

Abstract
Xanthomonas campestris pv. phaseoli OhrR belongs to a major family of multiple-cysteine-containing bacterial organic hydroperoxide sensors and transcription repressors. Site-directed mutagenesis and subsequent in vivo functional analyses revealed that changing any cysteine residue to serine did not alter the ability of OhrR to bind to the P1 ohrR-ohr promoter but drastically affected the organic hydroperoxide-sensing and response mechanisms of the protein. Xanthomonas OhrR requires two cysteine residues, C22 and C127, to sense and respond to organic hydroperoxides. Analysis of the free thiol groups in wild-type and mutant OhrRs under reducing and oxidizing conditions indicates that C22 is the organic hydroperoxide-sensing residue. Exposure to organic hydroperoxides led to the formation of an unstable OhrR-C22 sulfenic acid intermediate that could be trapped by 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole and detected by UV-visible spectral analysis in an oxidized C127S-C131S mutant OhrR. In wild-type OhrR, the cysteine sulfenic acid intermediate rapidly reacts with the thiol group of C127, forming a disulfide bond. The high-performance liquid chromatography-mass spectrometry analysis of tryptic fragments of alkylated, oxidized OhrR and nonreducing polyacrylamide gel electrophoresis analyses confirmed the formation of reversible intersubunit disulfide bonds between C22 and C127. Oxidation of OhrR led to cross-linking of two OhrR monomers, resulting in the inactivation of its repressor function. Evidence presented here provides insight into a new organic hydroperoxide-sensing and response mechanism for OhrRs of the multiple-cysteine family, the primary bacterial transcription regulator of the organic hydroperoxide stress response.
Xanthomonas campestris strains comprise a group of soil bacteria and plant pathogens. In the environment and during plant-microbe interactions, Xanthomonas spp. are exposed to reactive oxygen species including H2O2, lipid hydroperoxide, and superoxide anions generated as by-products of aerobic metabolism, from exposure to chemicals in the environment, and from plant active defense responses (6, 11).
Organic hydroperoxides are highly toxic due to their ability to directly oxidize macromolecules and participate in the generation of reactive lipid radicals. In many bacteria, organic hydroperoxide metabolism remains poorly characterized. In Xanthomonas and other bacteria, the best-characterized organic hydroperoxide detoxification systems involve peroxiredoxins, such as alkyl hydroperoxide reductase (AhpR), and organic hydroperoxide resistance (Ohr) thiol-dependent peroxidases (2, 4, 10, 18). Both enzymes directly catalyze the reduction of organic peroxides to less toxic organic alcohols. AhpR and Ohr have similar biochemical actions, although they differ in their physiological roles and gene expression patterns. The expression of ahpC (the peroxidatic component of AhpR) is regulated by OxyR, a peroxide sensor and transcription regulator (13, 21), whereas ohr is controlled by the organic peroxide-inducible transcription repressor OhrR (4, 15, 22).
The ability to sense and respond to changes in peroxide levels is crucial for bacterial survival under peroxide stress. Both OxyR and OhrR are involved in the sensing of organic hydroperoxide, but the latter is probably more sensitive to changes in organic hydroperoxide levels than the former. Oxidation of OxyR leads to changes in its structure and function from a transcription repressor, as in reduced OxyR, to an activator, as in oxidized OxyR. Reduced OhrR binds to its target site and represses gene expression, while organic hydroperoxide-dependent oxidation of OhrR results in the inactivation of its repressor function, resulting in transcription from the target promoter. Mechanisms by which Bacillus subtilis OhrR senses and is inactivated by organic hydroperoxide have been postulated, and they involve the oxidation of a sensing cysteine by hydroperoxide to a protein-cysteine sulfenic acid (C-SOH) that inactivates the repressor (5).
Here, we have classified OhrRs into two families based on the number of cysteine residues they contain. X. campestris pv. phaseoli OhrR belongs to the major family that contains multiple cysteine residues. In addition, we present evidence for a novel organic hydroperoxide-sensing and response mechanism for this major family of OhrRs.
MATERIALS AND METHODS
Materials.
Bacterial culture media were obtained from Difco. Restriction endonucleases, DNA modification enzymes, and isopropyl-1-thio-β-d-galactopyranoside were purchased from Promega. Organic solvents (high-pressure liquid chromatography [HPLC] grade) and water (optima grade) were obtained from Fisher. Acrylamide-bis (40%) solution was obtained from Bio-Rad. Cumene hydroperoxide, 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NDB chloride), and 4-vinylpyridine were obtained from Aldrich. l-1-(Tosylamino)-2-phenylethyl chloromethyl ketone-treated trypsin was obtained from Worthington. Pierce Biotechnology, Inc., supplied the immobilized Tris(2-carboxyethyl)phosphine disulfide-reducing gel, Gel Code Blue stain, and trifluoroacetic acid ampules. All other chemicals and antibiotics were purchased from Sigma. Purified proteins were concentrated using Millipore YM10 regenerated cellulose ultrafiltration membranes. The Amersham Biosciences Heparin FF, Q-Sepharose, and Sephadex 200 columns, connected to an Amersham Biosciences Explorer 10S Air fast performance liquid chromatography system, were used for protein purification. Hewlett-Packard HP-8452 and Beckman DU 7500 diode array spectrophotometers were used for spectroscopic measurements.
Bacterial strains and growth conditions.
All X. campestris strains were grown aerobically in Silva Buddenhagen medium (0.5% peptone, 0.5% yeast extract, 0.5% sucrose, and 0.1% glutamic acid, pH 7.0) containing the appropriate antibiotics at 28°C. Antibiotics were used at the following final concentrations: 15 μg ml−1 tetracycline, 30 μg ml−1 kanamycin, and 15 μg ml−1 gentamicin. The organic hydroperoxide induction experiments were done with exponential-phase X. campestris pv. phaseoli cultures (optical density at 600 nm of 0.6) treated with 100 μM cumene hydroperoxide (CHP) for 30 min.
β-Galactosidase assay.
Crude bacterial cleared lysates were generated as previously described (17). Cleared lysates were used for total protein determination and enzyme assays. β-Galactosidase was determined as described previously (17).
Site-directed mutagenesis of OhrR.
X. campestris. pv. phaseoli mutant OhrR proteins C127S, C131S, C22S-C127S, C22S-C131S, C127S-C131S, and C22S-C127S-C131S were constructed using a PCR-based site-directed mutagenesis method using primers complementary to the coding and noncoding sequences of the template OhrR but containing the desired mismatch to change a given C codon to S, as previously described (17). In order to generate the mutant C127S, the mutagenic forward primer BT355 (5′-CAGCTGTTTTCGGCATCGGC-3′), M13-Reverse and M13-Forward primers, and a mutagenic reverse primer, BT356 (5′-GCCGATGCCGAAAACACCTG-3′), were used in a PCR with pBBRohrR (17) as a DNA template. PCR products were digested with EcoRI plus SacI and cloned into the broad-host-range expression plasmid vector pBBR1 MCS-5 (8). The sequence of the mutated DNA was verified using an automated DNA sequencer. A similar protocol using different pairs of primers was used to produce other OhrR mutants. To generate the C131S mutation, the forward and complementary reverse primers used in the PCR were BT357 (5′-GCATCGGCCTCGTCGTTGGAC-3′) and BT358 (5′-GTCCAACGACGAGGCCGCCGCTGC-3′), respectively. C22S-C127S and C22S-C131S mutants were created using OhrR C22S plasmid (17) as the DNA template amplified with the BT355-BT356 and BT357-BT358 primer pairs, respectively. To create C127S-C131S and C22S-C127S-C131S mutants, BT357-BT358 primers were used to amplify OhrR C127S and C22S-C127S plasmids, respectively.
Purification of OhrR wild-type and mutant proteins.
The ohrR coding region was amplified from pBBRohrR with primers BT377 (5′-ATTCTCGAGTCCCGCGCCAAGGCT-3′) and BT378 (5′-CGAATTCGCCGATGGTCCC-3′), and the 590-bp NcoI- and XhoI-digested PCR product was ligated into the expression vector pETBlue-2 (Novagen), digested with the same enzymes, to create pETohrR. This created OhrR with a His tag fusion (two additional amino acid residues [leucine and glutamic acid] from the cloning vector and six histidine residues) with a calculated monomer molecular weight of 18,000.65. A similar protocol using different DNA templates was used to generate pETC22S and pETC127S,C131S for the high expression of OhrR(C22S) and OhrR(C127S-C131S), respectively. The PCR fragment sequence was confirmed by DNA sequencing. Escherichia coli BL21(DE3)/pLacI (Novagen) harboring pETohrR, pETC22S, or pETC127S,131S was cultured in a 10-liter BioFlo 2000 Biofermentor (New Brunswick Scientific) containing LB broth plus 50 μg ml−1 ampicillin at 37°C until the optical density at 600 nm reached 0.8. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce OhrR expression. The culture was grown for an additional 2 h before the cells were harvested by centrifugation at 5,000 × g for 10 min at 4°C. The bacterial pellet was disrupted using a Bead Beater (BioSpec Products). All steps of OhrR purification were performed at 4°C. The crude extract was subsequently treated with 2.5% (wt/vol) streptomycin sulfate to precipitate nucleic acids prior to being subjected to 20% and 65% (NH4)2SO4 precipitations. The 20% to 65% (NH4)2SO4 pellet was suspended in resuspension buffer (20 mM Tris, pH 8.0, 1 mM EDTA, pH 8.0, 5% glycerol, 0.1 mM phenylmethylsulfonyl fluoride, 2 mM dithiothreitol [DTT], and 25 mM NaCl) and applied to a HiPrep 16/10 Heparin FF column (Amersham Bioscience). Bound proteins were eluted with an NaCl gradient (0.025 to 1.0 M). OhrR in eluted fractions was identified using 15% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). Fractions containing OhrR were pooled and loaded onto a Q-Sepharose column (Amersham Bioscience). Bound proteins were eluted with the same NaCl gradient as described above. Fractions containing OhrR were pooled and concentrated by ultrafiltration using an Amico Ultra-10 centrifugal filter from Millipore. The purity of protein samples was determined using 15% SDS-PAGE. Finally, purified protein was aliquoted and stored at −20°C.
Molecular mass determination of native OhrR.
The apparent molecular mass of the native OhrR was determined by gel filtration on a Superdex 75 HR 10/30 column connected to a fast protein liquid chromatography system (Pharmacia Acta Purifier), equilibrated, and eluted in buffer A (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 50% glycerol, and 2.0 mM DTT). The column was calibrated with independent runs of the following markers (Bio-Rad): thyroglobin (670 kDa), γ-globulin (155 kDa), ovalbumin (43 kDa), myoglobin (18 kDa), and vitamin B12 (1.35 kDa). The elution of protein was monitored by the absorbance at 280 nm.
Mass spectrometric analysis.
Oxidized OhrR protein was prepared by treating purified protein with an equivalent concentration of CHP. Protein samples were extensively dialyzed in deionized water (6 liters) in a Slide-A-Lyzer cassette (Pierce) prior to analysis by electrospray ionization (ESI)-mass spectrometry (MS) (Quattro II triple quadrupole mass spectrometer equipped with a Z-spray source; Micromass, Manchester, United Kingdom) precalibrated with horse heart myoglobin. The protein sample (1 μM), in 50% acetonitrile and 1% formic acid, was injected at a flow rate of 300 μl/h, and positively charged ions in the m/z range of 800 to 1,800 were analyzed using MassLynx software (version 3.5; Micromass).
Trypsin digestion of OhrR.
Prior to performing trypsin digestion, the sulfhydryl groups in OhrR were modified by several different methods using 4-vinylpyridine, N-ethylmaleimide (NEM), or 2-bromoethylamine and NEM. In the case of 4-vinylpyridine treatments, reduced and oxidized OhrR samples (10 nmol) in MES buffer [250 mM of 2-(N-morpholino)ethanesulfonic acid (MES), pH 6.5, and 1 mM EDTA] were treated with 100 mM 4-vinylpyridine under denaturing conditions (8 M urea). In the case of 2-bromoethylamine treatments, protein samples (10 nmol) in TE buffer (100 mM Tris, pH 8.0, and 1 mM EDTA) were incubated overnight with 100 mM 2-bromoethylamine (pH 8.0) at room temperature under denaturing conditions (8 M urea). The free cysteine (R-CH2-SH) residues were alkylated to form S-2-aminoethylcysteines (R-CH2-S-CH2CH2NH3+) that are susceptible to trypsin digestion. In order to drop the pH of the mixture and promote S− formation on free cysteine residues, an equal volume of MES buffer was added to the reaction mixture. Blocking of unmodified cysteine residues was performed by the addition of 100 μM NEM and incubation at room temperature for 90 min. After dialysis and solvent removal, exhaustive trypsin digestion of OhrR, in either oxidized or reduced (DTT-treated) form, was carried out by incubation at an enzyme-to-substrate ratio of 1:60 at 37°C for 24 h. Trypsin digestion was carried out at pH 6.5 to minimize disulfide exchange (20).
Tryptic peptide separation using HPLC and mass spectrometric analysis.
Tryptic digest samples were analyzed by injection into a Hewlett-Packard 1100 HPLC system equipped with a 2.1- by 250-mm Vydac C18 column from which 15% of the eluant was directly injected into the mass spectrometer. Peptides were eluted with a 70-min gradient consisting of 0 to 100% solvent B in solvent A (0.05% trifluoroacetic acid in deionized, ultrapure H2O [solvent B was 100% acetonitrile with 0.04% trifluoroacetic acid in H2O]). The molecular mass of all fragments was determined by ESI-MS.
Protein sulfenic acid trapping with NBD chloride.
The formation of C-SOH as a reaction intermediate in the cysteine-dependent oxidation of wild-type and the mutant OhrRs C127S-C131S and C22S was detected by labeling these proteins with NBD chloride as previously described (3), with some modifications. NBD chloride reacts with both thiol and sulfenic acid forms of cysteine in proteins to form thioether (R-S-NBD) or sulfoxide [R-S(O)-NBD] products that can be distinguished by their UV-visible spectra with maxima at 420 nm and 347 nm, respectively. Each protein (60 μM) was treated with DTT (2 mM) for 1 h. Excess DTT was removed by ultrafiltration. Various proteins in phosphate buffer (pH 7.0) were stored in an equal volume of immobilized Tris(2-carboxyethyl)phosphine gel, an efficient reductant of disulfides over a wide pH range that is readily removed from protein samples just prior to analyses. Oxidized protein was prepared by treating the purified protein with an equivalent amount of CHP and incubating the protein at room temperature for 10 min. Both reduced and oxidized proteins were then treated with NBD chloride (20 equivalents) under denaturing conditions (4 M guanidine HCl) for 5 min at room temperature. TE buffer (pH 7.0) was added to the solution to give a final concentration of 2.0 M guanidine HCl. Excess NBD chloride was removed by ultrafiltration using an Apollo 7-ml high-performance centrifugal concentrator (Orbital Biosciences, Topsfield, MA), and the absorbance spectra (200 to 600 nm) of the protein samples were measured on a Beckman (Fullerton, CA) DU 7500 diode array spectrophotometer.
DTNB assay.
The free thiol content of reduced and oxidized wild-type and mutant OhrRs was determined using the DTNB (5,5′-dithiobis-2-nitrobenzoic acid) assay. First, excess DTT in purified protein samples was removed by ultrafiltration. The samples (10 μM) were then incubated with 100 μM DTNB in thiol assay buffer [0.1 M (NH4)2SO4, 0.05 M Tris, pH 8.0, 0.5 mM EDTA, pH 8.0] under denaturing conditions (4 M guanidine HCl). The 2-nitro-5-thiobenzoic acid generated by the reaction was detected by its absorbance at 412 nm (ɛ = 14,150 M−1 cm−1) (3).
Nonreducing SDS-PAGE.
In order to identify the disulfide linkages in reduced and oxidized OhrR, nonreducing SDS-PAGE was performed. Reduced protein was prepared by treating the purified OhrR (0.3 nmol) with 20 equivalents of DTT, and then excess DTT was removed by ultrafiltration. Oxidized OhrR (0.3 nmol) was prepared by treating the reduced protein with 1 equivalent of CHP for 10 min. The NEM-treated oxidized protein was prepared by treating the oxidized OhrR (0.3 nmol) with 100 μM of NEM. Protein samples were then subjected to electrophoresis on an SDS-polyacrylamide gel as described previously (9), with some modifications (23).
RESULTS
Two families of OhrR.
In bacteria, OhrR is the major sensor and regulator of organic hydroperoxide stress (5, 14, 17). Multiple alignments of OhrR deduced amino acid sequences from both gram-positive and gram-negative bacteria revealed an interesting pattern (Fig. 1). In all OhrRs, there was a highly conserved amino-terminal cysteine residue that corresponded to C22 of X. campestris pv. phaseoli and C15 of B. subtilis OhrR. This cysteine residue has been shown to be important for the repressor to respond to organic hydroperoxide in vivo and in vitro (5, 17). In B. subtilis, C15 has been shown to be the organic hydroperoxide-sensing residue that becomes oxidized by organic hydroperoxide, resulting in the inactivation of the repressor (5). Examination of other regions of these OhrRs revealed a striking difference. The majority of OhrRs had two or more additional cysteine residues located near their carboxy termini. The OhrRs from Caulobacter crescentus, Brucella melitensis, Vibrio cholerae, and Burkholderia mallei had cysteine residues at positions corresponding to C127 in Xanthomonas. The OhrRs from Acinetobacter calcoaceticus, Sinorhizobium meliloti, and Agrobacterium tumefaciens possessed a cysteine residue at position C131, while those from Azotobacter vinelandii, Clostridium acetobutylicum, and Pseudomonas aeruginosa contained an additional cysteine residue at C124 (Fig. 1). X. campestris pv. phaseoli and Erwinia carotovora OhrRs had cysteine residues at both positions 127 and 131. By contrast, a minor group of OhrRs consisting of those from B. subtilis, Oceanobacillus iheyensis, and Streptomyces coelicolor had only a single sensing cysteine residue, C22. These differences in the primary structure of OhrR raised the possibility that there could be differences in the mechanisms involved in sensing and responding to organic hydroperoxide between the different proteins.
FIG. 1.

Alignment of the deduced amino acid sequences of OhrRs from various bacteria. Xanthomonas, X. campestris (EMBL accession no. AAK62673); Caulobacter, C. crescentus (accession no. AAK22899); Brucella, B. melitensis (accession no. ALL53650), Vibrio, V. cholerae (accession no. AAF96901); Burkholderia, B. mallei (accession no. AAU46183); Erwinia, E. carotovora subsp. atroseptica (accession no. CAG76066); Acinetobacter, A. calcoaceticus (accession no. GAG69726); Sinorhizobium, S. meliloti (accession no. CAC45533); Agrobacterium, A. tumefaciens (accession no. AAL41860); Ralstonia, R. solanacearum (accession no. CAD18257); Pseudomonas, P. aeruginosa (accession no. AAG06237); Clostridium, C. acetobutylicum (accession no. AAK79536); Azotobacter, A. vinelandii (accession no. EAM06258); Bacillus, B. subtilis (accession no. CAA05594); Oceanobacillus, O. iheyensis (accession no. BAC15414); Streptomyces, S. coelicolor (accession no. CAB87337). Numbers indicate the positions of amino acid residues corresponding to X. campestris pv. phaseoli OhrR. Cysteine residues are shaded.
C22 and C127 are required for OhrR to sense and respond to organic hydroperoxide.
The studies thus far indicate that the reduced form of OhrR binds to target promoters and represses transcription (5, 15). In the presence of organic hydroperoxides, OhrR is inactivated and released from the promoter. It was of interest to know how X. campestris pv. phaseoli OhrR senses and responds to changes in organic hydroperoxide levels. Thus, the roles played by C22, C127, and C131 of X. campestris pv. phaseoli OhrR in the organic hydroperoxide-sensing and inactivation mechanisms were investigated. A series of site-directed mutagenesis experiments was performed to replace single and various combinations of the residues C22, C127, and C131 with serine residues. The ability of these mutant OhrRs to derepress and repress an OhrR-regulated promoter in response to the presence or absence of organic hydroperoxide was then evaluated in vivo by introduction of the plasmid-borne ohrR mutants (pBBR1MCS-5) (8) into strain XpP1lacZ, a mini-Tn5 P1lacZ chromosomal insertion mutant in X. campestris pv. phaseoli ohrR containing a promoterless lacZ transcriptionally fused downstream of the OhrR-regulated P1 promoter of the ohrR-ohr operon (17). Analysis of β-galactosidase activity revealed that XpP1lacZ cells containing plasmids carrying either the wild type or cysteine mutants of ohrR repressed P1 promoter activity under uninduced conditions to equal degrees (Fig. 2). However, CHP treatment of cells harboring various mutant ohrRs revealed novel and unexpected patterns. The mutation C22S in OhrR abolished the ability of the organic hydroperoxide CHP to derepress expression from the P1 promoter, resulting in constitutively low β-galactosidase levels in the presence or absence of CHP. This is consistent with previous observations that C22 is required for organic hydroperoxide induction (17). In addition, XpP1lacZ expressing ohrR(C127S) also failed to respond to CHP treatment, thereby implicating this residue in the sensing process (Fig. 2). By contrast, changing residue C131 in OhrR to S had no effect on CHP's ability to derepress the P1 promoter (Fig. 2).
FIG. 2.
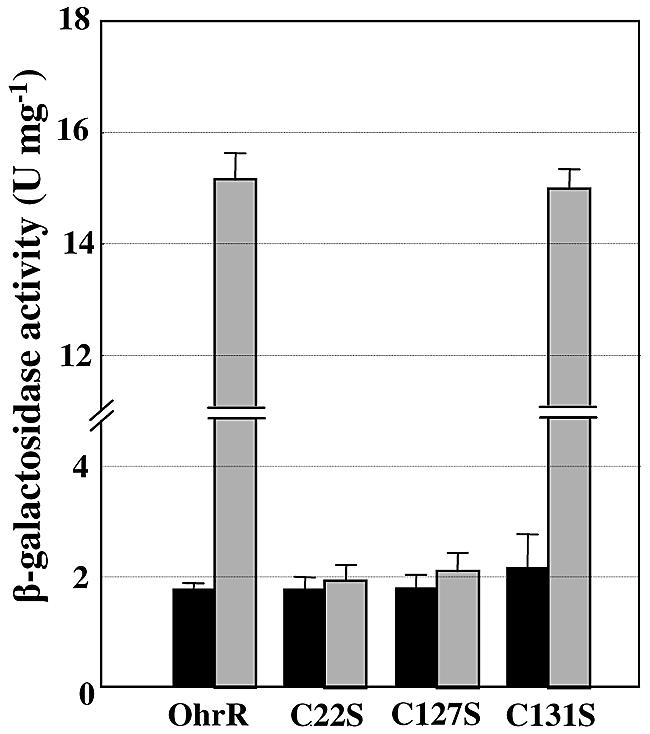
Organic hydroperoxide-dependent regulation of the X. campestris ohrR P1 promoter by wild-type and mutant OhrRs. β-Galactosidase activities of exponential-phase cultures of various XpP1lacZ transcriptional fusion strains (15, 17) expressing wild-type and C22S, C127S, and C131S mutant OhrRs were either induced with 100 μM CHP for 30 min (gray bars) or untreated (black bars). The values presented are the means and standard deviations of three independent experiments.
Analysis of sulfhydryl groups of reduced and oxidized OhrR.
The site-directed mutagenesis of ohrR revealed that residues C22 and C127 play essential roles in the protein's ability to sense and respond to organic hydroperoxide. However, the role played by each of these C residues, in the process by which OhrR senses and is inactivated by CHP, was not entirely clear. Thus, we attempted to determine the function of different C residues in their reduced and oxidized forms. Wild-type OhrR, along with the C22S and C127S-C131S mutants, was purified under reducing conditions in the presence of 2 mM DTT to prevent the overoxidation of free cysteine residues, as described in Materials and Methods. First, the number of free thiol groups in reduced and oxidized wild-type OhrR was determined by DTNB titration assay. The results showed that the thiol contents of OhrR in the reduced and oxidized (after CHP treatment) forms were 2.79 ± 0.22 and 0.82 ± 0.14 per subunit, respectively (Table 1). As expected, reduced OhrR had three free thiol groups, while upon CHP oxidation, only one free sulfhydryl group was detected (Table 1). Various mutant OhrRs were used to determine which cysteine residues lost thiol groups upon CHP oxidation. In contrast to wild-type OhrR, reduced and oxidized forms of C22S OhrR gave similar values for their thiol content, at around 1.8 of free sulfhydryl groups per subunit. Hence, in the absence of C22, the CHP treatment had no effect on the remaining cysteine residues. By contrast, the thiol content of reduced and oxidized C127S-C131S OhrR was 1.07 ± 0.04 and 0.01 ± 0.001 per subunit, respectively. The loss of the one free thiol group in the C127S-C131S mutant protein clearly indicated an important role for C22 in the CHP-mediated oxidation of OhrR.
TABLE 1.
DTNB analysis of free thiols in OhrR and mutant proteins under denaturing conditions
| OhrR (remaining cysteine[s]) | No. of free thiols per subunit
|
|
|---|---|---|
| Reduceda | Peroxide treatedb | |
| OhrR (C22, C127, C131) | 2.79 ± 0.22 | 0.82 ± 0.14 |
| C22S (C127, C131) | 1.89 ± 0.11 | 1.78 ± 0.16 |
| C127131S (C22) | 1.07 ± 0.04 | 0.010 ± 0.001 |
Proteins were prepared under reducing conditions.
Proteins were exposed to 1 equivalent of cumene hydroperoxide.
Oxidation of OhrR leads to generation of the sulfenic acid intermediate form of the sensing cysteine, C22.
The hydroperoxide oxidation of cysteine residues could lead to the formation of C-SOH or of a more highly oxidized product of cysteine, such as C-SO2H (3, 5). The observations to this point were consistent with a scenario where the exposure of OhrR to organic hydroperoxide led to the oxidation of C22 to form a metastable C-SOH intermediate. Experiments were carried out to obtain direct evidence of C-SOH formation in wild-type OhrR following CHP treatment by using NBD chloride to trap the highly labile C-SOH (3); however, this assay failed to detect an R-S(O)-NBD derivative in CHP-oxidized OhrR (data not shown). This could have been due to the subsequent rapid reaction of the protein sulfenic acid intermediate (R-SOH) with other C residues, as suggested by the results shown in Table 1, which indicated that two thiol groups were lost upon CHP oxidation of OhrR. To avoid the loss of C-SOH due to the subsequent disulfide formation step, the NBD chloride trapping experiment was repeated using CHP-oxidized C127S-C131S OhrR. We reasoned that without any other free thiol groups in the protein, the enhanced stability of the OhrR C22-SOH should allow for it to be trapped by NBD chloride. Indeed, the UV-visible absorbance spectrum of NBD-labeled, oxidized C127S-C131S OhrR exhibited a maximal absorbance at 347 nm, as is a typical of NBD adducts of R-SOH [R-S(O)-NBD] (Fig. 3, solid line). This species was clearly distinct from the NBD-modified thiol adduct (R-S-NBD) with a 420-nm peak (Fig. 3, dotted line), which was generated from NBD chloride treatment of reduced C127S-C131S OhrR. NBD chloride assays using C22S OhrR with or without organic peroxide treatment showed a dominant peak at 420 nm in the resulting spectra in both cases, indicating that neither C127 nor C131 is oxidized to C-SOH by organic hydroperoxide (data not shown). These analyses confirmed the conversion of the OhrR C22-thiolate (R-S−) to C22-sulfenic acid (R-SOH) upon oxidation by CHP (Fig. 4). These results were consistent with the results of the DTNB assays of OhrR (Table 1) that identified C22 as the sensing cysteine and indicated that neither of the other two OhrR cysteine residues, C127 and C131, can be directly oxidized by CHP.
FIG. 3.

Spectra of NBD chloride-treated OhrR mutant proteins. Reduced C127S-C131S OhrR was treated with (solid line) or without (dotted line) an equivalent amount of CHP prior to incubation with 20 molar equivalents of NBD chloride. Shown are the UV-visible absorbance spectra of the NBD-labeled proteins with absorbance maxima at 347 and 420 nm, respectively.
FIG. 4.

Disulfide bond formation between OhrR monomers. Purified OhrR samples prepared under various conditions were separated in nonreducing SDS-PAGE. R, reduced OhrR; O, oxidized OhrR; NEM, oxidized OhrR treated with N-ethylmaleimide; DTT, oxidized OhrR treated with DTT. Arrows indicate the positions of monomers (18 kDa) and dimers (37 kDa). Protein bands were stained with Coomassie blue. M represents protein molecular mass markers.
HPLC/ESI-MS peptide mapping of reduced and oxidized OhrR.
The loss of two free thiol groups following CHP treatment (Table 1) suggested the possible formation of a disulfide bond in oxidized OhrR. Thus, HPLC/ESI-MS was used to determine the existence and location of any disulfide bonds as well as the remaining free thiol group, in trypsin-digested reduced and oxidized OhrR. The free thiol groups in both reduced and oxidized OhrR were blocked with 4-vinylpyridine. The tryptic digest fragments of reduced and oxidized OhrR were analyzed by HPLC/ESI-MS. Tryptic peptides with molecular masses of 2,829.32 ± 0.51 and 2,276.31 ± 0.70 Da corresponding to the peptide surrounding C22 (residues 11 to 34) and the one including both C127 and C131 (residues 118 to 137), respectively, were observed in reduced OhrR (Table 2). These two peptides were not detected in tryptic digests of oxidized OhrR protein. Moreover, a new tryptic peptide with a mass of 4,892 ± 0.63 Da was detected in the oxidized protein. The molecular mass of this peptide corresponded to that of the peptide in which C22 formed a disulfide bond with either C127 or C131 (Table 2). Unfortunately, residues C127 and C131 are located in the same tryptic peptide fragment; thus, the MS analysis could not determine specifically which cysteine residues had formed a disulfide with C22. In order to identify the cysteine residue involved in the disulfide bond with C22, the sulfhydryl of the free cysteine residue in the oxidized protein was modified with 2-bromoethylamine to generate S-2-aminoethylcysteine that is susceptible to trypsin digestion (16). In order to prevent unmodified thiol groups from forming artifactual disulfide bonds or undergoing disulfide bond rearrangement after denaturation, the 2-bromoethylamine-treated oxidized OhrR was reacted with NEM to block any unmodified sulfhydryl groups. The tryptic map of this modified, oxidized OhrR was analyzed by HPLC/ESI-MS. Among the different peptides between oxidized and reduced OhrR, two important peptides from the oxidized OhrR, with molecular masses of 4,117.05 ± 0.24 and 731.34 ± 0.00 Da, that corresponded to the peptide containing a disulfide linkage between C22 and C127 and the peptide SLDELR (residues 132 to 137), respectively, were observed. This indicated that C131 was modified by 2-bromoethylamine and was rendered susceptible to trypsin digestion (Table 2). This evidence strongly indicated the presence of a disulfide linkage between C22 and C127 in oxidized OhrR.
TABLE 2.
Masses of trypsin digest fragments of OhrR under oxidizing and reducing conditions
 |
Z is free Cys modified with 4-vinylpyridine.
The location of the disulfide bond is deduced from the modification experiment with 2-bromoethylamine and NEM (see text).
C* is the S-2-aminoethylcysteine generated after treatment with 2-bromoethylamine; this residue is now a recognition site for trypsin that was cleaved on the C-terminal side of C*, proving the location of the disulfide bond between C22 and C127.
Intermolecular disulfide bonding in oxidized OhrR.
The native molecular weight of OhrR was determined by gel filtration as described in Materials and Methods. The reduced OhrR appeared to migrate as a dimer. Next, we extended the investigation by determining whether the disulfide bond that was formed in oxidized OhrR was inter- or intramolecular using nonreducing SDS-PAGE (23). The results reveal that the majority of the reduced OhrR existed as a monomer of 18.5 kDa (Fig. 4). After the protein was oxidized by CHP treatment, the amount of dimeric OhrR (37.0 kDa) significantly increased (Fig. 4, compare lanes R and O). When the oxidized protein was treated with the thiol-alkylating agent NEM prior to SDS-PAGE in order to block the rearrangement of disulfide bonds in the presence of free thiol groups upon denaturation, most of the protein was present as covalent dimers (Fig. 4, lane NEM). Consistent with the nonreducing SDS-PAGE results, ESI-MS analysis of reduced and oxidized OhrR detected proteins with masses of 18,000.47 ± 1.83 (monomer) and 35,997.97 ± 6.00 (dimer) Da, respectively, suggesting that the disulfide linkage formed in oxidized OhrR was intermolecular (calculated monomeric and dimeric masses of 18,000.65 and 35,998.65 Da, respectively). The reversibility of the disulfide bond was tested by reducing oxidized OhrR with DTT. This resulted in the conversion of essentially all of the dimeric OhrR to the monomeric form (Fig. 4, compare lanes O and DTT), indicating a reversible disulfide linkage.
DISCUSSION
The expression of ohr is regulated by OhrR, a transcriptional repressor in the MarR superfamily (4, 22). One of the major questions regarding the organic hydroperoxide stress response is how OhrR senses and responds to organic hydropreoxides. The data presented here indicate that the mechanism of sensing and responding to organic hydroperoxide proposed for B. subtilis OhrR, a member of the single-cysteine family of OhrRs, does not strictly apply to the majority of OhrRs. The analysis of OhrR primary amino acid sequence alignments clearly shows that OhrR can be divided into two groups, a minor group of single-cysteine OhrRs such as those in B. subtilis and a major group containing multiple-cysteine residues such as those in X. campestris pv. phaseoli. In the multiple-cysteine group, the sensing cysteine located near the amino terminus is absolutely conserved, while the second cysteine, near the carboxy terminus, is always located in the same general region of the protein, but its exact position varies. Mutational analysis of these cysteine residues in X. campestris pv. phaseoli OhrR proved that they are not required for binding of the repressor to the operator site. Nonetheless, in vivo functional analyses of cysteine mutants and wild-type OhrRs indicated that both C22 and C127 are required in order for the regulator to sense and respond to organic hydroperoxide. This is a major mechanistic difference from the sensing and responding mechanism of the single-cysteine OhrR family, where oxidation of the single sensing cysteine is enough to inactivate the repressor. DTNB assays to detect free thiol groups and NBD chloride trapping assays to detect the presence of cysteine sulfenic acid groups in wild-type OhrR and various cysteine mutants of OhrR have yielded important information concerning the mechanism of organic hydroperoxide sensing by OhrR and the roles of different cysteine residues in the process. The loss of two free thiol groups upon CHP oxidation of OhrR supports the idea that more than one C residue is involved in the response to organic hydroperoxide. Moreover, the role of C22 as the sensing residue for CHP is supported by the lack of an alteration in the number of free thiol groups in C22S OhrR and the loss of one free thiol group in C127S-C131S OhrR (containing only C22) after CHP treatment. Thus, C22 has to first be oxidized by CHP prior to forming a disulfide bridge with C127 that results in the inactivation of the protein. The initial oxidation of C22 was independent of the C127 and C131 residues that could not be directly oxidized by CHP. Thus, C127 and C131 do not function as the initial organic hydroperoxide-sensing residues. These in vitro results are consistent with the in vivo analyses of mutant OhrRs. The fact that the positions of the carboxy-terminal C residues are more varied but generally always in the same region of the protein suggests some structural flexibility in the region of OhrR that allows the C residue to react with the oxidized N-terminal sensing C residue. In a model of OhrR based on the MarR structure (1), the C residues in this region of the protein lie within an α-helical region, thus explaining their putative positions on the same face of the helix if they are located a turn or two away from C127 of Xanthomonas OhrR (3 to 4 residues/turn of the helix).
C-SOH in proteins has been reported to be the product of the reaction between cysteine thiols and peroxides such as H2O2, organic hydroperoxides, and peroxynitrite (3, 5, 19). The role of sulfenic acid in the redox-sensing pathways of prokaryotic cells involves the oxidation of specific transcription regulators such as E. coli OxyR and B. subtilis OhrR (5, 7). The detection of a cysteine sulfenic acid intermediate in oxidized C127S-C131S OhrR indicates that CHP oxidation of C22 leads to the formation of a protein-sulfenic acid intermediate. C-SOH is highly reactive and can, reversibly or irreversibly, generate other forms of modified cysteinyl groups. The irreversible oxidation of C-SOH gives rise to C-SO2H and C-SO3H, respectively (19). Nevertheless, C-SOH can be stabilized within the protein and recycled, via disulfide-bonded intermediates, back to C-SH by biological reductants (19). Condensation of C22-SOH in Xanthomonas OhrR with the proximal thiol group of residue C127 to form an intersubunit disulfide bond is the most likely explanation for the data presented here (19). The inability to detect a protein-sulfenic acid intermediate in wild-type OhrR indicated that C22-sulfenic acid is unstable and rapidly reacts with one of the C-terminal cysteines to form a disulfide bond.
Analysis of tryptic fragments of oxidized OhrR labeled with 4-vinylpyridine using HPLC/ESI-MS confirmed that disulfide formation between C22 and one of the other cysteine residues, C127 or C131, did indeed occur. Treatment of oxidized OhrR with 2-bromoethylamine and NEM, prior to trypsin digestion and HPLC/ESI-MS analysis, indicated that the disulfide linkage occurred between C22 and C127. Moreover, nondenaturing SDS-PAGE and ESI-MS analyses of reduced and oxidized OhrR indicated that the disulfide bond in oxidized OhrR is an intermolecular bond between C22 and C127 from different OhrR subunits. This is in good agreement with structural analyses of other MarR family members that suggested that reduced OhrR probably binds to its target site as a dimer (1, 12, 24). The results of nondenaturing SDS-PAGE of OhrR also indicated that the intermolecular disulfide linkage between C22 and C127 was easily reversed in the presence of the reducing agent DTT (Fig. 4).
The genetic and biochemical evidence presented here has led to the development of a model for Xanthomonas OhrR-mediated peroxide sensing and derepression of target promoters, such as the ohrR P1 promoter, that likely applies to other members of the multiple-cysteine family of OhrRs. Initially, exposure of promoter-bound dimeric OhrR to organic peroxide would result in the oxidation of the redox-sensing residue C22 to form a transient OhrR-C22-SOH intermediate. C22-SOH in each OhrR subunit rapidly reacts with the thiol group of residue C127 in the opposite subunit of the dimer to form intersubunit disulfide linkages. Disulfide bond formation between the two subunits induces a change in the conformation of the OhrR dimer such that it is no longer capable of binding DNA. The repressor is then released from the promoter, thus allowing transcription of ohrR. The fate of the covalently linked OhrR subunits is not known. However, the fact that the disulfide bonds are easily reversed by a reducing agent, combined with gel mobility shift data indicating that this rereduction restores DNA binding activity (17), raises the possibility that covalently linked OhrR subunits are recycled via reduction by cellular reducing agents. While many of the details as to how OhrRs sense organic peroxide remain to be elucidated, it is clear that the response mechanisms of the single- and multiple-cysteine families of OhrR are distinct.
Acknowledgments
We thank L. M. S. Baker for technical advice and J. M. Dubbs for a critical reading of the manuscript.
Research support was provided by a Research Team Strengthening Grant from BIOTEC and Senior Research Scholar Grant RTA4580010 from the Thailand Research Fund to S.M. and by grants from the ESTM through the Higher Education Development Project of the Commission of Higher Education, Ministry of Education. W.P. was supported by a postdoctoral fellowship from BIOTEC. Support from NIH RO1 GM50389 to L.B.P. is also acknowledged.
REFERENCES
- 1.Alekshun, M. N., S. B. Levy, T. R. Mealy, B. A. Seaton, and J. F. Head. 2001. The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 Å resolution. Nat. Struct. Biol. 8:710-714. [DOI] [PubMed] [Google Scholar]
- 2.Atichartpongkul, S., S. Loprasert, P. Vattanaviboon, W. Whangsuk, J. D. Helmann, and S. Mongkolsuk. 2001. Bacterial Ohr and OsmC paralogues define two protein families with distinct functions and patterns of expression. Microbiology 147:1775-1782. [DOI] [PubMed] [Google Scholar]
- 3.Baker, L. M. S., and L. B. Poole. 2003. Catalytic mechanism of thiol peroxidase from Escherichia coli: sulfenic acid formation and overoxidation of essential Cys61. J. Biol. Chem. 278:9203-9211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuangthong, M., S. Atichartpongkul, S. Mongkolsuk, and J. D. Helmann. 2001. OhrR is a repressor of ohrA, a key organic hydroperoxide resistance determinant in Bacillus subtilis. J. Bacteriol. 183:4134-4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuangthong, M., and J. D. Helmann. 2002. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc. Natl. Acad. Sci. USA 99:6690-6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jalloul, A., J. L. Montillet, K. Assigbetse, J. P. Agnel, E. Delannoy, C. Triantaphylides, J. F. Daniel, P. Marmey, J. P. Geiger, and M. Nicole. 2002. Lipid peroxidation in cotton: Xanthomonas interactions and the role of lipoxygenases during the hypersensitive reaction. Plant J. 32:1-12. [DOI] [PubMed] [Google Scholar]
- 7.Kim, S. O., K. Merchant, R. Nudelman, W. F. Beyer, Jr., T. Keng, J. DeAngelo, A. Hausladen, and J. S. Stamler. 2002. OxyR: a molecular code for redox-related signaling. Cell 109:383-396. [DOI] [PubMed] [Google Scholar]
- 8.Kovach, M. E., P. H. Elzer, D. S. Hill, G. T. Robertson, M. A. Farris, R. M. Roop II, and K. M. Peterson. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175-176. [DOI] [PubMed] [Google Scholar]
- 9.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 10.Lesniak, J., W. A. Barton, and D. B. Nikolov. 2002. Structural and functional characterization of the Pseudomonas hydroperoxide resistance protein Ohr. EMBO J. 21:6649-6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine, A., R. Tenhaken, R. Dixon, and C. Lamb. 1994. H2O2 from oxidative burst orchestrates the plant hypersensitive disease resistance response. Cell 79:583-593. [DOI] [PubMed] [Google Scholar]
- 12.Lim, D., K. Poole, and N. C. Strynadka. 2002. Crystal structure of the MexR repressor of the mexRAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Biol. Chem. 277:29253-29259. [DOI] [PubMed] [Google Scholar]
- 13.Loprasert, S., M. Fuangthong, W. Whangsuk, S. Atichartpongkul, and S. Mongkolsuk. 2000. Molecular and physiological analysis of an OxyR-regulated ahpC promoter in Xanthomonas campestris pv. phaseoli. Mol. Microbiol. 37:1504-1514. [DOI] [PubMed] [Google Scholar]
- 14.Mongkolsuk, S., and J. D. Helmann. 2002. Regulation of inducible peroxide stress responses. Mol. Microbiol. 45:9-15. [DOI] [PubMed] [Google Scholar]
- 15.Mongkolsuk, S., W. Panmanee, S. Atichartpongkul, P. Vattanaviboon, W. Whangsuk, M. Fuangthong, W. Eiamphungporn, R. Sukchawalit, and S. Utamapongchai. 2002. The repressor for an organic peroxide-inducible operon is uniquely regulated at multiple levels. Mol. Microbiol. 44:793-802. [DOI] [PubMed] [Google Scholar]
- 16.Okazaki, K., H. Yamada, and T. Imoto. 1985. A convenient S-2-aminoethylation of cysteinyl residues in reduced proteins. Anal. Biochem. 149:516-520. [DOI] [PubMed] [Google Scholar]
- 17.Panmanee, W., P. Vattanaviboon, W. Eiamphungporn, W. Whangsuk, R. Sallabhan, and S. Mongkolsuk. 2002. OhrR, a transcription repressor that senses and responds to changes in organic peroxide levels in Xanthomonas campestris pv. phaseoli. Mol. Microbiol. 45:1647-1654. [DOI] [PubMed] [Google Scholar]
- 18.Poole, L. B. 2005. Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch. Biochem. Biophys. 433:240-254. [DOI] [PubMed] [Google Scholar]
- 19.Poole, L. B., P. A. Karplus, and A. Claiborne. 2004. Protein sulfenic acids in redox signaling. Annu. Rev. Pharmacol. Toxicol. 44:325-347. [DOI] [PubMed] [Google Scholar]
- 20.Robertson, J. G., G. W. Adams, K. F. Medzihradszky, A. L. Burlingame, and J. J. Villafranca. 1994. Complete assignment of disulfide bonds in bovine dopamine β-hydroxylase. Biochemistry 33:11563-11575. [DOI] [PubMed] [Google Scholar]
- 21.Storz, G., and J. A. Imlay. 1999. Oxidative stress. Curr. Opin. Microbiol. 2:188-194. [DOI] [PubMed] [Google Scholar]
- 22.Sukchawalit, R., S. Loprasert, S. Atichartpongkul, and S. Mongkolsuk. 2001. Complex regulation of the organic hydroperoxide resistance gene (ohr) from Xanthomonas involves OhrR, a novel organic peroxide-inducible negative regulator, and posttranscriptional modifications. J. Bacteriol. 183:4405-4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veneziani, B. M., F. Giallauria, and F. Gentile. 1999. The disulfide bond pattern between fragments obtained by the limited proteolysis of bovine thyroglobulin. Biochimie 81:517-525. [DOI] [PubMed] [Google Scholar]
- 24.Wu, R. Y., R. G. Zhang, O. Zagnitko, I. Dementieva, N. Maltzev, J. D. Watson, R. Laskowski, P. Gornicki, and A. Joachimiak. 2003. Crystal structure of Enterococcus faecalis SlyA-like transcriptional factor. J. Biol. Chem. 278:20240-20244. [DOI] [PMC free article] [PubMed] [Google Scholar]