

Abstract
DNA polymerase η (PolH) is the product of the xeroderma pigmentosum variant (XPV) gene and a well-characterized Y-family DNA polymerase for translesion synthesis. Cells derived from XPV patients are unable to faithfully bypass UV photoproducts and DNA adducts and thus acquire genetic mutations. Here, we found that PolH can be up-regulated by DNA breaks induced by ionizing radiation or chemotherapeutic agents, and knockdown of PolH gives cells resistance to apoptosis induced by DNA breaks in multiple cell lines and cell types in a p53-dependent manner. To explore the underlying mechanism, we examined p53 activation upon DNA breaks and found that p53 activation is impaired in PolH knockdown cells and PolH-null primary fibroblasts. Importantly, reconstitution of PolH into PolH knockdown cells restores p53 activation. Moreover, we provide evidence that, upon DNA breaks, PolH is partially colocalized with phosphorylated ATM at γ-H2AX foci and knockdown of PolH impairs ATM to phosphorylate Chk2 and p53. However, upon DNA damage by UV, PolH knockdown cells exhibit two opposing temporal responses: at the early stage, knockdown of PolH suppresses p53 activation and gives cells resistance to UV-induced apoptosis in a p53-dependent manner; at the late stage, knockdown of PolH suppresses DNA repair, leading to sustained activation of p53 and increased susceptibility to apoptosis in both a p53-dependent and a p53-independent manner. Taken together, we found that PolH has a novel role in the DNA damage checkpoint and that a p53 target can modulate the DNA damage response and subsequently regulate p53 activation.
p53 is often referred to as the “guardian of the genome” (38, 43). In response to genotoxic or cellular stresses, such as DNA damage, oncoprotein activation, and hypoxia, p53 is activated to regulate a series of downstream target genes (18, 26, 32). These genes initiate a program of cell cycle arrest, apoptosis, DNA repair, cellular senescence, or antiangiogenesis (25, 39, 47, 67, 77).
p53 undergoes a series of posttranslational modifications when cells are exposed to stress signals, which leads to p53 stabilization and activation (8). Following stresses, several serine residues at both the amino terminus and the carboxyl terminus of p53 are phosphorylated (2, 31, 42). Among them, the best-characterized ones are serine 15 and serine 20. Serine 15 is phosphorylated by ataxia-telangiectasia (AT) mutated gene product ATM whereas serine 20 is phosphorylated by both ATM and its substrate checkpoint kinase 2 (Chk2) when DNA double-strand breaks (DSBs) are created (9, 27). Phosphorylation of serine 15 and serine 20 is shown to disrupt the association of p53 with Mdm2 and, consequently, prevent ubiquitin-dependent degradation of p53 by proteosome (3, 8, 52, 55, 57).
ATM activation is an early response to cellular stresses (5, 41). In unstressed cells, ATM is held inactive as a dimer or higher-order multimer. Cellular stresses induce rapid intermolecular autophosphorylation of serine 1981 that causes dimer disassociation and initiates ATM kinase activation (4). A previous report suggests that ATM does not need to bind to DSBs for its activation since a few DSBs can trigger extensive ATM phosphorylation in a very short time (4). However, several recent studies showed that ATM is recruited by NBS1 (nibrin) to the damage sites and that the Mre11-Rad50-NBS1 (MRN) complex is essential to ATM activation (21, 45, 46, 79). Histone H2AX at the ends of DSBs is phosphorylated by ATM and forms a typical punctate structure termed γ-H2AX foci. Phosphorylated ATM and some ATM substrates are recruited to γ-H2AX foci where ATM initiates a series of phosphorylation events that eventually lead to checkpoints or cell death. A few nuclear proteins have been shown to be able to affect ATM autophosphorylation and activity. For instance, both phosphorylation and activity of ATM triggered by ionizing radiation are impaired in cells from Nijmegen breakage syndrome (NBS) patients (24, 28, 40). However, phosphorylation of ATM substrates is not equally impaired in NBS cells. This is demonstrated by the discoveries of suppressed phosphorylation of BRCA1 and SMC1, but not p53, in NBS cells (28). The above evidence suggests that some proteins may preferentially influence ATM to phosphorylate certain substrates, which leads to differential cellular responses.
ATR is predominantly activated by replication stress, such as UV radiation. In contrast to ATM, ATR activation does not require autophosphorylation. After replication fork stalling, ATR and its partner, ATR-interacting protein (ATRIP), are recruited by single-strand DNA-binding protein, RPA, to the stalling sites. Independently, Rad9-Hus1-Rad1 (9-1-1) complex is loaded by Rad17 onto the nearby chromatin and recruits ATR substrates, such as Chk1 and p53, for phosphorylation. Phosphorylation of Chk1 by ATR also requires the assistance of claspin (69). Additionally, 53BP1, MDC1, and Mre11-Rad50-NBS1 (MRN) complex, which can facilitate ATM activation and promote ATM activity, also regulate ATR activity (59, 65, 74). Therefore, ATR activity is subject to multiple regulations. Phosphorylation of Chk1 and p53 by ATR leads to G1 arrest and prevention of mitotic entry. Loss of ATR function causes defects in checkpoints and premature entry of mitosis upon replication stress, which eventually leads to mitotic catastrophe and cell death (69).
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder characterized by sun sensitivity, cutaneous and ocular deterioration, and premature malignant skin cancers (7). According to various genes implicated in the disease, XP is categorized into eight groups: XPA to XPG and XPV (7, 14). It is now clear that the gene products XPA to XPG are involved in the repair of DNA lesions generated by UV radiation, such as cyclobutane pyrimidine dimers and 6-4 photoproducts (7, 14). This is called nucleotide excision repair (NER) (78). However, although XPV patients share clinical phenotypes with other XP patients, XPV cells have normal NER activity. Instead, XPV cells are deficient in DNA replication after UV exposure, namely, DNA translesion synthesis (TLS) (22, 37). Recently, several groups independently identified the XPV gene and found that the gene product, DNA polymerase η (PolH), is able to bypass T-T dimer in an error-free manner (34, 53). In addition to PolH, three more DNA polymerases (polymerases κ and ι and Rev1), categorized as the Y family, have been found to have TLS activity, but these polymerases are error prone and insert incorrect nucleotides opposite T-T (22). It is postulated that the early onset of multiple skin cancers is due to the replication of UV lesions executed by error-prone DNA polymerases in XPV patients who are deficient in PolH (72).
In this study, we identified PolH as a p53 target. We found that PolH can be induced by camptothecin (CPT)-induced DNA damage in a p53-dependent manner. We identified a potential p53-binding site in the promoter of the PolH gene, and this site is responsive to, and bound by, p53. We also found that knockdown of PolH gives cells resistance to CPT and ionizing radiation (IR)-induced apoptosis, which is p53 dependent. Surprisingly, p53 activation after DNA damage is impaired in both PolH knockdown and PolH-null cells and can be restored by reconstitution of PolH in PolH knockdown cells. Additionally, we found that PolH knockdown suppresses the activity of ATM in phosphorylating Chk2 and p53. Moreover, ATM and PolH are partially colocalized at γ-H2AX foci upon DNA damage, suggesting a mechanism for regulation of ATM activity by PolH. Finally, PolH knockdown gives cells resistance to UV-induced, p53-dependent apoptosis at early times, which is consistent with impaired p53 activation within this period. However, PolH knockdown delays recovery from UV-induced DNA damage and sustains p53 activation at late times after UV radiation.
MATERIALS AND METHODS
Plasmid.
PolH cDNA was generated by reverse transcription-PCR using total RNA purified from RKO cells treated with 300 nM CPT overnight with forward primer 5′ ACC ATG GCT ACT GGA CAG GAT CGA GTG G 3′ and reverse primer 5′ AGC CTG AGT GGG AGC AGT AAG AGA TGA AAG C 3′. To generate PolH cDNA whose transcript is resistant to degradation by PolH short interfering RNA (siRNA) when transfected into stable PolH knockdown cells, two nucleotides of the PolH cDNA in the specific PolH siRNA targeting region were mutated according to the alternative codon usage by directed-site mutagenesis (Stratagene). The primers for mutagenesis are as follows (mutated nucleotides shown in bold italic): sense, 5′ GGG GAT GCG AAA ACA AGG TCT ATT TCA ATG GCT CGA TTC T 3′, and antisense, 5′ AGA ATC GAG CCA TTG AAA TAG ACC TTG TTT TCG CAT CCC C 3′. The mutated PolH cDNA was cloned into pcDNA3 at BamHI and EcoRI sites. The resulting construct was designated pcDNA3-PolH. pcDNA3-p53 was described previously (49). To generate a construct that expresses PolH siRNA, DNA oligonucleotides were cloned into pBabe-U6 and the resulting construct was designated pBabe-U6-PolH. The siRNA oligonucleotides cloned in pBabe-U6-PolH (shown in boldface, derived from the PolH gene from +745 to +764) are as follows: sense, 5′ TCG AGG TCC GGC TTA TTT CAA TGG CTC GA ttcaagaga TCG AGC CAT TGA AAT AAG CCT TTT TG 3′, and antisense, 5′ GAT CCA AAA AGG CTT ATT TCA ATG GCT CGA tctcttgaa TCG AGC CAT TGA AAT AAG CCG GAC C 3′. The PolIII promoter-driven expression vector, pBabe-U6, was described previously (51). To generate a construct that expresses PolH siRNA under the control of tetracycline, one pair of oligonucleotides was cloned into pTer at HindIII and BglII sites and the resulting construct was designated pTer-PolH. pTer is a PolIII promoter-driven plasmid with a tetracycline operator sequence inserted before the transcriptional starting site (76). The siRNA oligonucleotides cloned in pTer-PolH (shown in boldface and the same as those designed for pBabe-U6-PolH) are as follows: sense, 5′ GAT CCC CGG CTT ATT TCA ATG GCT CGA ttcaagaga TCG AGC CAT TGA AAT AAG CCT TTT TGG AAA 3′, and antisense, 5′ AGC TTT TCC AAA AAG GCT TAT TTC AAT GGC TCG A tctcttgaa TCG AGC CAT TGA AAT AAG CCG GG 3′.
Cell lines.
Normal human fibroblasts (GM00495 and GM00024) and human fibroblasts derived from XPV patients (GM03617 and GM02359) were purchased from Coriell Cell Repositories (Camden, NJ) and grown in Eagle's minimal essential medium plus 10% fetal bovine serum at 37°C plus 5% CO2. H1299 cell lines that can inducibly express p53 were described previously (10, 81). RKO, MCF-7, LS174T, and SW480 were purchased from the American Type Culture Collection (ATCC). The p53-null-like cell lines RKO-E6 and MCF-7-E6 were described previously (49). HCT116 and HCT116-p53−/− were obtained from B. Vogelstein (Johns Hopkins University). To generate stable PolH knockdown RKO or MCF-7 cell lines, pBabe-U6-PolH was transfected into RKO or MCF-7 cells and the cells were split and plated at different dilutions on the second day following transfection. RKO or MCF-7 cells were cultured and later cloned in medium containing 1 μg/ml of puromycin. Individual clones were treated with 300 nM CPT overnight, and the cell extracts were collected for Western blot analysis to determine the PolH expression level. The resulting cell lines were designated RKO-PolH-KD and MCF-7-PolH-KD, respectively. Three representative RKO-PolH-KD clones (clones 14, 16, and 70) and two representative MCF-7-PolH-KD clones (clones 43 and 56) were selected for further experiments. The RKO clones that survived the selection but did not show reduction in PolH expression were designated as RKO control cell lines and used as a control in some experiments. To generate a stable H1299 cell line with PolH knockdown, transfections and selections were performed in the same way as those used to generate RKO-PolH-KD cell lines, except that the concentration of puromycin was 2 μg/ml. Individual clones were transfected with pcDNA3-p53. On the second day following transfection, cell extracts were collected to determine the PolH expression. The resulting cell line was designated H1299-PolH-KD, and one representative clone (clone 36) was used in the study. To generate RKO and MCF-7 stable cell lines with inducible PolH knockdown, an RKO and an MCF-7 cell line that can express tetracycline repressor were established using the T-REx system (Invitrogen) according to the manufacturer's instructions. The resulting cell line was designated RKO-TR-5 and MCF-7-TR-7, respectively. RKO-TR-5 and MCF-7-TR-7 were transfected with pTer-PolH, and cells were selected with 200 μg/ml of zeocin. Individual clones were cultured in the absence or presence of tetracycline and treated with 300 nM CPT overnight. PolH expression levels in uninduced and induced cells were determined by Western blotting. The resulting cell line was designated RKO-pTer-PolH and MCF-7-pTer-PolH, respectively. Two representative clones of RKO-pTer-PolH (clones 82 and 90) and one representative clone of MCF-7-pTer-PolH (clone 7) were selected for further experiments.
Affymetrix GeneChip assay and Northern blot analysis.
Total RNA was isolated from H1299 cells, which were uninduced or induced to express p53 or from RKO, RKO-E6, MCF-7, MCF-7-E6, HCT116, HCT116-p53−/−, LS174T, and SW480, which were untreated or treated with 300 nM CPT for 24 h. The U133-plus GeneChip, which contains oligonucleotides representing 47,000 unique human transcripts, was purchased from Affymetrix. GeneChip analysis was performed according to the manufacturer's instructions. Northern blots were prepared by using 10 μg total RNA. The PolH probe was prepared from a PolH expressed sequence tag clone (GenBank accession no. BG716631.1). The p21 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probes were prepared as previously described (50). The DDB2 probe was prepared from a DDB2 expressed sequence tag clone (GenBank accession no. BC000093).
Western blot analysis.
Cells were collected from plates, suspended with 2× sodium dodecyl sulfate sample buffer, and boiled for 10 min. Western blot analysis was performed as described previously (50). Antibodies against hemagglutinin (HA) epitope, PolH, p53, and p21 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phosphorylated serine 1981 ATM polyclonal antibody was purchased from Rockland (Philadelphia, PA). Antiactin polyclonal antibody was purchased from Sigma (St. Louis, MO). Anti-phosphorylated serine-15 p53 monoclonal antibody, anti-phosphorylated threonine-68 Chk2 polyclonal antibody, and anti-total Chk2 polyclonal antibody were purchased from Cell Signaling. Monoclonal antibodies against poly(ADP-ribose) polymerase (PARP), Mdm2, and Fas were purchased from Oncogene Science (Cambridge, MA). Anti-DR5 polyclonal antibody was a gift from T. Zhou (University of Alabama at Birmingham).
Trypan blue dye exclusion assay.
Cells were seeded at approximately 2 × 105 for RKO cells, 2 × 105 for H1299 cells, or 1 × 106 for MCF-7 cells in 60-mm-diameter plates. Twenty-four hours after plating, cells were untreated or treated with 500 nM CPT for 12 h, with 20 Gy of IR, or with 20 J/m2 of UV. At 72 h after treatment, both floating cells in the medium and live cells on the plates were collected and concentrated by centrifugation. After being stained with trypan blue dye (Sigma) for 15 min, both live (unstained) and dead (stained) cells were counted twice in a hemocytometer. The percentage of dead cells was the product of the number of dead cells divided by that of the total cells.
DNA histogram analysis.
Various RKO or H1299 cells were seeded at 2 × 105 per 90-mm-diameter plate. Twenty-four hours after plating, cells were untreated or treated with 500 nM CPT for 12 h. At 72 h after treatment, both floating cells in the medium and live cells on the plates were collected and DNA histogram analysis was performed as described previously (50). To determine apoptosis induced by UV irradiation, various RKO cells were irradiated by 12 J/m2 or 35 J/m2 of UV. Cells were collected at indicated time points, and DNA histogram analysis was performed.
Luciferase assay.
A DNA fragment containing a potential p53-responsive element located in the promoter of PolH (from −1992 to −1967) was generated by PCR amplification of human genomic DNA using forward primer 5′ GGA TCC GAG CTC GAG AAA TCA AGG CT 3′ and reverse primer 5′ AAG CTT AAA TCT GGA ACC ATC ACG CT 3′. The PCR product was cloned upstream of the minimum c-fos promoter in the luciferase reporter O-Fluc (33), and the resulting plasmid was designated O-Fluc-PolH. An 0.25-μg amount of O-Fluc-PolH was cotransfected with 0.25 μg of pcDNA3, pcDNA3-p53, and pcDNA3-p53(R175H), respectively. As an internal control, 5 ng of Renilla luciferase assay vector, pRL-CMV (Promega, Madison, Wis.), was also cotransfected. The dual luciferase assay was performed according to the manufacturer's instructions (Promega). The fold increase in relative luciferase activity is a product of the luciferase activity induced by wild-type or mutant p53 divided by that induced by pcDNA3.
ChIP assay.
The chromatin immunoprecipitation (ChIP) assay was performed essentially as described previously (51). RKO cells, which were untreated or treated with 300 nM CPT for 24 h, were cross-linked with 1% formaldehyde. Cell extracts were sonicated to generate 200- to 1,000-bp DNA fragments. One percent of cell extracts from each of the samples was taken as input. Protein-DNA complexes were immunoprecipitated with anti-p53 or anti-HA polyclonal antibody. After reverse cross-linking and phenol-chloroform extraction, DNA fragments bound by p53 were purified over a QIAGEN column. PCR was performed to visualize the enriched DNA fragments. Primers that were used to amplify the p53-responsive element within the PolH promoter were PolH-F (GAG CTC GAG AAA TCA AGG CT) and PolH-R (AAA TCT GGA ACC ATC ACG CT). Primers that were used to amplify the p53-responsive element 1 within the p21 promoter were described previously (51).
Immunofluorescence assay.
RKO cells were grown on chamber slides for 24 h and then treated with 300 nM CPT for 6 h. The untreated and treated cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS) for 30 min and permeabilized with 0.5% Triton X-100 for 5 min. After they were blocked with 5% bovine serum albumin (BSA) for 1 h, cells were then incubated with a mixture of anti-γ-H2AX monoclonal antibody and anti-PolH polyclonal antibody in PBS with 5% BSA, a mixture of anti-γ-H2AX and anti-p-S1981-ATM, or a mixture of anti-p-S1981-ATM and anti-PolH monoclonal antibody. After washing, appropriate fluorochrome-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were applied, and fluorescence was visualized using a Nikon microscope.
RESULTS
PolH is a p53 target gene.
p53 is able to regulate a plethora of target genes (18, 26), which mediate various p53 functions. However, although over 100 target genes have been identified, none of them is essential for a given p53 function (26). For example, as p53 targets, Bax, Puma, Noxa, Fas, PIG3, and Killer/DR5 are all capable of mediating p53-dependent apoptosis, but genetic studies have shown that p53 is still able to induce apoptosis in the absence of each individual one (71). This suggests that either p53 activates a redundant set of genes to fulfill its functions or some genes yet to be found may play an indispensable role. Thus, identification of novel p53 targets is still of great interest.
In order to identify novel target genes regulated by p53, the Affymetrix GeneChip assay was performed. Total RNA was isolated from H1299 cell lines that were uninduced and induced to express p53. cRNA was synthesized and used to hybridize the Affymetrix U133-plus GeneChip. We found that many known target genes, such as p21, MDM2, and PIG3, were induced by p53. We also found that some novel genes are potentially regulated by p53. Among these is the PolH gene, which has been shown to be mutated in XPV patients.
To confirm the regulation of PolH by p53, we performed Northern blot analysis. We found that PolH was significantly induced in two H1299 cell lines (H1299-p53-15 and H1299-p53-3) when p53 was induced by withdrawal of tetracycline (Fig. 1A, upper panel). As a control, we tested expression of p21 and found that p21 was induced by p53 (Fig. 1A, bottom panel). The level of GAPDH was also determined as a loading control (Fig. 1A, bottom panel).
FIG. 1.

PolH is a p53 target gene. (A) PolH is induced by p53. Northern blots were prepared using total RNA isolated from H1299 cells that were uninduced and induced to express p53 for 24 h. The blots were probed with cDNAs derived from the PolH, p21, and GAPDH genes, respectively. (B) PolH is induced in cells by CPT in a p53-dependent manner. The experiment was performed as in panel A. (C) PolH expression is up-regulated by p53. Western blots were prepared using cell extracts purified from H1299 cells that were uninduced and induced to express p53. The HA-tagged p53 was detected with anti-HA. PolH was detected with anti-PolH polyclonal antibody. Actin was determined as loading controls. (D) PolH expression is up-regulated in cells by CPT in a p53-dependent manner. (E) Schematic presentation of the PolH genomic structure. A potential p53-responsive element is located 1,965 nucleotides upstream of the PolH transcription starting site. The consensus p53-responsive element is shown for comparison. Mismatched nucleotides are indicated in italics. (F) The potential p53-binding site is responsive to p53 but not to p53(R175H). A DNA fragment containing the potential p53-binding site was cloned into the luciferase reporter O-Fluc. The resulting construct was designated O-Fluc-PolH. O-Fluc-PolH was cotransfected into H1299 cells with a pcDNA3 control vector or a vector that expresses p53 or p53(R175H). (G) Schematic presentation of the PolH and p21 promoters with the location of the transcriptional start site, p53-responsive elements, and primers used for ChIP assays. (H) p53 directly binds to the potential p53 response element within the PolH promoter. The ChIP assay was performed as described in Materials and Methods. p53-DNA complexes were captured with anti-p53. Anti-HA antibody was used as a control. The fragment containing the p53 response element in PolH was amplified by PCR with primers PolH-F and PolH-R. The fragment containing the p53 response element 1 in p21 was amplified by PCR as a control.
DNA damage stabilizes and activates p53, leading to induction of p53 target genes (25, 32, 39, 47). If PolH is a true p53 target, it would be induced by DNA damage in cells that contain an endogenous wild-type p53 gene. To this end, we tested eight cell lines untreated or treated with CPT, which is an inhibitor of topoisomerase I and can induce DSBs (61). We found that both PolH and p21 were induced in CPT-treated RKO, MCF-7, HCT116, and LS174T cells (Fig. 1B, PolH and p21 panels). These cell lines contain wild-type p53. In contrast, PolH was not induced in p53-null HCT116(p53−/−), p53-null-like MCF-7-E6 and RKO-E6 cells and SW480 cells in which p53 is mutant (Fig. 1B, PolH panel).
Next, we determined whether an increase in PolH transcripts correlates with an increase in PolH protein. Exogenous p53 was induced in H1299 cells by withdrawal of tetracycline. Endogenous wild-type p53 was induced in RKO, MCF-7, LS174T, and HCT116 cells by treatment with CPT. RKO-E6, MCF-7-E6, and HCT116(p53−/−) cells were similarly treated as a negative control. We found that the level of PolH protein was increased in H1299 cells by exogenous p53 (Fig. 1C, middle panel) and in RKO, MCF-7, LS174T, and HCT116 cells by endogenous p53 (Fig. 1D, middle panel). Levels of actin were determined as an equal loading control (Fig. 1C and D, bottom panels). In contrast, the level of PolH protein was not increased in RKO-E6, MCF-7-E6, HCT116(p53−/−), and SW480 cells (Fig. 1D, middle panel).
If PolH is transcriptionally regulated by p53, one or more p53-responsive elements should exist in the PolH gene. To do this, we searched the genomic locus encoding PolH and found one potential responsive element (TCACTTGAGCTCGAGAAATCAAGGCT), which is located between −1992 and −1967 of the PolH gene (Fig. 1E). This sequence contains six mismatches in the noncritical positions within the consensus p53-binding site (19).
To determine whether p53 can transactivate gene expression via this element, a 337-bp DNA fragment containing the responsive element (from nucleotide −1992 to −1656) was cloned into a luciferase reporter vector, O-Fluc. The resulting vector was designated O-Fluc-PolH. The reporter vector was cotransfected into H1299 cells with either a pcDNA3 control vector or a vector that expresses p53 or p53(R175H). We found that the luciferase activity for O-Fluc-PolH was markedly increased by wild-type p53 but not by mutant p53(R175H) (Fig. 1F). When the key C and G were mutated, the luciferase activity by wild-type p53 was reduced.
We then performed a ChIP assay to determine whether p53 can directly bind to the p53 response element in vivo. Endogenous p53 in RKO cells was induced by treatment with 300 nM CPT for 24 h. p53-DNA complexes were immunoprecipitated with anti-p53 or anti-HA. To visualize the enriched DNA fragments, PCR was performed to amplify the region of the PolH promoter containing the p53 response element as well as the upstream p53-responsive element within the p21 promoter (Fig. 1G). We found that the fragment, which contains the p53 response element in the PolH promoter, was significantly increased upon induction of p53 (Fig. 1H, PolH RE panel), indicating that p53 directly binds to the p53 response element. No DNA fragment was enriched by the control antibody, anti-HA (Fig. 1H, PolH RE panel). As a positive control, we found that the fragment containing the p53 response element in the p21 promoter was also enriched after DNA damage (Fig. 1H, p21 RE1 panel).
Knockdown of PolH gives cells resistance to CPT- and IR-induced apoptosis.
Previously identified p53 target genes are either p53 functional mediators, such as p21, Bax, and Puma, or p53 functional regulators, such as Mdm2, COP1, and PML (6, 15, 17, 20, 58, 60, 80). If PolH is a p53 functional mediator, overexpression of PolH may exert an effect similar to that of p53, such as induction of cell cycle arrest and apoptosis. To test this, we generated H1299 cell lines that can inducibly express PolH and found that overexpression of PolH alone did not have any effect on cell proliferation (data not shown). We then generated H1299 cell lines that can inducibly express p53 and are simultaneously deficient in PolH by PolH siRNA. We found that PolH knockdown had no effect on the activity of exogenous wild-type p53 (data not shown). Thus, we conclude that PolH itself does not appear to mediate p53 to induce cell cycle arrest or apoptosis.
Since PolH is up-regulated by DNA damage in a p53-dependent manner (Fig. 1B), we wanted to examine whether PolH plays a role in the DNA damage response. It is well documented that p53 is a key mediator of the DNA damage response and that loss of p53 function markedly attenuates the induction of apoptosis by DNA damage (63, 67, 68). To this end, PolH siRNA was used to generate stable PolH knockdown RKO cell lines. Parental RKO and two representative PolH knockdown cell lines (RKO-PolH-KD#16 and -#70) are shown in Fig. 2A and 2B. PolH was up-regulated in the parental cells but not in PolH knockdown cells upon CPT treatment or IR (Fig. 2A and 2B, PolH panel, compare lanes 1 to 3 with 4 to 6, respectively). To determine whether PolH knockdown directly affects DNA damage-induced cell death, we performed a trypan blue dye exclusion assay. Upon treatment with 500 nM CPT, PolH knockdown cells were less prone to cell death than were the parental cells (Fig. 2C). To examine whether PolH knockdown differentially affects cell death induced by various DNA damage agents, we treated the cells with IR, which directly generates DSBs. We found that PolH knockdown cells were more resistant to IR-induced cell death than were the parental cells (Fig. 2C). To confirm that the decreased cell death by knockdown of PolH is due to a decrease in apoptosis, we performed a DNA histogram assay to measure the number of cells with sub-G1 DNA content. The sub-G1 fraction is believed to represent apoptotic cells. We found that DNA damage-induced apoptosis was significantly reduced by knockdown of PolH in RKO cells (Fig. 2D). To further demonstrate that PolH knockdown impairs the normal apoptotic pathway, we examined PARP cleavage. Cleaved PARP was detectable in the parental cells 24 h after treatment with 500 nM CPT, but only minimal in PolH knockdown cells (Fig. 2E, upper panel, lanes 4 to 6). At 36 after CPT treatment, although PARP was increasingly cleaved in PolH knockdown cells (Fig. 2E, upper panel, lanes 7 to 9), the cleaved PARP was less than that in the parental cells. PARP cleavage was also reduced in PolH knockdown cells upon IR (Fig. 2F, lanes 4 to 9).
FIG. 2.

Knockdown of PolH gives cells resistance to CPT- and IR-induced apoptosis. (A and B) PolH is knocked down in RKO-PolH-KD cells. RKO or RKO-PolH-KD cells were untreated or treated with 300 nM CPT for 6 h (A) or with 10 Gy of IR (B). p53 was detected with anti-p53 polyclonal antibody. Actin was also examined as a loading control. (C) RKO-PolH-KD cells are less sensitive than RKO to cell death induced by various DNA damage agents. RKO and RKO-PolH-KD were treated with 500 nM CPT for 12 h or 20 Gy of IR. Three days following treatment, both floating cells in the medium and attached cells on the plates were collected. Cells were stained with trypan blue dye for 15 min. Unstained cells (live cells) and stained cells (dead cells) were counted separately. (D) RKO-PolH-KD cells are more resistant than the parental RKO cells to CPT-induced apoptosis. Cells were treated as in panel C and then collected and stained with propidium iodide for DNA histogram analysis. (E and F) PARP cleavage is decreased in RKO-PolH-KD cells after treatment with various DNA damage agents. Both RKO and RKO-PolH-KD cells were treated with 500 nM CPT (E) or 20 Gy of IR (F). At indicated times after treatment, cells were collected and intact PARP, cleaved PARP, and PolH were detected by Western blot analysis. (G) PolH is knocked down in MCF-7-PolH-KD cells. MCF-7 and MCF-7-PolH-KD cells were treated with 300 nM CPT for 6 h. p53, PolH, and actin were detected by Western blot analysis. (H) MCF-7-PolH-KD cells are less sensitive than the parental MCF-7 cells to CPT-induced cell death. Cells were treated and the trypan blue dye exclusion assay was performed as in panel C. (I) PARP cleavage is decreased in MCF-7-PolH-KD cells after DNA damage. Cells were treated and Western blot analysis was performed as in panel E.
To confirm the data established in RKO colorectal carcinoma cells, we repeated the experiment in MCF-7 breast adenocarcinoma cells. First, we used siRNA to generate PolH knockdown MCF-7 cell lines. After treatment with CPT, p53 was activated in both parental and PolH knockdown MCF-7 cells (Fig. 2G, upper panel). Although PolH was induced by p53 in the parental cells, PolH was undetectable in PolH knockdown cells (Fig. 2G, middle panel). Next, we examined the DNA damage response of these cells upon treatment with CPT and found that PolH knockdown cells were more resistant to DNA damage-induced cell death than were the parental cells (Fig. 2H). We also found that PARP cleavage was markedly decreased in PolH knockdown cells (Fig. 2I, upper panel). Thus, we conclude that the resistance to DNA damage-induced apoptosis by PolH knockdown is not cell type specific.
Suppression of DNA damage-induced apoptosis by PolH knockdown is p53 dependent.
DNA damage triggers the intrinsic apoptotic pathway predominantly by activation of p53 and thereby induces apoptotic genes, such as Bax, Puma, and Noxa (58, 60, 62, 80). These apoptotic proteins subsequently induce caspase activation (47, 60, 80). Thus, it is likely that p53 is involved in the suppression of apoptosis by PolH knockdown. To test this, we used a p53-null H1299 cell line and siRNA technique to generate PolH knockdown H1299 cell lines. The resulting cell line was designated H1299-PolH-KD, and one representative clone (clone 56) is shown in Fig. 3A. To demonstrate that PolH siRNA is capable of suppressing endogenous and/or p53-induced PolH, exogenous p53 was transiently expressed in the parental cells and PolH knockdown cells (H1299-PolH-KD#56). We found that p21 was induced in these cells, suggesting that the exogenous p53 is functionally active (Fig. 3A, p53 and p21 panels, lanes 2 and 4). However, PolH was up-regulated by p53 in the parental cells but not in PolH knockdown cells (H1299-PolH-KD#56 cells) (Fig. 3A, PolH panel). Next, the DNA damage response was examined in these cells treated with 500 nM CPT by trypan blue dye exclusion assay. We found that approximately 8% of these treated cells died regardless of the PolH status (Fig. 3B). To confirm this observation, DNA histogram analysis was performed and showed that the extent of apoptosis in PolH knockdown cells (H1299-PolH-KD#56) was comparable to that in the parental H1299 cells when they were treated with CPT (Fig. 3C). These data further demonstrated that PolH knockdown does not affect p53-independent apoptosis and that apoptosis suppression by PolH knockdown is dependent on p53.
FIG. 3.

Suppression of DNA damage-induced apoptosis by PolH knockdown is p53 dependent. (A) PolH is knocked down in H1299-PolH-KD#56 cells. Both H1299 and H1299-PolH-KD#56 cells were transfected with either pcDNA3 or pcDNA3-p53 for 24 h. p53, PolH, p21, and actin were detected by Western blot analysis. (B) H1299 cells are as sensitive as H1299-PolH-KD cells to DNA damage-induced cell death. Cells were treated and the trypan blue dye exclusion assay was performed as in Fig. 2C. (C) The extent of apoptosis induced by DNA damage in H1299 cells and that induced in H1299-PolH-KD cells were comparable. Cells were treated and the DNA histogram assay was performed as in Fig. 2D.
Knockdown of PolH impairs p53 activation and activity upon CPT- and IR-induced DNA damage.
To determine the underlying mechanism by which PolH regulates p53-dependent apoptosis induced by DNA damage, the extent of p53 accumulation was determined and compared among various RKO cells that are proficient or deficient in PolH. When the parental and siRNA control cells were treated with 100 nM CPT for 6 h, p53 accumulated and, subsequently, PolH was up-regulated (Fig. 4A, middle panel, lanes 1, 2, 5, and 6). In contrast, in PolH knockdown cells, p53 accumulation was substantially decreased (Fig. 4A, middle panel, compare lanes 5 and 6 with lanes 7 and 8). This finding suggests that PolH knockdown attenuates p53 stabilization upon DNA damage. To rule out the possibility that p53 stabilization is simply caused by clonal variation, we generated stable inducible PolH knockdown RKO cell lines in which PolH siRNA is inducibly expressed by the tetracycline-regulated H1 promoter. One representative clone, RKO-pTer-PolH#82, is shown in Fig. 4B. Under the uninduced condition, DNA damage-induced PolH was detected in RKO cells (Fig. 4B, compare lane 1 with lanes 3 and 5), which is consistent with the data above. However, when PolH siRNA was induced by tetracycline, DNA damage-induced PolH was suppressed (Fig. 4B, compare lanes 3 and 5 with lanes 4 and 6). Using this inducible PolH knockdown cell line, we found that p53 stabilization by CPT was also markedly reduced when PolH was inducibly knocked down (Fig. 4B, middle panels, compare lanes 3 and 5 with lanes 4 and 6, respectively). Similarly, when PolH was inducibly knocked down, IR-induced p53 was also significantly reduced (Fig. 4C, p53 panel, compare lanes 3 and 4). These observations were confirmed in another inducible PolH knockdown cell line (RKO-pTer-PolH#90) (data not shown). To demonstrate that the impairment in p53 stabilization is not cell type specific, we examined p53 accumulation in the parental MCF-7 cells and PolH knockdown MCF-7 cells (MCF-7-PolH-KD#43 and -#56). Similarly, p53 stabilization was markedly decreased in PolH knockdown cells compared to that in the parental cells (Fig. 4D and data not shown). These data firmly established that p53 stabilization requires PolH.
FIG. 4.

Knockdown of PolH impairs p53 activation and activity upon DNA damage. (A) p53 activation is impaired in RKO-PolH-KD cells. RKO and its derivatives were treated with 100 nM CPT for 6 h. Cell extracts were collected, and p53, PolH, and actin were detected by Western blot analysis. (B) p53 activation is decreased by inducible PolH knockdown in RKO cells treated with CPT. RKO cells that were uninduced or induced to knock down PolH were treated with CPT at indicated concentrations for 6 h. Cells were collected, and p53, PolH, and actin were detected by Western blot analysis. (C) p53 activation is decreased by inducible PolH knockdown in RKO cells following exposure to IR. RKO cells that were uninduced or induced to knock down PolH were irradiated with 2 Gy of IR. At 4 h after IR, cells were collected, and p53, PolH, and actin were detected by Western blot analysis. (D) Impaired p53 activation by PolH knockdown is not cell type specific. MCF-7 and MCF-7-PolH-KD#43 cells were treated with 100 nM CPT for 6 h. Cell extracts were collected, and p53, PolH, and actin were detected by Western blot analysis. (E) p53 activation is decreased in PolH-null human fibroblasts following exposure to IR. Normal human fibroblasts (GM00495 and GM00024) and human XPV fibroblasts (GM03617 and GM02359) were irradiated with 2 Gy of IR. At 10 h following IR, cells were collected and p53, PolH, and actin were detected by Western blot analysis. (F) The induction of p21 and Fas by DNA damage is decreased in PolH knockdown cells. RKO and its derivatives were treated with 300 nM CPT for 6 h. The expression of p53, PolH, p21, Fas, and actin was examined by Western blot analysis. (G) The induction of DR5 and Mdm2 by DNA damage is decreased when PolH is knocked down. RKO cells that were uninduced or induced to knock down PolH were treated with 50 nM CPT for 6 h. Cells were collected, and DR5, Mdm2, and actin were detected by Western blot analysis. (H) The induction of DDB2 by DNA damage is decreased in PolH knockdown cells. RKO and its derivatives were treated with 100 nM CPT for 0, 6, and 9 h. The expression of DDB2 and GAPDH was examined by Northern blot analysis.
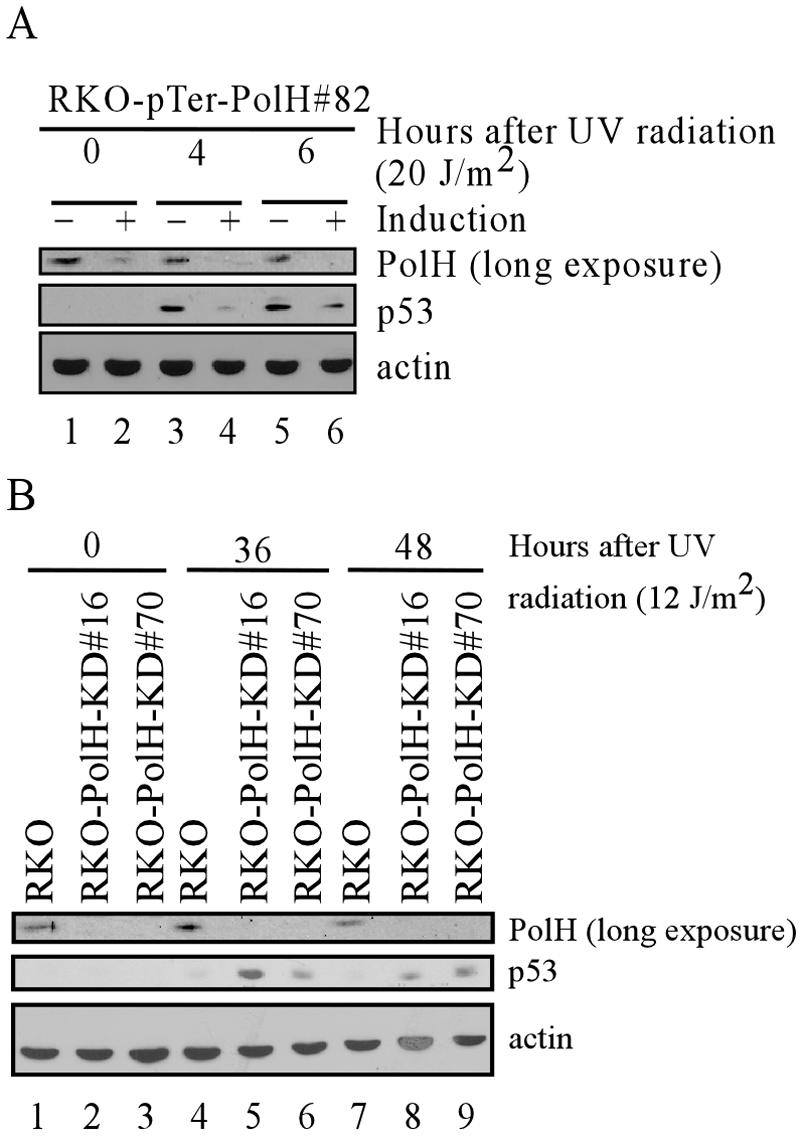
To rule out the possibility that the impaired p53 activation in PolH knockdown cells is simply caused by a defect in cancer cells, we compared the p53 stabilization by IR in human primary fibroblasts and primary fibroblasts derived from XPV patients. GM03617 and GM02359 fibroblasts carry a mutation in exon 2 and exon 10, respectively, in the PolH gene, which leads to translation of a severely truncated PolH protein (34). The normal fibroblasts, GM000495 and GM00024, were used as a control. We found that PolH was up-regulated by IR in normal fibroblasts (Fig. 4E, PolH panel, compare lanes 1 and 3 with 5 and 7). However, no PolH was detected in XPV fibroblasts either before or after IR (Fig. 4E, PolH panel, lanes 2, 4, 6, and 8). p53 was induced by IR in all four groups of fibroblasts. However, p53 activation in XPV cells was significantly less than that in normal cells (Fig. 4E, p53 panel, compare lanes 5 and 7 with 6 and 8). Thus, our findings demonstrated that PolH is essential for p53 activation in both normal and cancer cells upon DNA damage.
Next, we compared the induction of several well-defined p53 target genes in the parental and PolH knockdown RKO cells upon DNA damage. We found that the DNA damage-induced up-regulation of p21 and Fas was decreased in PolH knockdown cells compared to that in the parental cells, which correlates well with the impaired p53 activation in PolH knockdown cells (Fig. 4F). In addition, we examined the induction of DR5 and Mdm2 upon DNA damage in RKO cells uninduced or induced to knock down PolH. We found that induction of DR5 and Mdm2 was reduced in PolH knockdown cells (Fig. 4G). Fas and DR5 are major mediators of p53-dependent apoptosis. Thus, the decreased induction of Fas and DR5 may account for the resistance of PolH knockdown cells to DNA damage-induced apoptosis observed above.
Mutation of the DDB2 gene is responsible for XPE, and DDB2 has been shown to be up-regulated by p53 (29). Moreover, DDB2 deficiency impairs p53 activation (30). To examine whether p53 induction of DDB2 is affected by PolH, we measured the level of DDB2 transcripts. We found that DDB2 was up-regulated in both parental and PolH knockdown RKO cells following DNA damage (Fig. 4H). However, the extent of induction of DDB2 was decreased in PolH knockdown cells (Fig. 4H, compare lanes 4 and 7 with lanes 5, 6, 8, and 9).
Reconstitution of PolH in PolH knockdown cells restores p53 activation.
Since PolH knockdown impairs p53 activation upon DNA damage, overexpression of PolH might enhance p53 activation following DNA damage. However, we found that overexpression of PolH in RKO cells had no effect on p53 activation following DNA damage (data not shown). This suggests that endogenous PolH up-regulated by p53 may be sufficient for p53 activation. Thus, we reconstituted exogenous PolH, which is resistant to PolH siRNA degradation, into PolH knockdown cells (RKO-PolH-KD#16 and -#70). We found that the reconstituted PolH was able to restore p53 activation upon DNA damage (Fig. 5A and 5B, p53 panels, compare lane 3 with lane 4). However, the reconstituted PolH did not stabilize p53 in unstressed PolH knockdown cells (Fig. 5A and 5B, p53 panels, compare lane 1 with lane 2). These findings suggest that PolH is critical for facilitating DNA damage-induced p53 activation. To test this further, we examined the expression of p21 and Mdm2 and found that the induction of p21 and Mdm2 was increased after reconstitution of PolH (Fig. 5A and 5B, p21 and Mdm2 panels, lanes 3 and 4). Together, we conclude that the reconstitution of PolH in PolH knockdown cells is capable of increasing p53 activation and activity.
FIG. 5.
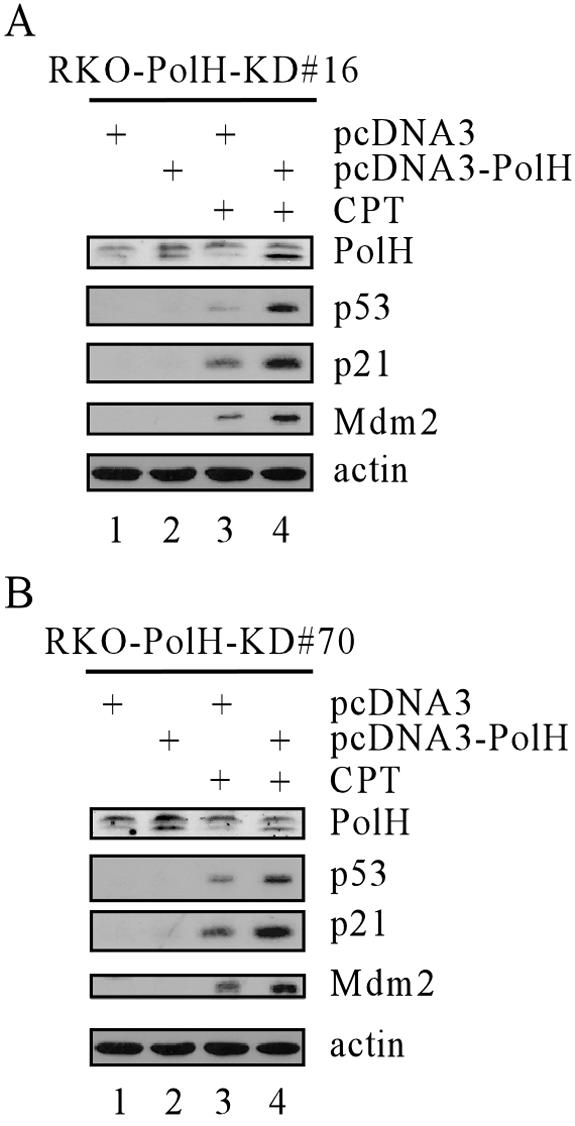
Reconstitution of PolH in PolH knockdown cells restores p53 activation and activities. RKO-PolH-KD#16 (A) or RKO-Pol-KD#70 (B) cells were transfected with either pcDNA3 or pcDNA3-PolH for 24 h. Cells were then untreated or treated with 100 nM CPT for 6 h. The expression of p53, PolH, p21, Mdm2, and actin was determined by Western blot analysis.
PolH knockdown impairs ATM's phosphorylation of Chk2 and p53 but not H2AX.
Since overexpression of PolH is unable to activate p53 in unstressed PolH-proficient and -deficient RKO cells, it is unlikely that PolH directly inhibits Mdm2-mediated p53 degradation. Thus, other mechanisms must be involved. It is well documented that, following DNA damage, serine 15 and serine 20 in p53 are subjected to phosphorylation by ATM and Chk2 (9, 27, 70, 75). This interrupts the association of Mdm2 and p53, thereby leading to p53 stabilization (3, 8, 52, 55, 57). A decrease in serine 15 phosphorylation has been shown to impair p53 activation (70, 75). To test this, we examined p53 phosphorylation upon CPT treatment in RKO cells uninduced or induced to knock down PolH. We found that both total p53 and serine 15-phosphorylated p53 were substantially decreased when PolH was inducibly knocked down, but the decrease in phosphorylation of serine 15 was more pronounced than that in total p53 (Fig. 6A, p53 and p-S15-p53 panels).
FIG. 6.

PolH knockdown impairs the ability of ATM to phosphorylate Chk2 and p53 but not H2AX. (A) RKO cells, which were uninduced or induced to knock down PolH, were treated with 100 nM CPT for 6 h. Cell extracts were then collected, and the level of PolH, p-S15-p53, p53, p-T68-Chk2, total Chk2, p-S1981-ATM, and actin was determined by Western blot analysis. (B) MCF-7 cells, which were uninduced or induced to knock down PolH, were treated with 2 Gy of IR. At 4 h after IR, cell extracts were then collected and the level of PolH, p-S15-p53, p53, p-T68-Chk2, total Chk2, p-S1981-ATM, and actin was determined by Western blot analysis. (C) Phosphorylation of p53 is decreased in human XPV fibroblasts. Fibroblasts were treated as in Fig. 4E. PolH, p53, p-S15-p53, and actin were determined by Western blot analysis. (D) Total p53 was normalized. The level of p53 and p-S15-p53 was determined by Western blot analysis. (E) Phosphorylation of H2AX is not affected by PolH knockdown. RKO cells, which were uninduced or induced to knock down PolH, were treated with CPT at indicated concentrations for 6 h. Phosphorylation of H2AX was determined by anti-γ-H2AX monoclonal antibody.
ATM and Chk2 are the major kinases responsible for p53 serine 15 phosphorylation in cells with DSBs (9, 27). When DSBs are generated in cells, ATM is induced to undergo autophosphorylation at serine 1981, which leads to ATM monomer formation and activation (4). ATM is able to phosphorylate Chk2 at threonine 68 and activate Chk2 (1, 54, 56). Thus, we examined the phosphorylation of ATM at serine 1981 and Chk2 at threonine 68. We found that phosphorylation of ATM was decreased only slightly by PolH knockdown upon DNA damage (Fig. 6A, p-S1981-ATM panel). However, the phosphorylation of Chk2 after DNA damage was substantially decreased in cells when PolH was inducibly knocked down whereas the total level of Chk2 remained unchanged (Fig. 6A, p-T68-Chk2 and Chk2 panels). These data strongly suggest that PolH knockdown affects the ability of ATM to phosphorylate Chk2 and p53. Defective phosphorylation of p53 then leads to failure in p53 stabilization and activation following DNA damage. To rule out cell type- and stress-specific effects of PolH knockdown on ATM activity, we also generated stable inducible PolH knockdown MCF-7 cell lines (Fig. 6B, PolH panel). p53, Chk2, and ATM phosphorylation was examined in these cells uninduced or induced to knock down PolH when exposed to IR. We found that, upon exposure to IR, p53 and Chk2 phosphorylation was significantly reduced whereas ATM phosphorylation was only marginally decreased (Fig. 6B, compare lanes 3 and 4). Furthermore, we examined p53 phosphorylation in normal human primary fibroblasts and XPV fibroblasts. We found that both total p53 and serine 15 phosphorylated p53 were substantially decreased in XPV fibroblasts, but the decrease in phosphorylation of serine 15 was more pronounced than that in total p53 (Fig. 6C, p53 and p-S15-p53 panels). To rule out the possibility that the decrease in p53 phosphorylation is simply caused by the reduction of the total p53 level in PolH knockdown cells, we examined p53 phosphorylation using equal amounts of total p53 from the parental and PolH-knockdown RKO cells. We found that phosphorylation of serine 15 decreased when PolH was knocked down, although the reduction is less dramatic than that prior to equalization of total p53 levels (Fig. 6D). Phosphorylation of another ATM substrate, H2AX, however, remained unaffected by PolH knockdown (Fig. 6E, middle panel). This finding indicates that PolH may preferentially influence ATM to phosphorylate selected substrates, such as Chk2 and p53.
PolH is partially colocalized with phosphorylated ATM at γ-H2AX foci after DNA damage.
Upon DNA damage, ATM is autophosphorylated to become active. The active ATM phosphorylates H2AX at the end of DSBs. γ-H2AX forms punctate particles termed γ-H2AX foci, which can recruit ATM substrates, such as Chk2 and p53, for phosphorylation (36). To explore the mechanism by which PolH affects ATM activity to phosphorylate Chk2 and p53 upon DNA damage, we examined the localization of phosphorylated ATM and PolH. In untreated RKO cells, the signals of γ-H2AX, phosphorylated ATM, and PolH were very low since H2AX and ATM were not activated and PolH was not up-regulated (Fig. 7A and C, − CPT panels). Upon CPT treatment, PolH was partially localized at γ-H2AX foci (Fig. 7A, + CPT panel). Phosphorylated ATM was also localized at γ-H2AX foci after CPT treatment (Fig. 7B). More importantly, phosphorylated ATM and PolH were partially colocalized at γ-H2AX foci (Fig. 7C, + CPT panel). These data suggest that PolH affects ATM activity to phosphorylate its substrate at γ-H2AX foci. However, we did not detect an interaction between phosphorylated ATM and PolH by immunoprecipitation assay (data not shown).
FIG. 7.

PolH is partially colocalized with phosphorylated ATM at γ-H2AX foci after DNA damage. (A) PolH is partially localized at γ-H2AX foci upon DNA damage. RKO cells were untreated or treated with 300 nM CPT for 6 h. Cells were then fixed with 4% formaldehyde in PBS, permeabilized with 0.5% Triton X-100 in PBS, and blocked with 5% BSA in PBS. Immunofluorescence staining was performed as described in Materials and Methods. PolH was detected with anti-PolH polyclonal antibody. γ-H2AX was detected with anti-γ-H2AX monoclonal antibody. Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole). (B) Phosphorylated ATM is localized at γ-H2AX foci upon DNA damage. RKO cells were treated as in panel A. p-S1981-ATM was detected with anti-p-S1981-ATM polyclonal antibody. γ-H2AX was detected with anti-γ-H2AX monoclonal antibody. (C) PolH is partially colocalized with phosphorylated ATM upon DNA damage. RKO cells were treated as in panel A. p-S1981-ATM was detected with anti-p-S1981-ATM polyclonal antibody. PolH was detected with anti-PolH monoclonal antibody.
Knockdown of PolH gives cells early resistance to UV-induced apoptosis but delays recovery from UV-induced DNA damage.
Upon UV exposure, DNA replication is stalled in XPV cells. As a result, stalled replication forks at the sites of CPDs and 6-4 photoproducts eventually collapse, which creates DSBs and subsequently causes cell death (48). This accounts for the sun sensitivity of XPV patients. We have found that knockdown of PolH gives RKO cells resistance to CPT- and IR-induced apoptosis. Thus, we wanted to investigate the response of PolH knockdown RKO cells to UV irradiation. We found that, within 12 h after 12 J/m2 of UV exposure, significantly more dead cells were detected in UV-irradiated parental cells than in UV-irradiated PolH knockdown cells (Fig. 8A), suggesting that PolH knockdown confers resistance to UV irradiation. We also found that, over a prolonged period following 12 J/m2 of UV exposure, the extent of apoptosis for UV-irradiated parental cells peaked at 24 h (16.3%) and gradually decreased over time (4.9% at 36 h and 2.5% at 48 h), suggesting that UV-induced damage was almost repaired at 48 h following UV exposure (Fig. 8A). In contrast, the extent of apoptosis for UV-irradiated PolH knockdown cells was increased in a time-dependent manner (Fig. 8A). This suggests that PolH knockdown suppresses DNA repair, which enhances UV-induced apoptosis over time. Next, we examined PARP cleavage in parental and PolH-KD RKO cells after UV irradiation. Cleaved PARP was detected in the parental RKO cells at 12 h post-UV irradiation, and its level was much higher than that in the PolH knockdown cells (Fig. 8B). However, over an extended period post-UV exposure, the level of cleaved PARP was higher in the PolH knockdown cells than in the parental cells (Fig. 8B). These data are consistent with the DNA histogram analysis and confirmed a delayed recovery from UV-induced damage in PolH knockdown cells.
FIG. 8.

Knockdown of PolH confers on cells early resistance to UV-induced apoptosis but defers recovery from UV-induced DNA damage. (A) PolH knockdown significantly decreased UV-induced apoptosis at early times but increased UV-induced apoptosis at late times. RKO cells that are proficient and deficient in PolH were radiated with 12 J/m2 of UV. At 0, 12, 24, 36, and 48 h after UV irradiation, cells were collected and fluorescence-activated cell sorting analysis was performed as in Fig. 2D to determine the ratio of apoptotic cells. (B) PARP cleavage is decreased in RKO-PolH-KD cells at early times but increased at late times after UV irradiation. RKO cells were treated as in panel A. Cells were collected, and uncleaved and cleaved PARP was detected with anti-PARP monoclonal antibody. (C) The parental and PolH knockdown RKO cells were treated as in panel A except that the cells were irradiated with 35 J/m2 of UV.
To examine whether PolH has any effect on the cellular response to high doses of UV irradiation, the extent of apoptosis was measured. We found that a stronger apoptotic response was detected in both parental cells and PolH knockdown cells irradiated with 35 J/m2 of UV than in cells irradiated with 12 J/m2 (Fig. 8C). Nevertheless, the extent of apoptosis was higher in UV-irradiated parental cells than in UV-irradiated PolH knockdown cells at the early stage within 24 h (Fig. 8C), suggesting that PolH knockdown confers resistance to UV irradiation regardless of UV dosage. At the late stage between 24 and 48 h, PolH knockdown made RKO cells more sensitive to UV-induced apoptosis (Fig. 8C). However, unlike exposure to low doses of UV, parental RKO cells were not able to recover from DNA damage following exposure to high doses of UV as irradiated cells continued to undergo apoptosis throughout the late stage (compare Fig. 8A and C).
Early resistance to UV-induced apoptosis by knockdown of PolH is p53 dependent.
UV induces apoptosis predominantly through activating p53. p53-deficient cells are resistant to UV-induced cell death (13). Above, we showed that suppression of CPT-induced apoptosis by PolH knockdown is p53 dependent (Fig. 4 to 6). It is likely that p53 is also involved in the early resistance of PolH knockdown cells to UV-induced cell death. To test this, p53-null H1299 cells and H1299-PolH KD cells, in which endogenous PolH was knocked down, were irradiated with 20 J/m2 of UV. At 12 h, 24 h, 48 h, and 72 h post-UV irradiation, dead cells were determined by trypan blue dye exclusion assay. We found that approximately 6% of cells died at 12 h following UV irradiation regardless of the PolH status (Fig. 9). This suggests that PolH knockdown does not affect p53-independent cell death at the early stage and that suppression of UV-induced early cell death by PolH knockdown in RKO and MCF-7 cells appears to be p53 dependent. For the UV-irradiated parental H1299, p53-independent cell death peaked at 48 h and started to decline. However, for the PolH knockdown H1299 cells, the extent of p53-independent cell death was increased in a time-dependent manner (Fig. 9), suggesting that PolH knockdown suppresses DNA repair and enhances UV-induced apoptosis.
FIG. 9.
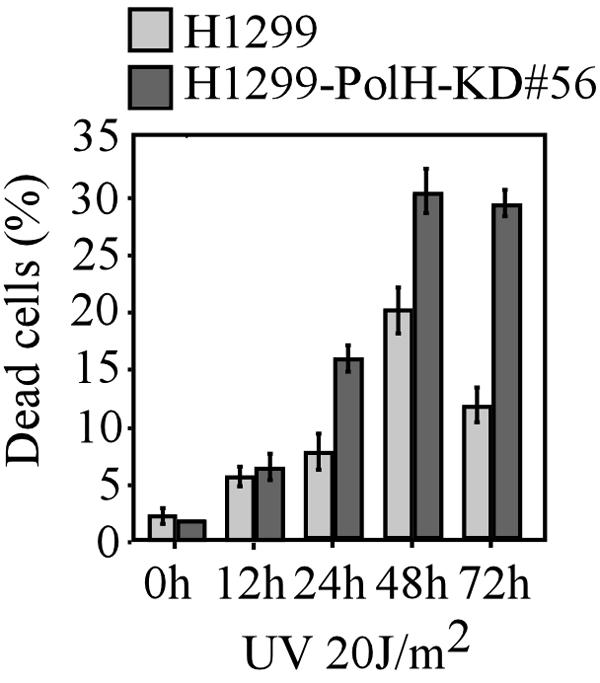
Early resistance to UV-induced apoptosis by knockdown of PolH is p53 dependent. H1299 and H1299-PolH-KD cells were irradiated with 20 J/m2 of UV. At the indicated time after UV irradiation, cells were collected and the trypan blue dye exclusion assay was performed to determine the ratio of dead cells.
p53 activation following UV irradiation is suppressed at the early time but enhanced at the late time by PolH knockdown.
To investigate the mechanism by which PolH knockdown confers cells early resistance to UV-induced cell death, we compared p53 activation by UV irradiation in RKO cells uninduced or induced to knock down PolH. At 4 and 6 h post-UV exposure, p53 stabilization was significantly reduced by PolH knockdown (Fig. 10A). This finding suggests that the impaired p53 activation by UV at the early time is responsible for the early resistance of PolH knockdown cells to UV-induced cell death. Furthermore, we found that, in contrast to CPT and IR, UV did not significantly up-regulate PolH (Fig. 10A).
FIG. 10.
PolH knockdown suppresses p53 activation at early times but enhances p53 activation at late times after UV irradiation. (A) p53 activation is decreased at early times after UV irradiation when PolH is knocked down. RKO cells that were uninduced or induced to knock down PolH were exposed to 20 J/m2 of UV. At the indicated times after UV exposure, cells were collected and PolH, p53, and actin were detected by Western blot analysis. (B) p53 activation is increased at late times after UV irradiation in PolH knockdown cells. The parental and PolH knockdown RKO cells were irradiated with 12 J/m2 of UV. At the indicated times, cells were collected and PolH, p53, and actin were detected by Western blot analysis.
Above, we showed that PolH knockdown delays cells' recovery from UV-induced DNA damage (Fig. 8). Thus, we wanted to determine the level of p53 over an extended period following UV irradiation in PolH-proficient and -deficient RKO cells. We found that, 36 and 48 h post-UV irradiation, the level of p53 was much higher in the PolH knockdown cells than in the parental cells (Fig. 10B). The high level of p53 in PolH knockdown cells is likely induced by excessive DSBs accumulated by the collapse of stalled replication forks over the extended period following UV irradiation (12). Therefore, the increased p53 activation contributes to the increased apoptotic response in PolH knockdown RKO cells at the late stage following UV irradiation.
DISCUSSION
PolH, a regulator of p53 activation but not a mediator of p53 growth suppression.
In this study, we found that PolH can be up-regulated by overexpressed p53. We also found that PolH is induced by DNA damage in a p53-dependent manner. Moreover, we identified a potential p53 response element in the promoter of PolH, and this element can be activated and bound by p53. All of these features suggest that PolH is a bona fide p53 target. Unexpectedly, when overexpressed in cells, PolH showed no effect on cell growth, suggesting that PolH alone is unable to induce cell cycle arrest and apoptosis. Next, we examined the possibility that PolH requires cooperation of other p53 targets to fulfill these functions. Thus, we generated dual inducible cell lines which can inducibly express p53 alone, PolH alone, or both (data not shown). We found that expression of PolH has no effect on the function of exogenous p53. We also generated cell lines that can inducibly express p53 and simultaneously are deficient in PolH. We found that knockdown of PolH has little, if any, effect on growth inhibition induced by exogenous p53. Taken together, we hypothesize that PolH is unlikely to be a functional mediator of p53 growth suppression.
PolH, a Y-family DNA polymerase, was identified as the product of the XPV gene (34, 53). XPV cells have normal NER but are deficient in the replication bypass of UV photoproducts and DNA adducts generated by various chemicals (66). However, although PolH is up-regulated by CPT treatment or IR, UV is unable to induce PolH. This suggests that p53, when stabilized by UV, does not play a role in TLS through PolH. Moreover, several studies showed that simian virus 40 T antigen- and human papillomavirus E6-transformed human fibroblasts are proficient in TLS (11, 44). Because p53 is defective in T antigen- or E6-transformed cells, we postulate that the basal level of PolH is sufficient for TLS. This prompts us to look for novel PolH functions. Quite surprisingly, we found that PolH knockdown gives cells resistance to apoptosis induced by DNA damage, which is p53 dependent. In addition, knockdown of PolH impairs p53 activation induced by DNA damage, and reconstitution of PolH into PolH knockdown cells restores p53 activation. Furthermore, we provide evidence that PolH regulates p53 activation by modulating ATM activity (see below for further discussion). To our knowledge, this is a novel function for PolH, and for the first time, we showed that a p53 target can positively affect p53 activation. Based on these observations, we propose a feed-forward model (Fig. 11): following DNA damage, PolH facilitates ATM to phosphorylate p53, leading to its stabilization and activation; p53 in turn up-regulates PolH expression; and finally elevated PolH enhances p53 activity via ATM and promotes p53-dependent apoptosis when DNA damage is irreparable.
FIG. 11.
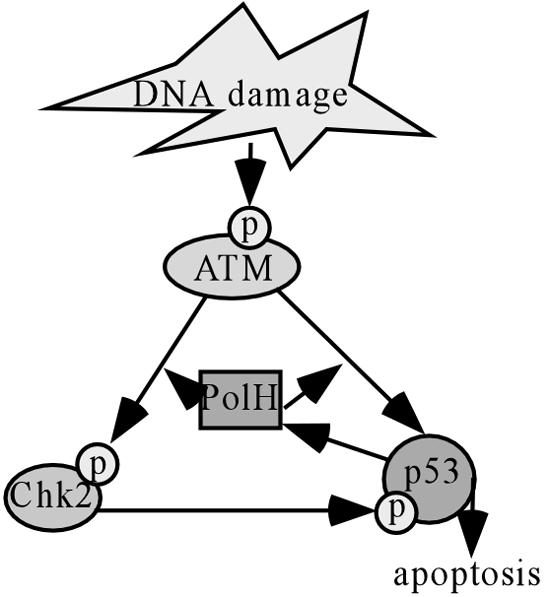
Schematic illustration of a feed-forward model for the role of PolH in ATM and p53 activation. Following DNA damage, PolH facilitates ATM's phosphorylation of p53, leading to its stabilization and activation; p53 in turn up-regulates PolH expression; and finally elevated PolH enhances p53 activity via ATM and promotes p53-dependent apoptosis when DNA damage is irreparable.
PolH and ATM activation and activity.
DNA damage, including DSBs, triggers ATM kinase activity (5, 41). However, the mechanism by which ATM is activated upon DNA damage remains unclear. An elegant study showed that ATM autophosphorylation at serine 1981 is induced by DNA damage, which leads to active monomer formation and ATM activation (4). Several BRCT domain-containing proteins, like 53BP1, NBS1, MDC1, and BRCA1, can affect ATM autophosphorylation and activation (16, 23, 28, 73). But none of these proteins are essential in ATM activation since ATM still undergoes autophosphorylation in the absence of each individual one, albeit decreased or delayed (16, 23, 28, 73). This indicates that redundant mechanisms participate in the regulation of ATM autophosphorylation. Furthermore, the specificity of ATM in phosphorylating its substrates is regulated by these BRCT-containing proteins (16, 23, 28). For example, phosphorylation of BRCA1 and SMC1, but not p53, is compromised in NBS cells (28). Here, we found that PolH is unable to regulate ATM autophosphorylation following DNA damage. However, PolH preferentially affects ATM to phosphorylate its substrates Chk2 and p53 but not H2AX. This suggests that PolH regulates ATM downstream of its activation. In addition, we provided evidence that PolH and phosphorylated ATM are colocalized at γ-H2AX foci after DNA damage, but we failed to detect an interaction between PolH and phosphorylated ATM. Thus, PolH may regulate ATM activity through its involvement in the interaction of phosphorylated ATM and some of its substrates at γ-H2AX foci. Thus, future studies are warranted to determine how PolH regulates ATM, which will provide an insight into the mechanism by which ATM is activated by DNA damage.
Interplay between PolH and ATR?
Following UV irradiation, normal DNA replication is stalled at replication forks. TLS polymerases, such as PolH, are then recruited (likely by PCNA) and take position to start the bypass replication (22, 37). Simultaneously, ATR is activated and initiates a phosphorylation cascade, including phosphorylation of p53, which eventually activates cell cycle checkpoints and apoptosis (69). How ATR is activated is under intensive investigation. Previous studies have shown that ATR activation requires association of ATR and ATRIP, RPA binding to single-strand DNA, Rad9-Hus1-Rad1 clamp complex loading to replication forks, and the assistance of claspin (64, 69). Because of the temporal and spatial similarity in TLS and ATR activation, it is not irrational to predict a mutual regulation of PolH functions and ATR activation. In fact, we found that PolH knockdown gives cells resistance to UV-induced apoptosis at early times, which is likely due to decreased p53 activation in PolH knockdown cells. ATR is the major kinase that phosphorylates and stabilizes p53 upon exposure to UV, which suggests that PolH directly regulates ATR activity. In addition, a recent study showed that a yeast Y-family polymerase, DinB, interacts with Hus1 and Rad1 (35). Thus, all of the evidence suggests that PolH plays a role in regulation of ATR activity.
The role of PolH in p53 activation—the physiological significance.
It is widely believed that multiple skin cancers in XPV patients are caused by error-prone TLS as the result of deficiency in error-free polymerase PolH (72). We have found that PolH participates in p53 activation by ATM after DNA damage. Since ATM and p53 are critical for maintaining genomic stability, we hypothesize that, in PolH-deficient cells, defective activation of p53 in response to DNA damage likely contributes to tumorigenesis. Thus, future studies to screen PolH mutations in human tumors may provide a clue about the role of PolH in other cancers in addition to its role in skin cancers.
Acknowledgments
We thank Cyrus Vaziri (Boston University) for the PolH cDNA construct and Wensheng Yan for the RKO tet-on cell line and Kelly Harms for the MCF-7 tet-on cell line.
This work was supported in part by an NIH grant (CA076069) from the National Cancer Institute and the Department of Defense Prostate Cancer Research Program under award number W81XWH-04-1-0079.
Views and opinions of, and endorsements by, the authors do not reflect those of the U.S. Army or the Department of Defense.
REFERENCES
- 1.Ahn, J. Y., J. K. Schwarz, H. Piwnica-Worms, and C. E. Canman. 2000. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 60:5934-5936. [PubMed] [Google Scholar]
- 2.Appella, E., and C. W. Anderson. 2001. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 268:2764-2772. [DOI] [PubMed] [Google Scholar]
- 3.Ashcroft, M., and K. H. Vousden. 1999. Regulation of p53 stability. Oncogene 18:7637-7643. [DOI] [PubMed] [Google Scholar]
- 4.Bakkenist, C. J., and M. B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499-506. [DOI] [PubMed] [Google Scholar]
- 5.Bakkenist, C. J., and M. B. Kastan. 2004. Initiating cellular stress responses. Cell 118:9-17. [DOI] [PubMed] [Google Scholar]
- 6.Barak, Y., T. Juven, R. Haffner, and M. Oren. 1993. mdm2 expression is induced by wild type p53 activity. EMBO J. 12:461-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berneburg, M., and A. R. Lehmann. 2001. Xeroderma pigmentosum and related disorders: defects in DNA repair and transcription. Adv. Genet. 43:71-102. [DOI] [PubMed] [Google Scholar]
- 8.Brooks, C. L., and W. Gu. 2003. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr. Opin. Cell Biol. 15:164-171. [DOI] [PubMed] [Google Scholar]
- 9.Chehab, N. H., A. Malikzay, M. Appel, and T. D. Halazonetis. 2000. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 14:278-288. [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, X., L. J. Ko, L. Jayaraman, and C. Prives. 1996. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 10:2438-2451. [DOI] [PubMed] [Google Scholar]
- 11.Cleaver, J. E., V. Afzal, L. Feeney, M. McDowell, W. Sadinski, J. P. Volpe, D. B. Busch, D. M. Coleman, D. W. Ziffer, Y. Yu, H. Nagasawa, and J. B. Little. 1999. Increased ultraviolet sensitivity and chromosomal instability related to P53 function in the xeroderma pigmentosum variant. Cancer Res. 59:1102-1108. [PubMed] [Google Scholar]
- 12.Cleaver, J. E., J. Bartholomew, D. Char, E. Crowley, L. Feeney, and C. L. Limoli. 2002. Polymerase eta and p53 jointly regulate cell survival, apoptosis and Mre11 recombination during S phase checkpoint arrest after UV irradiation. DNA Repair (Amsterdam) 1:41-57. [DOI] [PubMed] [Google Scholar]
- 13.Corbet, S. W., A. R. Clarke, S. Gledhill, and A. H. Wyllie. 1999. P53-dependent and -independent links between DNA-damage, apoptosis and mutation frequency in ES cells. Oncogene 18:1537-1544. [DOI] [PubMed] [Google Scholar]
- 14.de Boer, J., and J. H. Hoeijmakers. 2000. Nucleotide excision repair and human syndromes. Carcinogenesis 21:453-460. [DOI] [PubMed] [Google Scholar]
- 15.de Stanchina, E., E. Querido, M. Narita, R. V. Davuluri, P. P. Pandolfi, G. Ferbeyre, and S. W. Lowe. 2004. PML is a direct p53 target that modulates p53 effector functions. Mol. Cell 13:523-535. [DOI] [PubMed] [Google Scholar]
- 16.DiTullio, R. A., Jr., T. A. Mochan, M. Venere, J. Bartkova, M. Sehested, J. Bartek, and T. D. Halazonetis. 2002. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 4:998-1002. [DOI] [PubMed] [Google Scholar]
- 17.Dornan, D., I. Wertz, H. Shimizu, D. Arnott, G. D. Frantz, P. Dowd, K. O'Rourke, H. Koeppen, and V. M. Dixit. 2004. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429:86-92. [DOI] [PubMed] [Google Scholar]
- 18.el-Deiry, W. S. 1998. Regulation of p53 downstream genes. Semin. Cancer Biol. 8:345-357. [DOI] [PubMed] [Google Scholar]
- 19.el-Deiry, W. S., S. E. Kern, J. A. Pietenpol, K. W. Kinzler, and B. Vogelstein. 1992. Definition of a consensus binding site for p53. Nat. Genet. 1:45-49. [DOI] [PubMed] [Google Scholar]
- 20.el-Deiry, W. S., T. Tokino, V. E. Velculescu, D. B. Levy, R. Parsons, J. M. Trent, D. Lin, W. E. Mercer, K. W. Kinzler, and B. Vogelstein. 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75:817-825. [DOI] [PubMed] [Google Scholar]
- 21.Falck, J., J. Coates, and S. P. Jackson. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434:605-611. [DOI] [PubMed] [Google Scholar]
- 22.Fleck, O., and P. Schar. 2004. Translesion DNA synthesis: little fingers teach tolerance. Curr. Biol. 14:R389-R391. [DOI] [PubMed] [Google Scholar]
- 23.Foray, N., D. Marot, A. Gabriel, V. Randrianarison, A. M. Carr, M. Perricaudet, A. Ashworth, and P. Jeggo. 2003. A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J. 22:2860-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Girard, P. M., E. Riballo, A. C. Begg, A. Waugh, and P. A. Jeggo. 2002. Nbs1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene 21:4191-4199. [DOI] [PubMed] [Google Scholar]
- 25.Hansen, R., and M. Oren. 1997. p53; from inductive signal to cellular effect. Curr. Opin. Genet. Dev. 7:46-51. [DOI] [PubMed] [Google Scholar]
- 26.Harms, K., S. Nozell, and X. Chen. 2004. The common and distinct target genes of the p53 family transcription factors. Cell. Mol. Life Sci. 61:822-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirao, A., Y. Y. Kong, S. Matsuoka, A. Wakeham, J. Ruland, H. Yoshida, D. Liu, S. J. Elledge, and T. W. Mak. 2000. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287:1824-1827. [DOI] [PubMed] [Google Scholar]
- 28.Horejsi, Z., J. Falck, C. J. Bakkenist, M. B. Kastan, J. Lukas, and J. Bartek. 2004. Distinct functional domains of Nbs1 modulate the timing and magnitude of ATM activation after low doses of ionizing radiation. Oncogene 23:3122-3127. [DOI] [PubMed] [Google Scholar]
- 29.Hwang, B. J., J. M. Ford, P. C. Hanawalt, and G. Chu. 1999. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. USA 96:424-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itoh, T., C. O'Shea, and S. Linn. 2003. Impaired regulation of tumor suppressor p53 caused by mutations in the xeroderma pigmentosum DDB2 gene: mutual regulatory interactions between p48DDB2 and p53. Mol. Cell. Biol. 23:7540-7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jimenez, G. S., S. H. Khan, J. M. Stommel, and G. M. Wahl. 1999. p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene 18:7656-7665. [DOI] [PubMed] [Google Scholar]
- 32.Jin, S., and A. J. Levine. 2001. The p53 functional circuit. J. Cell Sci. 114:4139-4140. [DOI] [PubMed] [Google Scholar]
- 33.Johansen, F. E., and R. Prywes. 1994. Two pathways for serum regulation of the c-fos serum response element require specific sequence elements and a minimal domain of serum response factor. Mol. Cell. Biol. 14:5920-5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson, R. E., C. M. Kondratick, S. Prakash, and L. Prakash. 1999. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 285:263-265. [DOI] [PubMed] [Google Scholar]
- 35.Kai, M., and T. S. Wang. 2003. Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev. 17:64-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang, J., D. Ferguson, H. Song, C. Bassing, M. Eckersdorff, F. W. Alt, and Y. Xu. 2005. Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol. Cell. Biol. 25:661-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kannouche, P., and A. Stary. 2003. Xeroderma pigmentosum variant and error-prone DNA polymerases. Biochimie 85:1123-1132. [DOI] [PubMed] [Google Scholar]
- 38.Kastan, M. B., O. Onyekwere, D. Sidransky, B. Vogelstein, and R. W. Craig. 1991. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 51:6304-6311. [PubMed] [Google Scholar]
- 39.Ko, L. J., and C. Prives. 1996. p53: puzzle and paradigm. Genes Dev. 10:1054-1072. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi, J., A. Antoccia, H. Tauchi, S. Matsuura, and K. Komatsu. 2004. NBS1 and its functional role in the DNA damage response. DNA Repair (Amsterdam) 3:855-861. [DOI] [PubMed] [Google Scholar]
- 41.Kurz, E. U., and S. P. Lees-Miller. 2004. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair. (Amsterdam) 3:889-900. [DOI] [PubMed] [Google Scholar]
- 42.Lakin, N. D., and S. P. Jackson. 1999. Regulation of p53 in response to DNA damage. Oncogene 18:7644-7655. [DOI] [PubMed] [Google Scholar]
- 43.Lane, D. P. 1992. Cancer p53, guardian of the genome. Nature 358:15-16. [DOI] [PubMed] [Google Scholar]
- 44.Laposa, R. R., L. Feeney, and J. E. Cleaver. 2003. Recapitulation of the cellular xeroderma pigmentosum-variant phenotypes using short interfering RNA for DNA polymerase H. Cancer Res. 63:3909-3912. [PubMed] [Google Scholar]
- 45.Lee, J. H., and T. T. Paull. 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308:551-554. [DOI] [PubMed] [Google Scholar]
- 46.Lee, J. H., and T. T. Paull. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304:93-96. [DOI] [PubMed] [Google Scholar]
- 47.Levine, A. J. 1997. p53, the cellular gatekeeper for growth and division. Cell 88:323-331. [DOI] [PubMed] [Google Scholar]
- 48.Limoli, C. L., E. Giedzinski, W. M. Bonner, and J. E. Cleaver. 2002. UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA double-strand breaks, gamma-H2AX formation, and Mre11 relocalization. Proc. Natl. Acad. Sci. USA 99:233-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu, G., and X. Chen. 2002. The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene 21:7195-7204. [DOI] [PubMed] [Google Scholar]
- 50.Liu, G., S. Nozell, H. Xiao, and X. Chen. 2004. ΔNp73β is active in transactivation and growth suppression. Mol. Cell. Biol. 24:487-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu, G., T. Xia, and X. Chen. 2003. The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J. Biol. Chem. 278:17557-17565. [DOI] [PubMed] [Google Scholar]
- 52.Lohrum, M. A., and K. H. Vousden. 1999. Regulation and activation of p53 and its family members. Cell Death Differ. 6:1162-1168. [DOI] [PubMed] [Google Scholar]
- 53.Masutani, C., R. Kusumoto, A. Yamada, N. Dohmae, M. Yokoi, M. Yuasa, M. Araki, S. Iwai, K. Takio, and F. Hanaoka. 1999. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399:700-704. [DOI] [PubMed] [Google Scholar]
- 54.Matsuoka, S., G. Rotman, A. Ogawa, Y. Shiloh, K. Tamai, and S. J. Elledge. 2000. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 97:10389-10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meek, D. W. 1999. Mechanisms of switching on p53: a role for covalent modification? Oncogene 18:7666-7675. [DOI] [PubMed] [Google Scholar]
- 56.Melchionna, R., X. B. Chen, A. Blasina, and C. H. McGowan. 2000. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat. Cell Biol. 2:762-765. [DOI] [PubMed] [Google Scholar]
- 57.Michael, D., and M. Oren. 2003. The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 13:49-58. [DOI] [PubMed] [Google Scholar]
- 58.Miyashita, T., and J. C. Reed. 1995. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293-299. [DOI] [PubMed] [Google Scholar]
- 59.Nakada, D., Y. Hirano, and K. Sugimoto. 2004. Requirement of the Mre11 complex and exonuclease 1 for activation of the Mec1 signaling pathway. Mol. Cell. Biol. 24:10016-10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakano, K., and K. H. Vousden. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7:683-694. [DOI] [PubMed] [Google Scholar]
- 61.Nelson, W. G., and M. B. Kastan. 1994. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol. Cell. Biol. 14:1815-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053-1058. [DOI] [PubMed] [Google Scholar]
- 63.Oren, M. 1999. Regulation of the p53 tumor suppressor protein. J. Biol. Chem. 274:36031-36034. [DOI] [PubMed] [Google Scholar]
- 64.Parrilla-Castellar, E. R., S. J. Arlander, and L. Karnitz. 2004. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amsterdam) 3:1009-1014. [DOI] [PubMed] [Google Scholar]
- 65.Pichierri, P., and F. Rosselli. 2004. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 23:1178-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prakash, S., R. E. Johnson, M. T. Washington, L. Haracska, C. M. Kondratick, and L. Prakash. 2000. Role of yeast and human DNA polymerase eta in error-free replication of damaged DNA. Cold Spring Harbor Symp. Quant. Biol. 65:51-59. [DOI] [PubMed] [Google Scholar]
- 67.Prives, C., and P. A. Hall. 1999. The p53 pathway. J. Pathol. 187:112-126. [DOI] [PubMed] [Google Scholar]
- 68.Schmitt, C. A., J. S. Fridman, M. Yang, E. Baranov, R. M. Hoffman, and S. W. Lowe. 2002. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 1:289-298. [DOI] [PubMed] [Google Scholar]
- 69.Shechter, D., V. Costanzo, and J. Gautier. 2004. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amsterdam) 3:901-908. [DOI] [PubMed] [Google Scholar]
- 70.Shieh, S. Y., J. Ahn, K. Tamai, Y. Taya, and C. Prives. 2000. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 14:289-300. [PMC free article] [PubMed] [Google Scholar]
- 71.Slee, E. A., D. J. O'Connor, and X. Lu. 2004. To die or not to die: how does p53 decide? Oncogene 23:2809-2818. [DOI] [PubMed] [Google Scholar]
- 72.Stary, A., and A. Sarasin. 2002. Molecular mechanisms of UV-induced mutations as revealed by the study of DNA polymerase eta in human cells. Res. Microbiol. 153:441-445. [DOI] [PubMed] [Google Scholar]
- 73.Stucki, M., and S. P. Jackson. 2004. MDC1/NFBD1: a key regulator of the DNA damage response in higher eukaryotes. DNA Repair (Amsterdam) 3:953-957. [DOI] [PubMed] [Google Scholar]
- 74.Takata, H., Y. Tanaka, and A. Matsuura. 2005. Late S phase-specific recruitment of Mre11 complex triggers hierarchical assembly of telomere replication proteins in Saccharomyces cerevisiae. Mol. Cell 17:573-583. [DOI] [PubMed] [Google Scholar]
- 75.Tibbetts, R. S., K. M. Brumbaugh, J. M. Williams, J. N. Sarkaria, W. A. Cliby, S. Y. Shieh, Y. Taya, C. Prives, and R. T. Abraham. 1999. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13:152-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van de Wetering, M., I. Oving, V. Muncan, M. T. Pon Fong, H. Brantjes, D. van Leenen, F. C. Holstege, T. R. Brummelkamp, R. Agami, and H. Clevers. 2003. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 4:609-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vogelstein, B., D. Lane, and A. J. Levine. 2000. Surfing the p53 network. Nature 408:307-310. [DOI] [PubMed] [Google Scholar]
- 78.Wood, R. D. 1996. DNA repair in eukaryotes. Annu. Rev. Biochem. 65:135-167. [DOI] [PubMed] [Google Scholar]
- 79.You, Z., C. Chahwan, J. Bailis, T. Hunter, and P. Russell. 2005. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 25:5363-5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu, J., L. Zhang, P. M. Hwang, K. W. Kinzler, and B. Vogelstein. 2001. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 7:673-682. [DOI] [PubMed] [Google Scholar]
- 81.Zhu, J., W. Zhou, J. Jiang, and X. Chen. 1998. Identification of a novel p53 functional domain that is necessary for mediating apoptosis. J. Biol. Chem. 273:13030-13036. [DOI] [PubMed] [Google Scholar]