

Abstract
The role in virulence of the Shigella flexneri ospB-phoN2 operon has been evaluated. Here we confirm that OspB is an effector and show that apyrase, the product of phoN2, may be a virulence factor, since it is required for efficient intercellular spreading. Apyrase may be important in a deoxynucleoside triphosphate-hydrolyzing activity-independent manner, suggesting that it may act as an interaction partner in the process of IcsA localization.
ospB and phoN2 (apy) are genes located on the virulence plasmid (pINV) of Shigella species and of related enteroinvasive Escherichia coli (EIEC) strains (3, 6, 35). Indirect evidence indicates that ospB and phoN2 may contribute to pathogenicity (1, 3, 5, 8, 17, 23, 25, 35). ospB encodes a protein of unknown function (OspB) surmised to be an effector secreted by the type III secretion (TTS) apparatus of Shigella flexneri (6, 20). The two genes are organized in a single highly conserved bicistronic operon. Although phoN2 encodes a periplasmic protein (5), their expression is regulated by the VirF-VirB cascade (35) and by MxiE in concert with IpgC so that they are expressed in a VirB-dependent, MxiE-independent manner under conditions of nonactivated secretion and are up-regulated, in a MxiE-dependent manner, under conditions of activated secretion (8, 17, 20, 26, 30), indicating that these genes may be important in postinvasion events related to virulence.
phoN2 encodes apyrase, a periplasmic enzyme which belongs to the family of ATP-hydrolyzing enzymes (3, 5, 18) able to sequentially hydrolyze nucleoside triphosphates to diphosphates and then to monophosphates. However, it is not active against monophosphates, a distinguishing feature from acid phosphatases (1, 3, 41). Due to its catalytic activity and primary structure (apyrase presents an exposed N-terminal poly-proline sequence), it has been implicated in the decrease of host cell intracellular ATP, a characteristic metabolic event following S. flexneri infection, as well as in the S. flexneri-induced actin-polymerization process (1, 3, 5, 25, 35). Thus, these findings strongly suggest that OspB and apyrase may be virulence factors. The purpose of this study was to investigate the role of both genes in the mechanism of pathogenicity of S. flexneri.
OspB is an effector secreted by the TTS apparatus of S. flexneri.
To confirm and extend previous reports indicating OspB as an effector secreted by the TTS apparatus (6, 20), we transduced a nonpolar deletion encompassing mxiA into the S. flexneri M90T derivative strain HND549 (ospB::3×FLAG), thus generating HND5311 (mxiA ospB::3×FLAG) (Table 1). C-terminal 3× FLAG tagging was achieved essentially as described by Uzzau et al. (42) using the specific primer pair shown in Table S1 in the supplemental material. Whole-cell extracts and supernatants of exponentially growing bacteria were analyzed by immunoblotting using monoclonal antibodies against the 3× FLAG epitope (Sigma) and mouse polyclonal antibodies preparation directed against apyrase (Fig. 1). Apyrase antiserum was obtained by immunizing BALB/c mice with purified His6-tagged apyrase. Monoclonal antibodies against IpaB and IpaC (gifts of A. Phalipon) were used to monitor the activity of the TTS system (2, 27). In whole-cell extracts, a protein of the same size as 3× FLAG-tagged OspB was present in HND549 and HND5311 before exposure to Congo red, indicating that ospB is expressed even when the TTS system was not active. 3× FLAG-tagged OspB, IpaB, and IpaC proteins were found in the supernatant of the Congo red-induced HND549 strain but not in the mxiA mutant. Secretion was restored by complementing HN5311 with plasmid pHN111(PBADmxiA) only when expression of the cloned mxiA gene was induced by l-arabinose. As expected, being a periplasmic protein, apyrase was found only in whole-cell extracts. These results clearly demonstrate that OspB is secreted by the MxiA-Spa secretion apparatus and that an active TTS system is required for its secretion.
TABLE 1.
Bacterial strains and plasmidsa
| Strain or plasmid | Relevant genotype or characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| M90T | Wild-type S. flexneri serotype 5a | 32 |
| HND115 | M90T ΔphoN2; susceptible | This work |
| HND201 | M90T ΔospB; susceptible | This work |
| HND215 | M90T ΔospBphoN2; susceptible | This work |
| HND43 | M90T ΔmxiA; susceptible | This work |
| HND53 | M90T ΔvirB::aphA-3; Kmr | This work |
| HND549 | M90T ospB-3×FLAG; susceptible | This work |
| HND5311 | M90T ΔmxiA ospB-3×FLAG; susceptible | This work |
| SC560 | M90T ΔicsA::Ω; Smr | 10 |
| M15[pREP4] | E. coli K-12 strain carrying plasmid pREP4 | Qiagen |
| Plasmids | ||
| pGEM-T | Cloning vector, Apr | Promega |
| pBluescript SK | Cloning vector, Apr | Stratagene Inc., La Jolla, CA |
| pBAD28 | Arabinose-inducible PBAD expression vector, Apr Cmr | 13 |
| pSUB11 | Template plasmid carrying a 3×FLAG epitope and Kmr cassette | 42 |
| pKD46 | λ Red helper plasmid; oriR101 repA101(Ts) P-araB-gam-bet-exo Apr | 7 |
| pKD4 | Template plasmid carrying a Kmr gene with FLP recognition target sequence | 7 |
| pCP20 | FLP helper plasmid; pSC101 replicon (Ts) bla cat Flp (λRp) cI857 Apr Cmr | 7 |
| pREP4 | lacI, Kmr | Qiagen |
| pQE30 | Cloning vector; Apr | Qiagen |
| pHN301 | pQE30 carrying His6-phoN2; Apr | This work |
| pHN28 | pBAD28 carrying phoN2; Apr Cmr | This work |
| pHN111 | pBAD28 carrying mxiA; Apr Cmr | This work |
| pFB1-4 | pBluescript SK carrying a mutated phoN2 gene of EIEC strain HN280 presenting the R192P amino acid substitution | 36 |
Apr, ampicillin resistance; Cmr, cloramphenicol resistance; Kmr, kanamycin resistance.
FIG. 1.
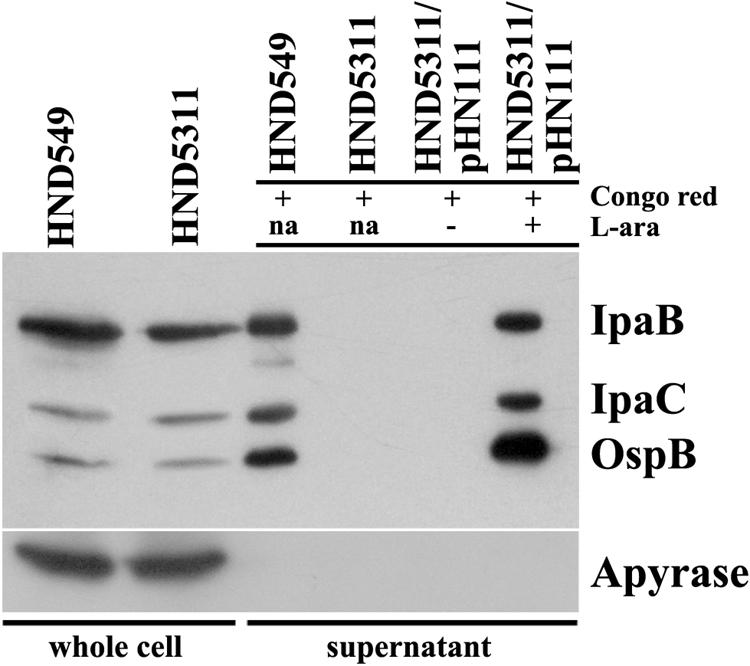
Secretion of OspB by the TTS machinery. Western blot analysis with anti-FLAG, anti-IpaB, anti-IpaC, and anti-apyrase antibodies of whole-cell extracts and culture supernatants of HND549 (ospB-3×FLAG), of HND5311 (mxiA ospB::3×FLAG), and of HND5311 [(mxiA ospB::3×FLAG)/pHN111(PBADmxiA)] in the absence (MxiA−) or in the presence (MxiA+) of 0.2% of the inducer l-arabinose (L-ara). A plus sign denotes culture supernatant extracts of bacteria resuspended in phosphate-buffered saline and incubated at 37°C for 30 min in the presence of 7 μg/ml of Congo red in order to induce activation of TTS-mediated secretion of effector proteins. The locations of 3× FLAG-tagged OspB recombinant protein as well as of IpaB, IpaC, and apyrase proteins are indicated. na, not added.
Characterization of the ospB, phoN2, and ospB phoN2 mutants.
Nonpolar deletions of ospB, phoN2, and of the entire ospB-phoN2 operon (HND201 [ospB], HND115 [phoN2], and HND205 [ospB-phoN2]) (Table 1) were introduced into S. flexneri strain M90T by the PCR-based gene disruption method reported by Datsenko and Wanner (7) to assess their role in virulence (Table S1 in the supplemental materials shows the primers used to generate mutants). Like the wild-type strain, all mutants were able to bind Congo red, produced contact-mediated hemolysis (34), and were able to synthesize and secrete IpaB and IpaC (data not shown), indicating that they all still have a functional TTS apparatus. Consistent with these observations, HND201, HND115, and HND205 were able to invade HeLa or Caco-2 cell monolayers (14) and to form protrusions as efficiently as the wild-type M90T (data not shown), indicating that they are proficient in host cell invasion and in cell-to-cell dissemination (17).
To determine whether intracellular secretion of Ipa proteins was affected, the ability of the three mutants to induce apoptosis in macrophages was evaluated by using the AnnexinV-PE Apoptosis Detection kit (Medical & Biological Laboratories Co., Ltd, Japan) and a Becton Dickinson FACScan flow cytometer (Becton Dickinson, La Jolla, CA). Data were analyzed with CellQuest (Becton Dickinson). J774 macrophages infected with the three mutants showed signs of the onset of apoptosis that were similar to those displayed by wild-type M90T (data not shown).
The death of macrophages infected with S. flexneri is preceded by a rapid loss of the intracellular levels of ATP, and apyrase has been implicated in this decrease (19, 25, 33). To ascertain whether phoN2 plays any role in this process, we infected nonconfluent monolayers of different cultured cells (J774 macrophages, HeLa cells, and Caco-2 cells) and measured intracellular ATP by using the ViaLight Plus kit (Cambrex), as recommended by the manufacturer. During the 3-h course of monolayer infections with wild-type M90T and with HND115 (phoN2), a similar sharp decrease of the intracellular ATP levels was induced in all cell lines used while cells infected with the noninvasive mutant HND53 (virB) (Table 1) maintained stable ATP levels indistinguishable from those of noninfected control cells (data not shown). These results clearly indicate that apyrase is not responsible for the decrease of the intracellular ATP pool, even though, as a periplasmic enzyme, it is likely that it affects to some minor extent the intracellular ATP/ADP levels, as these small molecules may passively diffuse through the outer membrane and enter the periplasmic space.
Next, mutants were tested for plaque formation on HeLa and Caco-2 cells (29). All three mutants gave rise to a number of plaques similar to that observed with the wild-type strain. However, in spite of their wild-type ability to invade cultured cells and to form protrusions, the size of the plaques formed by HND115 (phoN2) and HND205 (ospB-phoN2) was smaller than that of the plaques formed by M90T (after 48 and 72 h of incubation, the size of the plaques on HeLa and Caco-2 cells was reduced by approximately 65% and 45%, respectively), while those formed by HND201 (ospB) were indistinguishable from those of wild-type M90T (Fig. 2, which shows only plaques formed on Caco-2 cells, and Table 2). These results indicate that phoN2 expression, but not ospB expression, is required for wild-type cell-to-cell spreading. To determine whether the small plaques observed were indeed caused by the lack of apyrase, we performed complementation tests by introducing pHN28(PBADphoN2) into both strains and by adding the inducer l-arabinose (0.2% final concentration) within the agarose layer. Under these conditions the plaque size increased to an extent similar to that observed in wild-type M90T-infected cells (Fig. 2, Table 2), indicating that the absence of apyrase is responsible for the dissemination defect of HND115 and HND205.
FIG. 2.

phoN2 mutant strain HND115 forms small plaques on Caco-2 cell monolayers. Plaque formation by wild-type strain M90T, HND115 (phoN2), and HND115/pHN28(PBADphoN2) supplemented with 0.2% of the inducer l-arabinose (L-ara) (apyrase+) within the agarose layer is shown. Dishes were photographed after 48 h of infection.
TABLE 2.
Formation of plaques on confluent monolayers of HeLa and Caco-2 cellsa
| Strain | Description | HeLa at 72 h (mm) | Caco-2 at 48 h (mm) |
|---|---|---|---|
| M90T | Wild type | 1.2 | 2.5 |
| HND115 | phoN2 | 0.4 | 1.35 |
| HND115/pHN28 | phoN2/PBADphoN2 | 0.45 | 1.35 |
| HND115/pHN28 | phoN2/PBADphoN2 + l-arabinose | 0.9 | 1.9 |
| HND115/pFB1-4 | phoN2/pBluescript SKapyR192P | 0.95 | 2.15 |
| HND201 | ospB | 1.3 | 2.35 |
| HND205 | ospB-phoN2 | 0.45 | 1.35 |
| HND205/pHN28 | ospB-phoN2/PBADphoN2 | 0.45 | 1.3 |
| HND205/pHN28 | ospB-phoN2/PBADphoN2 + l-arabinose | 0.9 | 1.95 |
Monolayers infected by the various strains were overlaid with gentamicin-containing soft agar supplemented with 0.2% l-arabinose or not, incubated for 48 h or 72 h at 37°C, fixed, and stained with Giemsa. The diameter of the plaques are indicated in millimeters and is the average of at least 10 plaques.
Previous studies have shown that mutations of genes involved in the biosynthesis of the lipopolysaccharide (LPS) of S. flexneri affect the establishment or the maintenance of IcsA at one pole of the bacterium, resulting in defective cell-to-cell spread (31, 38, 43). To determine whether the small-plaque phenotype could be due to altered LPS biosynthesis, LPS preparations of M90T, HND115, and HND205 were prepared as described by Hitchcock and Brown (16), and aliquots were subjected to tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis and visualized by silver staining (21, 40). Examination of silver-stained gels showed that the mutants produce the typical LPS structure (a complete core and repeating O-antigen subunits) seen in the wild-type M90T strain (data not shown), thus ruling out that the inability to produce wild-type plaques is due to LPS alterations.
To evaluate whether the inability of the phoN2 mutants to produce wild-type plaques could have resulted from a growth defect in the intracellular environment, we compared the growth of HND115 and HND205 to that of the wild-type strain M90T in broth cultures and inside HeLa cells. In Luria-Bertani medium there was no difference between the optical density of M90T and that of HND115 and HND205, and the multiplication kinetics of both strains within the HeLa cell cytoplasm were similar to that of the wild type (data not shown), indicating that the failure to form full-size plaques is not due to a reduced intracellular growth rate.
Taken together, these data indicate that while phoN2 is required for maximal S. flexneri cell-to-cell spread, neither ospB nor phoN2 is required for invasion, intracellular multiplication, induction of macrophage apoptosis, decrease of intracellular ATP levels, or LPS biosynthesis. The finding that no specific function related to virulence was found for ospB indicates that the virulence assays used in this study were probably not suited to uncover specific postinvasion events influenced by this gene. This issue is the subject of ongoing investigation.
Role of phoN2 in the surface localization of IcsA.
Plaque formation is a complex phenotype that measures several distinct aspects of the virulence of S. flexneri, including its ability to invade, to multiply, and to move intra- and intercellularly. Genetic defects in one or more of the above processes may reflect reduced-size plaques. The ability of S. flexneri to move within the cytoplasm of infected cells and to spread to adjacent cells relies entirely on the production and unipolar localization of IcsA on the bacterial surface (4, 12, 22, 24). IcsA is inserted into the outer membrane as a 120-kDa protein which is subsequently cleaved by IcsP (SopA), releasing IcsA*, a 95-kDa secreted polypeptide into culture supernatants (11). To determine whether HND115 (phoN2) was able to produce both the membrane-bound IcsA and the related IcsA*, whole cells and culture supernatants of HND115 and of wild-type strain M90T were analyzed by Western blotting using an anti-IcsA polyclonal antiserum (gift of L. Bernardini), as previously described (9, 11). Equal amounts of IcsA and IcsA* were detected in whole-cell extracts and in culture supernatants of HND115 and of the wild-type strain, while these proteins were absent from the control icsA mutant strain SC560 (Fig. 3). These results indicate that the phoN2 mutation does not influence the delivery of IcsA to the bacterial surface and its subsequent cleavage.
FIG. 3.
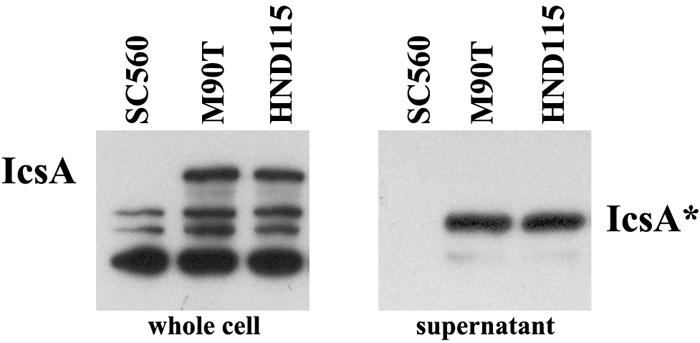
IcsA expression and secretion in phoN2 mutant strain HND115. Western analysis with anti-IcsA antibodies of whole-cell extracts and culture supernatants of wild-type M90T and of its derivatives SC560 (icsA) and HND115 (phoN2). IcsA and IcsA* are indicated.
We next wanted to determine whether the small-plaque phenotype was due to an aberrant localization of IcsA on the bacterial surface. To this end, by immunolabeling with anti-IcsA antiserum, we analyzed the localization of IcsA at the surface of exponentially grown HND115, HND115/pHN28(PBADphoN2), and wild-type M90T strains (Fig. 4). IcsA was detected at the bacterial surface of approximately 50% of wild-type M90T and of 20% of HND115, but its distribution on the surface of HND115 was distinctly different from the typical unipolar distribution seen on the wild-type strain. On average, more than 90% of M90T bacteria presenting IcsA on the bacterial surface showed IcsA localized exclusively at the pole, while the great majority (90%) of labeled HND115 cells presented IcsA distributed over the entire bacterial surface, albeit with some polar reinforcement. Polar localization of IcsA on HND115 cells was noticed only in less than 10% of the bacterial population, and in these cases the intensity of staining was considerably reduced compared to that of the wild-type strain. Introduction of pHN28 into HND115 restored unipolar localization of IcsA only if bacteria were grown in the presence of 0.2% l-arabinose. In these experimental conditions, approximately 45% of the bacterial cells presented IcsA on the bacterial surface, and of these more than 85% showed IcsA localized at the pole, indicating that a functional phoN2 gene is required for polar localization of IcsA. Thus, the altered surface distribution of IcsA on HND115 may account for the small-plaque phenotype produced by this strain. As expected, HND205 (ospB-phoN2) behaved in the same way as HND115, while HND201 (ospB) presented a wild-type unipolar IcsA localization (data not shown), indicating that ospB, although transcriptionally linked to phoN2, is not required for proper IcsA localization.
FIG. 4.

Indirect immunofluorescence labeling of IcsA. Panels show IcsA localization using an anti-IcsA antiserum in wild-type strain M90T, in phoN2 deletion strain HND115, and in HND115/pHN28(PBADphoN2) and their corresponding phase-contrast images. In the panels of M90T and HND115/pHN28(PBADphoN2) supplemented with the inducer l-arabinose (L-ara) (PhoN2+), arrows point to representative bacteria exhibiting polar IcsA localization, while in HND115 (apyrase−) arrows point to bacteria presenting IcsA apparently localized over the entire bacterial surface.
The phoN2 mutant forms altered actin tails.
To determine whether the absence of apyrase impaired the ability to form actin tails, we examined by staining with tetramethyl rhodamine isothiocyanate-phalloidin the ability of HND115 to form polar actin tails and to accumulate polymerized actin in infected HeLa cells. Wild-type M90T-infected cells displayed long polar-orientated actin tails with no staining along the sides of the bacterium (Fig. 5). In contrast, actin polymerization by HND115 frequently extended far up the lateral sides of the bacterium, a phenotype that was rarely detected with M90T. A population of HND115 appeared to be entirely surrounded by actin filaments, possibly because of IcsA being distributed over the entire surface of the bacterium. Other bacteria did form actin tails, but they were not as focused as those formed by the wild-type strain and extended over the pole of the bacterium. Representative examples are shown in Fig. 5. The alteration in actin polymerization displayed by the intracellular phoN2 mutant HND115 is consistent with the altered distribution of IcsA on the surface of HND115 bacteria (Fig. 4) and may reduce the overall efficiency of intercellular spreading of HND115. Partial restoration of wild-type actin tail patterns was obtained by complementing HND115 with pHN28(PBADphoN2) in the presence of 0.2% l-arabinose only (Fig. 5). As expected, HND205 (ospB-phoN2) produced alterations of actin polymerization indistinguishable from those produced by HND115 (alterations partially complemented by the introduction of pHN28), while HND201 (ospB) produced wild-type actin tails (data not shown). These results are in accordance with the localization of IcsA reported above and further strengthen our finding that phoN2 may be required for S. flexneri motility within host cells.
FIG. 5.

Fluorescence images of actin tail formations inside HeLa cells infected by phoN2 deletion strain HND115. HeLa cells were infected with wild-type strain M90T, HND115, or HND115/pHN28(PBADphoN2) in the presence of 0.2% of the inducer l-arabinose (L-ara) (apyrase+). Actin was stained with rhodamine-conjugated phalloidin. Nuclei and bacterial DNA were counterstained in blue (4′,6′-diamidino-2-phenylindole). Images were recorded with a Leica camera and processed using Qwin software (Leica). Arrows in M90T and in HND115/pHN28 (apyrase+) panels point to representative bacteria exhibiting normal actin tail formation at a pole of a bacterium moving within the cytoplasm. Arrows in the HND115 (apyrase−) panel point to representative bacteria impaired in their ability to form normal actin tails (altered phenotype).
The deoxynucleoside triphosphate (dNTP)-hydrolyzing activity of phoN2 is not necessary for wild-type cell-to-cell spread.
Although the process of IcsA localization has not been fully elucidated (15), the current model suggests that, once transported across the inner membrane, IcsA directly targets the old pole where it anchors to the outer membrane by its β-domain. Successively, it diffuses laterally in the membrane where the exposed passenger domain is cleaved by IcsP at all sites, leading to the typical unipolar distribution (15). Accordingly, IcsA translocation across the outer membrane does not appear to depend on energy coupling (neither ATP nor GTP molecules are present in the periplasm) but rather on the correct folding of the passenger domain (12, 15, 37). Thus, it is unlikely that apyrase may influence IcsA localization by virtue of its ability to hydrolyze dNTP and deoxynucleoside diphosphate molecules. To test the importance of the catalytic activity of apyrase and thus to get insights into the mechanism of action, we transformed plasmid pFB1-4 (Table 1) into HND115 and assayed for plaque formation on HeLa and Caco-2 cell monolayers. pFB1-4 is pBluescript SK vector carrying a randomly generated mutant of the apy gene, the orthologue of phoN2 in the EIEC strain HN280 (35) which encodes a recombinant apyrase presenting a single amino acid substitution (R192P) located within the putative active site which inactivates its dNTP-hydrolyzing activity (3, 36). We used pFB1-4 to assess the role of the catalytic activity of apyrase on the ability of cell-to-cell spreading of S. flexneri. As evidenced by Western immunoblotting, the introduction of pFB1-4 into HND115 induces the synthesis of the recombinant apyrase to an extent similar to that of the wild-type apyrase produced by M90T (Fig. 6A). Under these experimental conditions, while the introduction of pFB1-4 into M90T does not affect plaque size (data not shown) the introduction of pFB1-4 into HND115 increases plaque size to an extent similar to that observed with cells infected with wild-type strain M90T (Fig. 6B, which shows only plaques formed on Caco-2 cells, and Table 2) or with HND115 complemented with pHN28(PBADphoN2) (Fig. 2, Table 2). As expected, the introduction of pFB1-4 into HND115 also restored the polar localization of IcsA (Fig. S1 of the supplemental material). These results clearly indicate that the catalytic activity of apyrase is not necessary for wild-type cell-to-cell spread and for polar localization of IcsA on the S. flexneri surface.
FIG. 6.

phoN2 mutant strain HND115 forms plaques of approximately wild-type size on Caco-2 cell monolayers when complemented with plasmid pFB1-4. pFB1-4 (Table 1) contains a randomly generated mutant of the apy gene, the orthologue of phoN2 in the EIEC strain HN280, encoding a recombinant apyrase presenting the R192P substitution which inactivates its dNTP-hydrolyzing activity. (A) Western blot analysis with anti-apyrase antibodies of whole-cell extracts of M90T, HND115 (phoN2), and HND115/pFB1-4 (pBluescript SKapyR192P). (B) Plaques formed by M90T, HND115 (phoN2), and HND115/pFB1-4 (pBluescript SKapyR192P). Dishes were photographed after 48 h of infection.
Although more studies are needed to fully clarify the mechanism of action of apyrase, it is worth noting that, as in the case of ActA of Listeria monocytogenes, apyrase possesses an exposed poly-proline sequence, ILPPPPAE, at its N-terminal domain (1, 28). Proline-rich motifs have been reported to be often involved in protein-protein interactions and to be essential for the virulence of various intracellular pathogens (28, 39). Although the role of the proline-rich motif of apyrase is unknown, considering that apyrase does not influence intercellular spreading and IcsA targeting by virtue of its catalytic activity, we would like to speculate that it may act as an interaction partner in protein-protein interactions, likely with protein(s) directly or indirectly involved in the polar localization of IcsA. Preliminary copurification experiments, performed using 3× FLAG-tagged apyrase, failed to detect interactions between apyrase and IcsA (M. Nicoletti, personal communication), suggesting that either the two proteins do not interact directly or that a weaker protein-protein interaction exists.
Future work will precisely assess the function of apyrase. However, the discovery that apyrase is involved in actin-based motility of S. flexneri adds knowledge and provides a new and useful key to the understanding of the complex and not fully elucidated mechanism of unipolar IcsA localization.
Supplementary Material
Acknowledgments
We thank M. L. Bernardini and A. Phalipon for the gift of anti-IcsA, anti-IpaB, and IpaC antibodies, respectively, and G. Micheli for critical reading of the manuscript. We are indebted to C. Fiorentini and A. Fabbri for help in flow cytometry experiments. We also thank A. Calconi for expert technical assistance.
This work was supported by MURST PRIN projects “Espressione di determinanti di virulenza batterica in relazione a segnali ambientali e dell'ospite” and “Effettori di virulenza in patogeni enterici: caratteristiche e studio delle loro interazioni,” granted to M.N., and by Faculty 60% funds granted to M.N. and to F.B.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Babu, M. M., S. Kalamalakkannan, Y. V. B. K. Subrahmanyam, and K. Sankaran. 2002. Shigella apyrase-a novel variant of bacterial acid phosphatases? FEBS Lett. 512:8-12. [DOI] [PubMed] [Google Scholar]
- 2.Baharani, F. K., P. J. Sansonetti, and C. Parsot. 1997. Secretion of Ipa proteins by Shigella flexneri: inducer molecules and kinetics of activations. Infect. Immun. 65:4005-4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berlutti, F., M. Casalino, C. Zagaglia, P. A. Fradiani, P. Visca, and M. Nicoletti. 1998. Expression of the virulence plasmid-carried apyrase gene (apy) of enteroinvasive Escherichia coli and Shigella flexneri is under the control of H-NS and the VirF and VirB regulatory cascade. Infect. Immun. 66:4957-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernardini, M. L., J. Mounier, H. d'Hauteville, M. Coquis-Rondon, and P. J. Sansonetti. 1989. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interactions with F-actin. Proc. Natl. Acad. Sci. USA 86:3867-3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhargava, T., S. Datta, V. Ramakrishnan, R. K. Roy, K. Sankaran, and Y. V. B. K. Subrahmanyam. 1995. Virulent Shigella codes for a soluble apyrase: identification, characterization and cloning of the gene. Curr. Sci. 68:293-300. [Google Scholar]
- 6.Buchrieser, C., P. Glaser, C. Rusniok, H. Nedjari, H. d'Hauteville, F. Kunst, P. J. Sansonetti, and C. Parsot. 2000. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 38:760-771. [DOI] [PubMed] [Google Scholar]
- 7.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demers, B., P. J. Sansonetti, and C. Parsot. 1998. Induction of type III secretion in Shigella flexneri is associated with different control of transcription of genes encoding secreted proteins. EMBO J. 17:2894-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.d'Hauteville, H., R. Dufourcq-Lagelouse, F. Nato, and P. J. Sansonetti. 1996. Lack of cleavage of IcsA in Shigella flexneri causes aberrant movement and allow demonstration of a cross-reactive eukaryotic protein. Infect. Immun. 64:511-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.d'Hauteville, H., and P. J. Sansonetti. 1992. Phosphorylation of IcsA by cAMP-dependent protein kinase and its effect on intracellular spread of Shigella flexneri. Mol. Microbiol. 6:833-841. [DOI] [PubMed] [Google Scholar]
- 11.Egile, C., H. d'Hauteville, C. Parsot, and P. J. Sansonetti. 1997. SopA, the outer membrane protease responsible for polar localization of IcsA in Shigella flexneri. Mol. Microbiol. 23:1063-1073. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg, M. B., O. Barzu, C. Parsot, and P. J. Sansonetti. 1993. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. J. Bacteriol. 175:2189-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hale, T. L., and S. B. Formal. 1981. Protein synthesis in HeLa or Henle 407 cells infected with Shigella dysenteriae 1, Shigella flexneri 2a, or Salmonella typhimurium W118. Infect. Immun. 32:137-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson, I. R., F. Navarro-Garcia, M. Desvaux, R. C. Fernandez, and D. Ala'Aldeen. 2004. Type V protein secretion pathway: the autotransporter story. Microbiol. Mol. Biol. Rev. 68:692-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hitchcock, P. J., and T. M. Brown. 1983. Morphological enterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 154:269-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kane, C. D., R. Schuch, W. A. Day, and A. T. Maurelli. 2002. MxiE regulates intracellular expression of factor secreted by the Shigella flexneri 2a type III secretion system. J. Bacteriol. 184:4409-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komoszynki, M., and A. Wojtczak. 1996. Apyrase (ATP diphosphohydrolase, EC 3.6.1.5): function and relationship with ATPases. Biochim. Biophys. Acta 1310:233-241. [DOI] [PubMed] [Google Scholar]
- 19.Koterski, J. F., M. Nahvi, M. M. Venkatasen, and B. Haimovich. 2005. Virulent Shigella flexneri causes damage to mitochondria and triggers necrosis in infected human monocyte-derived macrophages. Infect. Immun. 73:504-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Gall, T., M. Mavris, M. C. Martino, M. L. Bernardini, E. Denamour, and C. Parsot. 2005. Analysis of virulence plasmid gene expression defines three classes of effectors in the type III secretion system of Shigella flexneri. Microbiology 151:951-962. [DOI] [PubMed] [Google Scholar]
- 21.Lesse, A. J., A. A. Campagnari, W. E. Bittner, and M. A. Apicella. 1990. Increased resolution of lipopolysaccharides and lipooligosaccharides utilizing tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis. J. Immunol. Methods 126:109-117. [DOI] [PubMed] [Google Scholar]
- 22.Lett, M. C., C. Sasakawa, N. Okada, T. Sakai, S. Makino, M. Yamada, K. Komatsu, and M. Yoshikawa. 1989. virG, a plasmid-coded virulence gene of Shigella flexneri: identification of the virG protein and determination of the complete coding sequence. J. Bacteriol. 171:353-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucchini, S., H. Liu, Q. Jin, J. C. D. Hinton, and J. Yu. 2005. Transcriptional adaptation of Shigella flexneri during infection of macrophages and epithelial cells: insights into the strategies of a cytosolic bacterial pathogen. Infect. Immun. 73:88-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makino, S., C. Sasakawa, K. Kamata, T. Kurata, and M. Yoshikawa. 1986. A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in Shigella flexneri 2a. Cell 46:551-555. [DOI] [PubMed] [Google Scholar]
- 25.Mantis, N., M. C. Prevost, and P. J. Sansonetti. 1996. Analysis of epithelial cell stress response during infection by Shigella flexneri. Infect. Immun. 64:2474-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mavris, M., P. J. Sansonetti, and C. Parsot. 2002. Identification of the cis-acting site involved in activation of promoters regulated by activity of the type III secretion apparatus in Shigella flexneri. J. Bacteriol. 184:6751-6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menard, R., P. J. Sansonetti, and C. Parsot. 1994. The secretion of the Shigella flexneri Ipa invasins is activated by epithelial cells and controlled by IpaB and IpaD. EMBO J. 13:5293-5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neibuhr, K., F. Ebel, R. Frank, M. Reinhard, E. Domann, U. D. Carl, U. Walter, F. B. Gertier, J. Weheland, and T. Chakraborty. 1997. A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASp family. EMBO J. 16:5433-5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oaks, E. V., M. E. Wingfield, and S. B. Formal. 1985. Plaque formation by virulent Shigella flexneri. Infect. Immun. 48:124-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Penno, C., P. J. Sansonetti, and C. Parsot. 2005. Frameshifting by transcriptional slippage is involved in production of MxiE, the transcriptional activator regulated by the activity of the type III secretion apparatus in Shigella flexneri. Mol. Microbiol. 56:204-214. [DOI] [PubMed] [Google Scholar]
- 31.Sadlin, R. C., K. A. Lampel, S. P. Keasler, M. B. Goldberg, A. L. Stolzer, and A. T. Maurelli. 1995. Avirulence of rough mutants of Shigella flexneri: requirement of O antigen for correct unipolar localization of IcsA in the bacterial outer membrane. Infect. Immun. 63:229-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sansonetti, P. J., D. J. Kopecko, and S. B. Formal. 1982. Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect. Immun. 35:852-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sansonetti, P. J., and J. Mounier. 1987. Metabolic events mediating early killing of host cells infected by Shigella flexneri. Microb. Pathogens 3:53-61. [DOI] [PubMed] [Google Scholar]
- 34.Sansonetti, P. J., A. Ryter, P. Clerc, A. T. Maurelli, and J. Mounier. 1986. Multiplication of Shigella flexneri within HeLa cells: lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect. Immun. 51:461-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santapaola, D., M. Casalino, A. Petrucca, C. Presutti, C. Zagaglia, F. Berlutti, B. Colonna, and M. Nicoletti. 2002. Enteroinvasive Escherichia coli virulence plasmid-carried apyrase (apy) and ospB genes are organized as a bicistronic operon and are subjected to differential expression. Microbiology 148:2519-2529. [DOI] [PubMed] [Google Scholar]
- 36.Sarli, S., M. Nicoletti, S. Schippa, F. Del Chierico, D. Santapaola, P. Valenti, and F. Berlutti. 2005. Ala160 and His116 residues are involved in activity and specificity of apyrase, an ATP-hydrolyzing enzyme produced by enteroinvasive Escherichia coli. Microbiology 151:2853-2860. [DOI] [PubMed] [Google Scholar]
- 37.Steinhauer, J., R. Agha, T. Pham, A. W. Varga, and M. B. Goldberg. 1999. The unipolar Shigella surface protein IcsA is targeted directly to the bacterial old pole: IcsP cleavage of IcsA occurs over the entire bacterial surface. Mol. Microbiol. 32:367-377. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki, T., H. Miki, T. Takenawa, and C. Sasakawa. 1998. Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 17:2767-2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki, T., and C. Sasakawa. 2001. Molecular basis of the intracellular spreading of Shigella. Infect. Immun. 69:5959-5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai, C., and C. D. Frasch. 1982. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem. 119:115-119. [DOI] [PubMed] [Google Scholar]
- 41.Uchiya, K. I., M. Tohsuji, T. Nikai, H. Sugihara, and C. Sasakawa. 1996. Identification and characterization of phoN-Sf, a gene on the large plasmid of Shigella flexneri 2a encoding a nonspecific phosphatase. J. Bacteriol. 178:4548-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uzzau, S., N. Figueroa-Bossi, S. Rubino, and L. Bossi. 2001. Epitome tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. USA 98:15264-15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Den Bosch, L., P. A. Manning, and R. Morona. 1997. Regulation of O-antigen chain length is required for Shigella flexneri virulence. Mol. Microbiol. 23:765-775. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.