

Abstract
The sequencing of genomes of several organisms and advances in high throughput technologies for transcriptome and proteome analysis has allowed detailed mechanistic studies of transcription and translation using mathematical frameworks that allow integration of both sequence-specific and kinetic properties of these fundamental cellular processes. To understand how perturbations in mRNA levels affect the synthesis of individual proteins within a large protein synthesis network, we consider here a genome-scale codon-wide model of the translation machinery with explicit description of the processes of initiation, elongation, and termination. The mechanistic codon-wide description of the translation process and the large number of mRNAs competing for resources, such as ribosomes, requires the use of novel efficient algorithmic approaches. We have developed such an efficient algorithmic framework for genome-scale models of protein synthesis. The mathematical and computational framework was applied to the analysis of the sensitivity of a translation network to perturbation in the rate constants and in the mRNA levels in the system. Our studies suggest that the highest specific protein synthesis rate (protein synthesis rate per mRNA molecule) is achieved when translation is elongation-limited. We find that the mRNA species with the highest number of actively translating ribosomes exerts maximum control on the synthesis of every protein, and the response of protein synthesis rates to mRNA expression variation is a function of the strength of initiation of translation at different mRNA species. Such quantitative understanding of the sensitivity of protein synthesis to the variation of mRNA expression can provide insights into cellular robustness mechanisms and guide the design of protein production systems.
INTRODUCTION
Translation is a central cellular process in every living organism. It is a complex template biopolymerization process in which the information encoded in the mRNA sequence is translated into the corresponding protein using ribosomes as catalysts. Protein synthesis comprises three steps: 1), initiation; 2), elongation; and 3), termination (Fig. 1). Initiation involves the series of reactions during which the ribosome binds reversibly at the ribosomal binding site on the mRNA and forms the initiation complex around the start codon assisted by GTP hydrolysis. Elongation consists of a cycle of reactions in which a charged tRNA is recruited by the ribosome in a codon-specific manner, the corresponding amino acid is added to the growing polypeptide chain, and the ribosome translocates on the mRNA template one codon at a time. This process is also assisted by GTP hydrolysis and elongation factors which act as cofactors for the different steps of the elongation cycle. Termination involves the recognition of the termination codon by the ribosome assisted by the termination factors and subsequent hydrolysis of the peptidyl tRNA. The completed peptide chain and the ribosome are finally released and the ribosomes can reinitiate translation. In the time between the initiation of the synthesis and the complete formation of single protein molecule, multiple initiation events can take place if the previous ribosome has moved sufficiently far away from the initiation site. This results in the loading of each mRNA molecule with more than one ribosome actively synthesizing proteins. These mRNA-ribosome structures are called polysomes (or polyribosomes), and they have been visually observed (1).
FIGURE 1.

Schematic of the translational machinery. Translation comprises three key steps: initiation, elongation, and termination. The value n is the length of the mRNA template; γ and β are the scaled initiation and termination rate constants, respectively; λ is the ribosomal binding affinity; and L is the number of codons occupied by a ribosome.
In recent years, there have been significant advances in high-throughput technologies to monitor the various components of the mRNA and protein synthesis machineries. DNA microarrays (2–4) allow the estimation of the copy-number for every mRNA species within a single cell and the fold changes in expression between two different physiological conditions. Two-dimensional gel electrophoresis in concert with tandem mass spectrometry enables simultaneous measurement of specific protein levels for thousands of proteins in the cell (5,6). Other high-throughput techniques allow monitoring of polysomes, i.e., the amount of ribosomes that actively translate individual proteins from their corresponding mRNAs (7,8) and the monitoring of levels of all the tRNAs in a cell (9). Most of the studies on the mRNA or protein expression patterns within a cell are perturbation studies: they investigate the relative (fold) changes in the levels of mRNA or protein in response to environmental changes, such as temperature shock and amino acid starvation (10), and genetic modifications, such as gene deletions. Recent experiments on the relative changes in mRNA and protein expression in the galactose utilization pathway in yeast (11), genetic and environmental perturbations in Escherichia coli (12), and other absolute measurements of mRNA and protein levels in human liver cells (13) and yeast (14) all demonstrate a lack of correspondence between expression of mRNA and the expression of the corresponding proteins.
The complexity and the large size of the translation machinery make mathematical modeling and simulation an attractive framework to aid in understanding the design principles and functional properties of this system. We have recently developed a genome-wide model for the translation machinery in E. coli (15) that provides mapping between changes in mRNA levels and changes in protein levels in response to environmental or genetic perturbations. We identified the key parameters that affect this mapping as
Changes in the concentration of free and total ribosomes in response to the perturbation.
Changes in initiation and elongation kinetics due to competition for aminoacyl tRNAs.
Changes in termination kinetics.
Average changes in total mRNA levels in response to the perturbation.
Changes in protein stability.
Based on these studies, we concluded that the presence of polysomes and the kinetics of the elongation process necessitate consideration of codon-dependent elongation and codon usage in experimental and theoretical studies. Such considerations will require developments in high-throughput analytical techniques and mathematical modeling and computational frameworks that take into account such codon-dependent variability.
In this study, we have used a mechanistic model for translation that takes into account such codon-dependent variability (16–18). The model, presented in detail in Methods, describes the processes of initiation and termination, and the elongation process for every single codon in every mRNA species in the system as individual steps. The variables of the model are the states of each codon on the mRNAs: being occupied by a ribosome or being free. This formulation introduces a large number of parameters and variables. For example, in a small-genome organism with 1000 protein-coding genes of an average protein size of 40 kDa, i.e., an average of 400 codons per mRNA species, the corresponding genome-wide, codon-dependent model of protein synthesis will involve 400,000 coupled nonlinear equations, each of them representing the dynamics of the state of each codon. The large size of translation networks, along with the strong nonlinearities in the kinetics of the various steps, present a significant computational challenge in solving for the dynamics and the steady state of the system. We have developed a computational framework for efficiently deriving the steady-state solution of the problem, and we used it to study the impact of over- and underexpression of mRNAs on system responses, and to identify the system parameters and conditions that underlie these responses.
METHODS
Mathematical model
The mechanistic model for the translation we consider is based on the lattice model first proposed by MacDonald and Gibbs (17,18) and later extended by Heinrich and Rapoport (16). We have further extended these models to include an additional mechanistic detail of the initiation step: the reversible binding of the ribosome around the Shine-Dalgarno sequence, an mRNA sequence upstream of the start codon which is complementary to a ribosomal RNA and thus allows for recognition and reversible binding of the ribosome. The Shine-Dalgarno sequence is different among different mRNA species, and therefore, the binding affinity of ribosome to mRNA is different among the mRNA species. This is one of the mechanistic origins of the different initiation rates in protein synthesis.
In this mathematical formulation, ribosomes are assumed to be hard bodies that can occupy L codons. They bind first at the ribosomal binding site and occupy L sites around the start codon, and they move independently along the mRNA chain. The mathematical model considers the mass balance of the codons occupied by the front of a ribosome, 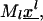 where
where  is the vector of probabilities of each codon on mRNA species l being occupied by the front of a ribosome, and Ml is the number of copies of the lth mRNA species. For each mRNA species l with length nl codons, the model consists of (nl + 1) differential equations of the form
is the vector of probabilities of each codon on mRNA species l being occupied by the front of a ribosome, and Ml is the number of copies of the lth mRNA species. For each mRNA species l with length nl codons, the model consists of (nl + 1) differential equations of the form
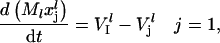 |
(1) |
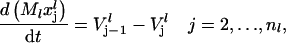 |
(2) |
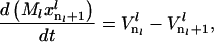 |
(3) |
where 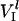 is the rate of initiation,
is the rate of initiation, 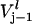 and
and 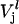 are the rates of ribosome movement from codon j−1 to j and from j to j+1, respectively, and
are the rates of ribosome movement from codon j−1 to j and from j to j+1, respectively, and  is the rate of termination of translation for the lth mRNA species. These equations assume no cellular growth and they do not account for the dilution due to growth. The one additional variable (with subscript l) in the model corresponds to the ribosomal binding site for each mRNA. The initiation rate is described by the equation
is the rate of termination of translation for the lth mRNA species. These equations assume no cellular growth and they do not account for the dilution due to growth. The one additional variable (with subscript l) in the model corresponds to the ribosomal binding site for each mRNA. The initiation rate is described by the equation
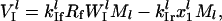 |
(4) |
where 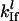 is the rate constant for initiation complex formation for mRNA l, Rf is the number of free ribosomes,
is the rate constant for initiation complex formation for mRNA l, Rf is the number of free ribosomes, 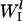 is the probability that the initiation site is free,
is the probability that the initiation site is free,
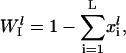 |
(5) |
and 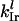 is the initiation complex dissociation rate constant. The number of free ribosomes, Rf, is a function of the total number of ribosomes, RT, the number of copies of each mRNA species, and the occupancy probabilities for each codon on every mRNA,
is the initiation complex dissociation rate constant. The number of free ribosomes, Rf, is a function of the total number of ribosomes, RT, the number of copies of each mRNA species, and the occupancy probabilities for each codon on every mRNA,
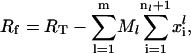 |
(6) |
where 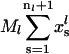 is the total number of ribosomes bound on the lth mRNA species. This formulation for the initiation process that allows explicit description of the reversible binding has not been used in the earlier models.
is the total number of ribosomes bound on the lth mRNA species. This formulation for the initiation process that allows explicit description of the reversible binding has not been used in the earlier models.
The rates of movement of ribosomes during the elongation steps are described by the equations
 |
(7) |
where 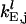 is the elongation constant for codon j on mRNA species l and
is the elongation constant for codon j on mRNA species l and 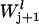 denotes the conditional probability that codon j+1 is free given that codon j is occupied and is formulated as (17,18),
denotes the conditional probability that codon j+1 is free given that codon j is occupied and is formulated as (17,18),
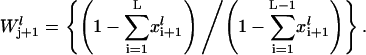 |
(8) |
The steady-state rate of protein synthesis is equal to the rate of termination,
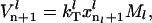 |
(9) |
where  is the termination rate constant.
is the termination rate constant.
Equations 1–9 describe the rates of the key steps of translation for a particular mRNA species. The differences in the performance among the different mRNA species arise from the differences in their sequences, which in turn determine the kinetic parameters of the various steps and the size of the mRNA molecules. For example, differences in complementarity between the Shine-Dalgarno sequence and the 16S ribosomal RNA subunit among different mRNAs can lead to different initiation rate constants (19,20). Similarly, different codons have different elongation rate constants. These differences in the kinetic parameters and the length lead to varying polysome size and positional ribosome distributions on the mRNAs in the cell.
To minimize the complexity of the mathematical analysis in the following studies, we assumed that the elongation rate constants for each codon of the lth mRNA species are equal to a characteristic rate constant, 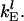 However, the algorithmic framework, discussed below, is fully adaptable to different elongation rate constants for different codons. The above equations can be scaled taking the elongation rate constant as the characteristic scaling factor for the rate constants of each mRNA species. The scaled parameters and variables are defined as
However, the algorithmic framework, discussed below, is fully adaptable to different elongation rate constants for different codons. The above equations can be scaled taking the elongation rate constant as the characteristic scaling factor for the rate constants of each mRNA species. The scaled parameters and variables are defined as
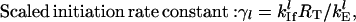 |
(10) |
 |
(11) |
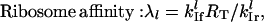 |
(12) |
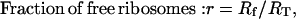 |
(13) |
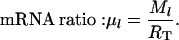 |
(14) |
The scaled initiation rate constant denotes the ratio of the maximum forward initiation rate to the maximum elongation rate and the scaled termination rate constant denotes the ratio of the maximum termination rate to the maximum elongation rate. The scaled rates of initiation, elongation, and termination for the lth mRNA species are
 |
(15) |
where
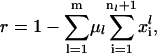 |
(16) |
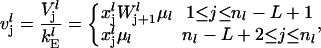 |
(17) |
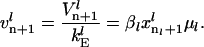 |
(18) |
The rate of translation of a single mRNA species depends on the kinetic parameters of initiation, elongation, and termination, the availability of free ribosomal binding sites, and the availability of free ribosomes. The rates of initiation and elongation also depend on the supply of charged (aminoacylated) tRNAs. These functional relationships can be represented as
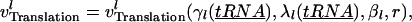 |
(19) |
where 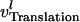 represents any of the rate expressions in Eqs. 15, 17, and 18.
represents any of the rate expressions in Eqs. 15, 17, and 18.
Under the assumption that the tRNA concentrations are not limiting, all parameters and variables affecting the initiation rate at a particular mRNA species, except the fraction of free ribosomes, are specific to that particular mRNA. The fraction of free ribosomes is a quantity shared between all mRNA species (Eq. 16) and therefore changes in free ribosomes can impact protein synthesis from every mRNA in a cell.
Algorithm description and framework
We have developed a bilevel nonlinear programming approach for the steady-state solution of Eqs. 1–3, which provides a quantitative mapping between mRNA and protein expression levels. The formulation of the problem as a bilevel programming problem allows the time of the problem solution to scale linearly with the number of mRNA species. The proposed formulation involves p polynomial-time problems of size q, where p is the number of mRNA species and q is the average size (number of codons) of the mRNA species. The equivalent single nonlinear programming problem would be a polynomial time problem of size (p × q). The algorithmic framework (for all mRNA species) is based on the observation that in the current formulation, the variable that couples the components of the system, i.e., each mRNA species with the rest of the mRNA species, is the fraction of free ribosomes, r (Eq. 16). The first level, outer problem, thus involves the estimation of the fraction of free ribosomes in the cell at steady state. The second level, inner problems, involve the estimation of the distribution of ribosomes on each mRNA at steady-state, given the fraction of free ribosomes determined in the outer problem.
Analysis of the model has shown that the functional dependence between the total number of ribosomes and the number of free ribosomes is monotonic. Since the total number of ribosomes in the cell is an input parameter to the problem, we could use the conservation relationship (Eq. 16) to formulate the outer problem as follows:
Objective
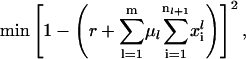 |
(20) |
subject to
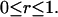 |
(21) |
The objective function for the outer problem (Eq. 20, see Fig. 2) is convex; hence, we employ a bisection subroutine to estimate the number of free ribosomes.
FIGURE 2.

The objective of the outer problem, 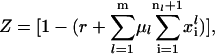 is a convex function of the fraction of free ribosomes, r. Convexity of the objective allows use of bisection method to estimate the fraction of free ribosomes.
is a convex function of the fraction of free ribosomes, r. Convexity of the objective allows use of bisection method to estimate the fraction of free ribosomes.
At steady state in protein synthesis, the scaled rates of initiation, elongation, and termination, as described by Eqs. 14–18, are equal 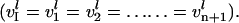 So, the inner problem involves solving a set of coupled nonlinear algebraic equations for each mRNA. The following equations describe the formulation of the inner problem:
So, the inner problem involves solving a set of coupled nonlinear algebraic equations for each mRNA. The following equations describe the formulation of the inner problem:
Objective
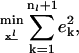 |
(22) |
subject to
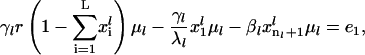 |
(23) |
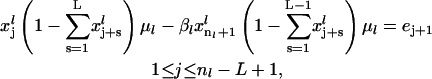 |
(24) |
 |
(25) |
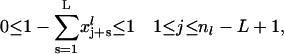 |
(26) |
 |
(27) |
The following equality and inequality constraints have been formulated for every inner problem:
Linear equality constraints: One linear equality constraint (Eq. 23) guarantees that the rate of initiation of translation at steady state is equal to the rate of protein synthesis, i.e., rate of termination. A set of linear equality constraints (Eq. 25) ensure that the rate of ribosome movement through the last L codons at steady state is equal to the steady-state rate of protein synthesis.
Nonlinear equality constraints: A set of nonlinear equality constraints (Eq. 24) enforces the rates of ribosome movement through the ribosomal binding site and the first nl − L codons to be equal to the rate of protein synthesis.
Linear inequality constraints: The set of linear inequality constraints (Eq. 26) ensure that no L-codon window on an mRNA template is occupied by more than one ribosome (volume exclusion).
Boundary constraints: The boundary constraints (Eq. 27) ensure that value of the variable xj (probability that a codon is occupied by the front of a ribosome) is bounded between 0 and 1.
We used a nonlinear programming software, KNITRO, for solving the inner problems as described by Eqs. 22–27. KNITRO implements both state-of-the-art interior-point and active-set methods for solving nonlinear optimization problems (21–23). More details about the software can be found in the KNITRO 4.0 reference manual (24).
Our bilevel algorithmic procedure consists of the following steps (see Fig. 3):
Step 1: We assume that the fraction of free ribosomes in the system is 0.5.
Step 2: The number of free ribosomes in the system uniquely determines the number of bound ribosomes on each mRNA, and the steady-state distribution of ribosomes on each mRNA species is then determined based on the formulation of the inner problem as discussed above (Eqs. 22–27).
Step 3: The objective function for the outer problem (Eq. 20) is evaluated and the sign of the error
 is determined.
is determined.Step 4: If the value of the objective function is greater than our chosen tolerance, a new value of fraction of free ribosomes is calculated based on the sign of the error. A negative sign of error indicates that the current guess for fraction of free ribosomes is greater than the optimal value, hence the interval between 0 and the current value of free ribosomes is bisected to estimate the new guess for free ribosomes. A positive sign on the other hand indicates underprediction of the fraction of free ribosome hence a new value for free ribosomes is chosen by bisecting the interval between the current guess for free ribosomes and 1.
Step 5: If the value of the outer objective is less than the chosen tolerance, the procedure is stopped and the current fraction of free ribosomes and the distribution of ribosomes on the mRNA species gives the steady-state solution to the system described by Eqs. 20–27.
FIGURE 3.

Diagrammatic description of the bilevel algorithmic framework.
Definition and characterization of the system
We considered a model organism that consists of 400 mRNA species expressing proteins. The mRNAs were assigned lengths randomly based on the mRNA length distribution of the E. coli genome, and they were also randomly assigned copy numbers between 2 and 5 such that the total number of mRNA copies is ∼1400 and the total number of ribosomes in the system is ∼14,000 (25). In the most general case, these values could be estimated from genome-scale mRNA expression measurements using DNA microarrays (2–4).
Most of the currently used models describe rate of protein synthesis, Vs,p, using a single expression of the form
 |
(28) |
where Rf is the concentration of free ribosomes and M is the concentration of the free ribosomal binding sites. Expressions like Eq. 28 implicitly assume that initiation is the rate-limiting step, since they consider protein synthesis as a function of the free ribosome and the ribosomal binding site alone, thereby ignoring the elongation and termination steps and the states of the codons of the mRNA species. In the mathematical formalism discussed above, the rate-limiting step of protein synthesis is determined by the initiation, elongation, and termination rate constants, as well as the total number of ribosomes in the system. To quantify the rate-limiting step, the sensitivity of protein synthesis rate to the scaled initiation rate constant, ribosome affinity and scaled termination rate constants, can be calculated based on the definitions (16,26),
 |
(29) |
where Vl is the rate of protein synthesis from the lth mRNA species, and 
 and
and 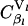 are the sensitivity coefficients to scaled initiation rate constant, ribosome affinity, and scaled termination rate constant, respectively, also known as control coefficients within the metabolic control analysis (MCA) framework (26,27). The value
are the sensitivity coefficients to scaled initiation rate constant, ribosome affinity, and scaled termination rate constant, respectively, also known as control coefficients within the metabolic control analysis (MCA) framework (26,27). The value 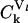 denotes the percentage change in the rate of protein synthesis from lth mRNA species for 1% change in a particular rate constant k. The sensitivity of protein synthesis rate to initiation, elongation, and termination rate constants can be calculated from Eq. 29 and using the summation theorem (26) as
denotes the percentage change in the rate of protein synthesis from lth mRNA species for 1% change in a particular rate constant k. The sensitivity of protein synthesis rate to initiation, elongation, and termination rate constants can be calculated from Eq. 29 and using the summation theorem (26) as
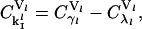 |
(30) |
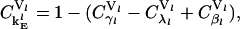 |
(31) |
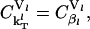 |
(32) |
where the subscript 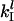 is the net control coefficient of protein synthesis from the lth mRNA species with respect to both the rate constants of initiation complex formation and dissociation.
is the net control coefficient of protein synthesis from the lth mRNA species with respect to both the rate constants of initiation complex formation and dissociation.
For our computational studies, the kinetic parameters were chosen such that most mRNAs follow initiation-limited kinetics based on the experimental evidence that most control in translation is at the initiation process (8,28). We performed analysis on single mRNA species of variable lengths to identify the parameter space of initiation and termination rate constants that can lead to initiation-limited protein synthesis conditions (results presented below), using Eqs. 29–32. This analysis, in addition to theoretical considerations of mean-field lattice models for protein synthesis (18), allowed us to identify the parameter regimes in our large-scale studies that would guarantee initiation limited conditions. The scaled initiation rate constants, 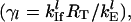 were thus randomly assigned to mRNA species to vary between 0.005 and 0.256. These parameters also represent a wide range of translation efficiencies for the various mRNA species. For some of the larger values of the scaled initiation rate constants, elongation can become as important as initiation in determining the protein synthesis rate. Based on similar considerations as above, the scaled termination rate constants,
were thus randomly assigned to mRNA species to vary between 0.005 and 0.256. These parameters also represent a wide range of translation efficiencies for the various mRNA species. For some of the larger values of the scaled initiation rate constants, elongation can become as important as initiation in determining the protein synthesis rate. Based on similar considerations as above, the scaled termination rate constants, 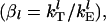 for each mRNA were assigned values between 0.011 and 0.334 to ensure initiation or elongation limitation.
for each mRNA were assigned values between 0.011 and 0.334 to ensure initiation or elongation limitation.
RESULTS AND DISCUSSION
We derived the steady state of the system as described above and found the fraction of free ribosomes to be ∼30%, which is very close to the experimentally observed free ribosome fraction of 20% (25). We next investigated the control distribution between the rate constants and the effects of changes in the concentration of mRNA species on the protein synthesis rate of the individual species, as well as of the overall system.
Control distribution and specific protein synthesis rate
To quantitatively characterize the rate-limiting steps of the individual mRNA species, we estimated the sensitivities of protein synthesis rates from mRNAs to their respective rate constants allowing the levels of the free ribosomes to also change in response to changes in the corresponding rate constants (Eq. 29). In Fig. 4, the distribution of the sensitivities of protein synthesis rates of all mRNAs with respect to their initiation, elongation and termination rate constants are shown. The sensitivities of the protein synthesis rates with respect to initiation and elongation rate constants are found to be three orders-of-magnitude higher than the sensitivities with respect to termination rate constants. In these studies, the elongation rate constants are assumed equal for each codon on an mRNA, and therefore, the sensitivities to elongation rate constants of each codon are distributed among the codons without any one of them individually having significant impact on the protein synthesis rate. However, in a more realistic situation, individual codons or sets of codons can exert significant control on the protein synthesis rate. Ninety-five percent of the mRNA species we considered were initiation-limited, whereas the remaining 5% were elongation-limited.
FIGURE 4.

Distribution of sensitivities of protein synthesis rates from all mRNA species to their initiation (a), elongation (b), and termination (c) rate constants, respectively.
Previous experimental studies have established a link between the mRNA association with ribosomes and the behavior of the cellular translation machinery (8). To quantify the effect of ribosome density of the mRNA on its translational behavior, we studied the specific rate of protein synthesis from each mRNA species as a function of the ribosome density, 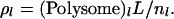 We found that there exists a critical value of ρl of 0.46, beyond which the mRNA species are always limited by their elongation rate (Fig. 5 a). This critical value of ρl, which marks the transition from initiation to elongation-limited regime, holds for every mRNA irrespective of its length. Interestingly, the specific protein synthesis rates, i.e., protein synthesis rate per mRNA molecule, from elongation-limited mRNA species, are higher than the initiation-limited mRNA species (Fig. 5 b). This suggests that elongation can play an important control in the efficiency of protein synthesis.
We found that there exists a critical value of ρl of 0.46, beyond which the mRNA species are always limited by their elongation rate (Fig. 5 a). This critical value of ρl, which marks the transition from initiation to elongation-limited regime, holds for every mRNA irrespective of its length. Interestingly, the specific protein synthesis rates, i.e., protein synthesis rate per mRNA molecule, from elongation-limited mRNA species, are higher than the initiation-limited mRNA species (Fig. 5 b). This suggests that elongation can play an important control in the efficiency of protein synthesis.
FIGURE 5.

The elongation-limited mRNA species have higher protein synthesis rates than the initiation-limited mRNA species. (a) Sensitivity of the protein synthesis rates from every mRNA species to their initiation (+), elongation (□), and termination (•) rate constants as a function of ribosome density, ρ (the fraction of the mRNA covered by ribosomes). (b) Specific scaled protein synthesis rates from every mRNA species as a function of ribosome density, ρ.
Effect of over- and underexpression of groups of mRNA species on global and local system response
We studied next the effect of changes in mRNA expression patterns on the protein synthesis rate from each mRNA and on the number of bound ribosomes on each mRNA. These properties can be thought of as local system properties because they are specific to each mRNA. To better understand the system level responses of the translation network, we studied the changes in fraction of free ribosomes and the changes in the rate of total protein synthesis from all mRNAs in response to changes in mRNA expression. These properties can be thought of as global system properties.
A genetic or environmental perturbation to the cellular environment can lead to over- and/or underexpression of groups of mRNA species within the cell. Extensive experimental studies on the environmental stress response (10) have identified sets of genes which are overexpressed in response to environmental stress, and sets of genes that are simultaneously underexpressed. For example, sudden heat shock leads to concurrent induction of protein folding chaperones localized to the cytoplasm, mitochondria and ER, and repression of genes involved in growth-related processes, various aspects of RNA metabolism, nucleotide biosynthesis, secretion, and other metabolic processes (10,29,30). Other experimental studies have also identified sets of mRNA species that are simultaneously over- and underexpressed in colon carcinomas (31) and prostate cancer (32) relative to the reference—healthy tissue cells.
To quantify the effect of changes in mRNA expression, we considered the model system at a reference physiological state as discussed above (see Methods) and applied to it two types of perturbations: 1) all the mRNA species with the lowest expression levels were overexpressed; and 2) all the mRNA species with highest expression levels were underexpressed, while keeping the kinetic parameters and the concentrations of the rest of the mRNA species at their reference values. Over- and underexpression involved increasing or decreasing the abundance of the mRNA species by fivefold from their reference values. We studied the effect of the perturbations on the global system response by estimating the relative change in the fraction of free ribosomes, 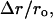 where ro is the fraction of free ribosomes at reference state and Δr is the change in the fraction of free ribosomes. To systematically quantify the local system responses, we studied how sensitive the rates of protein synthesis from individual mRNA species are to changes in the concentration of other groups of mRNA species by estimating the relative changes in the protein synthesis rates from each unperturbed mRNA species,
where ro is the fraction of free ribosomes at reference state and Δr is the change in the fraction of free ribosomes. To systematically quantify the local system responses, we studied how sensitive the rates of protein synthesis from individual mRNA species are to changes in the concentration of other groups of mRNA species by estimating the relative changes in the protein synthesis rates from each unperturbed mRNA species, 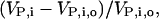 where VP,i,o and VP,i are the scaled rates of synthesis of protein i at a reference state and perturbed state, respectively. Fig. 6 shows the effect of changes in mRNA expression on the relative change in protein synthesis rate from all the mRNA species which were not perturbed. In response to an overexpression of the mRNA species with the lowest expression levels at the reference state (corresponding to a 55% increase in the total cellular mRNA levels), the fraction of free ribosomes in the system decreased by 33.3%, whereas the protein synthesis rates from the unperturbed mRNA species decreased by 18–33%. Similarly, underexpression of the mRNA species with the highest expression levels at the reference state (corresponding to a 27% decrease in total cellular mRNA levels) led to a 25.4% increase in the fraction of free ribosomes and a 12–25% increase in the synthesis of proteins from the unperturbed mRNA species.
where VP,i,o and VP,i are the scaled rates of synthesis of protein i at a reference state and perturbed state, respectively. Fig. 6 shows the effect of changes in mRNA expression on the relative change in protein synthesis rate from all the mRNA species which were not perturbed. In response to an overexpression of the mRNA species with the lowest expression levels at the reference state (corresponding to a 55% increase in the total cellular mRNA levels), the fraction of free ribosomes in the system decreased by 33.3%, whereas the protein synthesis rates from the unperturbed mRNA species decreased by 18–33%. Similarly, underexpression of the mRNA species with the highest expression levels at the reference state (corresponding to a 27% decrease in total cellular mRNA levels) led to a 25.4% increase in the fraction of free ribosomes and a 12–25% increase in the synthesis of proteins from the unperturbed mRNA species.
FIGURE 6.

Distribution of fold changes in protein synthesis rate from unperturbed mRNA species in response to (a) overexpression by fivefold of the mRNA species with low expression levels, and (b) underexpression by fivefold of the mRNA species with high expression levels.
These computational studies suggest that the protein synthesis rate from mRNA species, whose copy number remains unchanged between two different conditions, can change significantly due to changes in the copy number of the rest of the mRNA species. The competition of the mRNA species for the ribosomes seems to have a significant impact on the responses of the overall system to changes on a part of the system as reflected by the changes in the fraction of free ribosomes. The sensitivity of the protein synthesis rates from individual mRNA species thus depends on both the changes in cellular conditions, i.e., free ribosomes, and the specific properties of different mRNA species such as the kinetic and/or sequence properties. This complex interplay is investigated in the following sections.
Effect of changes in the concentration of individual mRNA species on global and local system response
We studied next the effect of an increase or decrease in the number of individual mRNA species by one copy on the global system response by estimating the relative change in the fraction of free ribosomes, 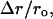 as above (Fig. 7). Most mRNA species (>80%) have a small impact (<0.1%) on the fraction of free ribosomes (Fig. 7, a and b) in the system. But a system perturbation involves increase or decrease of multiple copies for each mRNA species, and therefore the response of protein synthesis to changes in a single mRNA species could be several times higher. Under such conditions, the much higher changes in the concentration of an mRNA species will have a significant impact on system behavior.
as above (Fig. 7). Most mRNA species (>80%) have a small impact (<0.1%) on the fraction of free ribosomes (Fig. 7, a and b) in the system. But a system perturbation involves increase or decrease of multiple copies for each mRNA species, and therefore the response of protein synthesis to changes in a single mRNA species could be several times higher. Under such conditions, the much higher changes in the concentration of an mRNA species will have a significant impact on system behavior.
FIGURE 7.

Distribution of relative changes in the number of free ribosomes to changes in mRNA concentration estimated by increasing (a) or decreasing (b) the number of copies of each mRNA species by 1. (c) Relative changes in number of free ribosomes to increase (□) or decrease (×) in mRNA concentration is a linear function of the number of ribosomes associated with each mRNA.
It has been experimentally observed that protein synthesis in E. coli is limited by the concentration of free ribosomes (33), and thus it is a key determinant of the response of the translational machinery to a perturbation. Further analysis of the above results suggests that the relative changes in the fraction of free ribosomes to changes in the concentration of mRNA species is a linear function of the number of ribosomes bound to that mRNA species (polysome) (Fig. 7 c). Therefore, the mRNA species with the highest polysome size has maximum impact on the global system response.
To systematically quantify the local system responses, we studied how sensitive the rates of protein synthesis from individual mRNA species are to changes in the concentration of the other individual mRNA species. We increased and decreased by one copy each mRNA species and we ranked them from least to most influential, based on the magnitude of the relative changes in the protein synthesis rates from each of the rest of the mRNA species in response to these copy-number changes. We found that the identity of the most influential mRNA species was the same for all the other mRNA species in the system. Moreover, the influence ranking of each mRNA species was the same across the rest of the mRNA species. Based on conclusions from the previous studies, we hypothesized that the most influential mRNA species should have the highest polysome size. Fig. 8 shows the total relative change in the total protein synthesis rates from all unperturbed mRNA species,
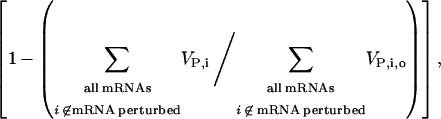 |
as a function of the polysome size of the perturbed mRNA. We observed that the total relative change in protein synthesis rate from all unperturbed mRNA species is a linear function of the polysome size of the perturbed mRNA species, and that the mRNA species with the highest polysome size induces maximum systemwide change in the rate of protein synthesis, thus confirming our hypothesis.
FIGURE 8.

 |
These computational studies suggest that the systemic responses to mRNA expression variation are strong functions of the translation state (number of ribosomes bound) of the mRNA species and provide guidance toward identifying mRNA species that could potentially cause maximum systemwide changes in protein expression in response to a genetic or an environmental perturbation. Such studies thus allow the development of a ranking criterion for different mRNA species based on their effect on the cellular response. The modulation of the concentration of mRNA species with the higher number of bound ribosomes can also be potentially used as a strategy to control the production of heterologous proteins and assist in the interpretation of physiological responses in biotechnological and medical problems. Our results thus have implications in the design of protein production systems, wherein quantitative knowledge of global system response to changes in the cellular environment can be used to optimize a cellular system toward the production of a protein of interest, and they further suggest a new potential mechanism of a systemic translational regulation.
Effect of the parameters of the individual mRNA species on system behavior
The rate of protein synthesis from different mRNA species is a function of the sequence specific properties (such as mRNA length), the kinetic properties of translation (rate constants of initiation, elongation and termination) of individual mRNA species, and the number of free ribosomes in the cell available for initiating translation. The studies above have shown that the mRNA with the highest polysome size exerts maximum control over the rate of synthesis of proteins from different mRNA species. The strength of control, though, was observed to be different for different mRNA species, suggesting that the magnitude of this control might be related to one or more characteristic properties of each mRNA. An analysis of both the sequence-specific and kinetic parameters of the translation machinery showed that relative response of the rate of protein synthesis of each mRNA species, 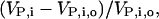 to the mRNA with the highest polysome size, is a function of its initiation rate constant (Fig. 9 a). The protein synthesis rates from mRNA species with the higher translation initiation rate constants are more robust to changes in concentration of different mRNA species. These mRNA species with high initiation rate constants can also recruit more ribosomes per codon and achieve higher ribosome densities (Fig. 9 c) at reference state, which corresponds to a higher polysome size. Further analysis of the local properties of each mRNA species showed that the mRNA species with the highest polysome size has the maximum impact on the translation state (number of ribosomes bound) of each mRNA and the magnitude of this control is also a function of the initiation rate constant of the particular mRNA (Fig. 9 b). These computational studies thus allow quantification of the link between system response and the translation state and kinetic parameters of individual mRNA species.
to the mRNA with the highest polysome size, is a function of its initiation rate constant (Fig. 9 a). The protein synthesis rates from mRNA species with the higher translation initiation rate constants are more robust to changes in concentration of different mRNA species. These mRNA species with high initiation rate constants can also recruit more ribosomes per codon and achieve higher ribosome densities (Fig. 9 c) at reference state, which corresponds to a higher polysome size. Further analysis of the local properties of each mRNA species showed that the mRNA species with the highest polysome size has the maximum impact on the translation state (number of ribosomes bound) of each mRNA and the magnitude of this control is also a function of the initiation rate constant of the particular mRNA (Fig. 9 b). These computational studies thus allow quantification of the link between system response and the translation state and kinetic parameters of individual mRNA species.
FIGURE 9.

Relative changes in rate of protein synthesis from all mRNA species (a), and relative changes in the polysome size of each mRNA species (b), in response to a change in the concentration of the mRNA species with highest polysome size are a function of the scaled initiation rate constant, γ. Relative changes are calculated by increasing (I, ○) or decreasing (II, ⋄) the number of the mRNA species with highest polysome size by one copy. The ribosome density of each mRNA species is an increasing function of its scaled initiation rate constant, γ (c).
CONCLUDING REMARKS
The studies presented here suggest that local changes in the expression patterns of a small set of mRNA species can have a significant impact on both local protein expression patterns, as measured by the protein synthesis rate of individual mRNA species, and global system behavior, as measured by the fraction of free ribosomes. The consideration of protein synthesis in the context of translation state of the mRNAs, i.e., polysome size, provides insights into quantifying and interpreting systemwide responses to perturbations in the mRNA expression levels. Polysomes introduce nonlinear effects that can have significant impact in the way we understand and interpret the relationship between mRNA and protein expression. The mRNA species with the greatest number of bound ribosomes exerts maximum control on the system response but at the same time, the mRNA species, which can recruit more ribosomes per unit length (per codon) due to their higher initiation efficiencies, are less sensitive to perturbations in mRNA concentrations.
These results suggest that protein synthesis does not follow the molecular democracy model suggested by Kacser and Burns (34). According to this model, based on the analysis of metabolic reaction networks, the control, or the extent of the responses, of metabolic fluxes is distributed among the enzymes in the pathway irrespectively of the level of the fluxes. In protein synthesis, it appears that the mRNA species with kinetic parameters that support high protein synthesis rates have the maximum control over the protein synthesis rates from the rest of the mRNA species, while their protein synthesis rate is less sensitive to the changes in the level of mRNA species with a lower protein synthesis rate.
Previous experimental studies on relative changes in mRNA and protein levels in response to an environmental and/or genetic perturbation (11,12) have shown a nonlinear, not one-to-one, relation between mRNA and protein expression. Our studies predict a monotonic response of protein expression to changes in mRNA expression when the mRNAs in the cell compete for ribosomes and the tRNAs are abundant. However, we found that the experimentally observed nonlinear mapping between mRNA and protein expression is only possible when there is systemwide competition for the tRNAs, in addition to the competition for the ribosomes, and our analysis can be expanded to take into account the consideration of tRNA limitation.
Although some of the conclusions drawn from our studies might be as expected to those experienced with protein synthesis, the proposed computational framework provides a quantitative verification and allows the formulation of hypotheses for the origins of the observed phenomena that mental simulations alone cannot offer. The objective of the studies presented here were the study of the responses of protein synthesis to changes in the mRNA levels under a constant amount of ribosomes. These studies provide insights for further ongoing investigations of the steady-state responses to simultaneous changes in the mRNA expression levels, in the total amount of ribosomes, and in the values of any of the parameters of the system. The proposed model and solution algorithm can also be used to study the steady-state responses of protein synthesis to simultaneous changes in the mRNA expression levels, in the total amount of ribosomes, and in the values of any of the parameters of the system. However, the finding that elongation-limited mRNA species can sustain higher specific protein synthesis rates is not obvious, and it has not been suggested before. This finding suggests a more important role for protein elongation than has been considered previously.
The proposed modeling framework and the solution algorithm can be further used for the study of smaller cellular systems in the context of cellular environment. Modeling and analysis of cellular subsystems is often carried out without taking into account the fact that the mRNA species of the subsystem compete for catalytic resources and amino acids with the rest of the cellular processes. Using the methods presented here it will be possible to augment models of cellular subsystems with larger networks of background mRNA species and proteins, whose average properties will reflect the average properties of the overall system, and study the properties of the subsystems of interest in the context of a larger system. Exploiting the efficiency of the computational algorithm, we are currently performing exhaustive parametric studies to derive the rules and the scaling properties that govern the performance of single mRNA species within a large network of mRNA species. These rules and scaling properties will provide the criteria for evaluating the conclusions drawn from small-scale models of cellular subsystems that assume a constant background environment and identify the properties that are most sensitive to this assumption.
With the current advances in high-throughput technologies in genomics, transcriptomics, and proteomics, mathematical modeling frameworks will provide the tools for the integration and analysis of the large amounts of data from such sources. Algorithmic frameworks like the one presented here will allow the estimation of the various parameters of the translation machinery from transcriptomic and proteomic data and will provide insights into mechanisms of translational regulation and optimal design of artificial protein production systems. Our studies on the identification of these parameters suggest that two levels of information are needed for parameter identification: 1), translation state (polysome size); and 2), mRNA copy numbers. High throughput methods for obtaining such information have been recently developed (8,35) and the proposed framework can used for genome scale determination of the kinetic parameters based on this information.
Acknowledgments
The authors thank Drs. Richard Waltz and Jorge Nocedal for providing access to KNITRO solver and for their helpful suggestions. The authors also thank the two anonymous reviewers for their constructive comments.
This research has been supported by the National Science Foundation through the Quantitative Systems Biotechnology Initiative (grant No. BES 0132014), and DuPont through a DuPont Young Professor Award to V.H.
References
- 1.Miller, O. L., Jr., B. A. Hamkalo, and C. A. Thomas, Jr. 1970. Visualization of bacterial genes in action. Science. 169:392–395. [DOI] [PubMed] [Google Scholar]
- 2.Brown, P. O., and D. Botstein. 1999. Exploring the new world of the genome with DNA microarrays. Nat. Genet. 21:33–37. [DOI] [PubMed] [Google Scholar]
- 3.Lockhart, D. J., and E. A. Winzeler. 2000. Genomics, gene expression and DNA arrays. Nature. 405:827–836. [DOI] [PubMed] [Google Scholar]
- 4.Selinger, D. W., K. J. Cheung, R. Mei, E. M. Johansson, C. S. Richmond, F. R. Blattner, D. J. Lockhart, and G. M. Church. 2000. RNA expression analysis using a 30-basepair resolution Escherichia coli genome array. Nat. Biotechnol. 18:1262–1268. [DOI] [PubMed] [Google Scholar]
- 5.Anderson, N. L., and N. G. Anderson. 1998. Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis. 19:1853–1861. [DOI] [PubMed] [Google Scholar]
- 6.Lahm, H. W., and H. Langen. 2000. Mass spectrometry: a tool for the identification of proteins separated by gels. Electrophoresis. 21:2105–2114. [DOI] [PubMed] [Google Scholar]
- 7.Zong, Q., M. Schummer, L. Hood, and D. R. Morris. 1999. Messenger RNA translation state: the second dimension of high-throughput expression screening. Proc. Natl. Acad. Sci. USA. 96:10632–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arava, Y., Y. L. Wang, J. D. Storey, C. L. Liu, P. O. Brown, and D. Herschlag. 2003. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 100:3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dittmar, K. A., E. M. Mobley, A. J. Radek, and T. Pan. 2004. Exploring the regulation of tRNA distribution on the genomic scale. J. Mol. Biol. 337:31–47. [DOI] [PubMed] [Google Scholar]
- 10.Gasch, A. P., P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen, G. Storz, D. Botstein, and P. O. Brown. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 11:4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ideker, T., V. Thorsson, J. A. Ranish, R. Christmas, J. Buhler, J. K. Eng, R. Bumgarner, D. R. Goodlett, R. Aebersold, and L. Hood. 2001. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 292:929–934. [DOI] [PubMed] [Google Scholar]
- 12.Lee, P. S., L. B. Shaw, L. H. Choe, A. Mehra, V. Hatzimanikatis, and K. H. Lee. 2003. Insights into the relation between mRNA and protein expression patterns: II. Experimental observations in Escherichia coli. Biotechnol. Bioeng. 84:834–841. [DOI] [PubMed] [Google Scholar]
- 13.Anderson, L., and J. Seilhamer. 1997. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis. 18:533–537. [DOI] [PubMed] [Google Scholar]
- 14.Gygi, S. P., Y. Rochon, B. R. Franza, and R. Aebersold. 1999. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19:1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehra, A., K. H. Lee, and V. Hatzimanikatis. 2003. Insights into the relation between mRNA and protein expression patterns: I. Theoretical considerations. Biotechnol. Bioeng. 84:822–833. [DOI] [PubMed] [Google Scholar]
- 16.Heinrich, R., and T. A. Rapoport. 1980. Mathematical-modeling of translation of messenger-RNA in eukaryotes—steady-states, time-dependent processes and application to reticulocytes. J. Theor. Biol. 86:279–313. [DOI] [PubMed] [Google Scholar]
- 17.MacDonald, C. T., J. H. Gibbs, and A. C. Pipkin. 1968. Kinetics of biopolymerization on nucleic acid templates. Biopolymers. 6:1–25. [DOI] [PubMed] [Google Scholar]
- 18.MacDonald, C. T., and J. H. Gibbs. 1969. Concerning kinetics of polypeptide synthesis on polyribosomes. Biopolymers. 7:707–725. [Google Scholar]
- 19.Jacob, W. F., M. Santer, and A. E. Dahlberg. 1987. A single base change in the Shine-Dalgarno region of 16S rRNA of Escherichia coli affects translation of many proteins. Proc. Natl. Acad. Sci. USA. 84:4757–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hui, A., and H. A. de Boer. 1987. Specialized ribosome system: preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Natl. Acad. Sci. USA. 84:4762–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrd, R. H., M. E. Hribar, and J. Nocedal. 1999. An interior point algorithm for large-scale nonlinear programming. SIAM J. Optim. 9:877–900. [Google Scholar]
- 22.Byrd, R. H., J. C. Gilbert, and J. Nocedal. 2000. A trust region method based on interior point techniques for nonlinear programming. Math. Progr. 89:149–185. [Google Scholar]
- 23.Byrd, R. H., N. I. M. Gould, J. Nocedal, and R. A. Waltz. 2004. An algorithm for nonlinear optimization using linear programming and equality constrained subproblems. Math. Progr. 100:27–48. [Google Scholar]
- 24.Waltz, R. A. 2004. KNITRO 4.0 User's Manual. Ziena Optimization, Evanston, IL.
- 25.Bremer, H., and P. P. Dennis. 1996. Modulation of chemical composition and other parameters of the cell by growth rate. In Escherichia coli and Salmonella. F. C. Neidhart, editor. ASM Press, Washington, DC. 1553–1569.
- 26.Kacser, H., and J. A. Burns. 1973. The control of flux. Symp. Soc. Exp. Biol. 27:65–104. [PubMed] [Google Scholar]
- 27.Fell, D. A. 1992. Metabolic control analysis—a survey of its theoretical and experimental development. Biochem. J. 286:313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hershey, J. 1987. Protein synthesis. In Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. F. Neidhart, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter, and H. E. Umbarger, editors. American Society for Microbiology, Washington, DC.
- 29.Helmann, J. D., M. F. Wu, P. A. Kobel, F. J. Gamo, M. Wilson, M. M. Morshedi, M. Navre, and C. Paddon. 2001. Global transcriptional response of Bacillus subtilis to heat shock. J. Bacteriol. 183:7318–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richmond, C. S., J. D. Glasner, R. Mau, H. Jin, and F. R. Blattner. 1999. Genome-wide expression profiling in Escherichia coli K-12. Nucleic Acids Res. 27:3821–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Notterman, D. A., U. Alon, A. J. Sierk, and A. J. Levine. 2001. Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 61:3124–3130. [PubMed] [Google Scholar]
- 32.Magee, J. A., T. Araki, S. Patil, T. Ehrig, L. True, P. A. Humphrey, W. J. Catalona, M. A. Watson, and J. Milbrandt. 2001. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 61:5692–5696. [PubMed] [Google Scholar]
- 33.Vind, J., M. A. Sorensen, M. D. Rasmussen, and S. Pedersen. 1993. Synthesis of proteins in Escherichia coli is limited by the concentration of free ribosomes. Expression from reporter genes does not always reflect functional mRNA levels. J. Mol. Biol. 231:678–688. [DOI] [PubMed] [Google Scholar]
- 34.Kacser, H., and J. A. Burns. 1979. Molecular democracy: who shares the controls? Biochem. Soc. Trans. 7:1149–1160. [DOI] [PubMed] [Google Scholar]
- 35.Iyer, V., and K. Struhl. 1996. Absolute mRNA levels and transcriptional initiation rates in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 93:5208–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]