J., G. Randall, A. Higginbottom, P. Monk, C. M. Rice, and J. A. McKeating. 2004. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 78:1448-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong, J., P. Gastaminza, G. Cheng, S. B. Kapadia, T. Kato, D. R. Burton, S. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]