

Abstract
Bilayers of palmitoyl-oleoylphosphatidylnucleoside derivatives (1-palmitoyl-2-oleoyl-phosphatidyl-adenosine and 1-palmitoyl-2-oleoyl-phosphatidyl-uridine) were synthesized and investigated in the low-water content regime by a combination of neutron diffraction and Fourier transform infrared linear dichroism (LD-FTIR). Attention was focused on the modulation of structural properties operated by the presence of nucleic acid bases (either adenosine or uridine, a purine and a pyrimidine that are complementary in RNA). Base substitution causes major differences in phase behavior of the phospholipids, i.e., water sorption from a controlled humidity atmosphere and smectic periodicity. The profile of scattering length density can be inferred from five diffraction orders for 1-palmitoyl-2-oleoyl-phosphatidyl-uridine lamellar phase. 1-Palmitoyl-2-oleoyl-phosphatidyl-adenosine is characterized by lower and less ready hydration, giving rise to a powder-like sample. A linear dichroism FTIR investigation on the same lamellar phases was undertaken with the purpose of gathering details at the submolecular level on different portions of the molecule. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayers were also investigated with the same technique for the sake of comparison. Besides a confirmation of the diffraction data interpretation, FTIR has provided evidence that the same chemical groups at the bilayer interface (namely the sugar-phosphate) have a different orientation depending on whether the base is a purine or a pyrimidine. A very simple geometrical optimization agrees with this observation. This indicates that a different pattern of base interaction is operating in the two cases and that base substitution acts as a modulator of the phase properties.
INTRODUCTION
Self-organization achieved through weak interactions can be considered a fingerprint in living organisms, some examples being protein folding and hydrophobic self-organization of phospholipids that provides the structural scaffolding of cell membranes. One of the most important classes of biopolymers, DNA and RNA, represents an unrivalled example where supramolecular chemistry is used to store, transmit, and replicate information in a challenging environment (water) with a limited number of structural units (adenine, cytosine, guanine, thymine, and uracil).
Chemistry can borrow nature's strategy to design “bottom-up” molecular machines, built by noncovalent interactions, where self-assembly is responsible for expression of specific functions, coded in molecular subunits (1–7). This can be achieved in aqueous media by ingeniously employing multiple noncovalent interactions such as hydrogen bonds, stacking interactions, and appropriate molecular and supramolecular architectures (8). Soft-matter science and noncovalent build-up according to a nucleobase-like pattern started to merge about 12 years ago, when the investigation of self-organization of some nucleic functionalized amphiphiles arranged at the air/water interface (8) stimulated many groups working in the field of molecular films.
In this research field, our group has focused attention on a class of compounds with a chemical relationship with nucleic acids, which hold biological relevance and are also promising molecular devices in medical and pharmaceutical applications and possibly in the near future for nonviral gene therapy (9–12). These products, called phosphatidylnucleosides, are composed of a phospholipid backbone and a nucleotidic polar head, and reproduce the chemistry and the charge of each DNA monomer.
In self-assemblies of these biosurfactants, responsiveness to soft external stimuli, dictated by the dynamic noncovalent nature of autoorganization forces, is integrated and enriched by the presence of additional energetic contributions due to the presence of smart nucleic polar heads. Therefore, besides its traditional hydrophobic effects, molecular recognition can modulate aggregation and phase behavior.
Phospholipid membranes decorated with molecular DNA functions represent interesting structures for engineering surfaces able to display biological functionalities acting, for instance, as biocompatible vectors of complementary DNA, RNA, or PNA.
The geometry of spontaneous self-assembly and the interfacial film properties are the result of a delicate balance between hydrophobic forces and polar head interactions. Therefore, base-base interaction patterns in a bidimensional confinement, triggered by aggregation, are themselves responsible for structural modulations on the nanoscale. In this respect, a structural characterization is essential for a complete and full understanding of base-base properties in the aggregates.
This article reports a structural characterization of the swelling behavior of 1-palmitoyl-2-oleoyl-phosphatidyl-uridine (POPU) and 1-palmitoyl-2-oleoyl-phosphatidyl-adenosine (POPA) in the Lα phase region. The role of the different nature of nucleic base headgroup is stressed and highlighted in terms of alteration of the structural properties of the membrane.
POPA and POPU form liposomes (2,3,13) whose spectroscopic properties indicate base interaction through stacking and H-bonding modes. It is therefore important to monitor structural characteristics of such bilayers in the low water content regime, i.e., in their lyotropic phase. This investigation is accomplished by a combined use of neutron diffraction and infrared linear dichroism to gather information on, respectively, the global array of bilayers (spacing and thickness) and the local orientation (order parameter) of chain group and headgroup in the membranes.
Molecular orientation in self-assembled structures was investigated through determination of order parameters for vibrational modes in the alkyl chain region of the bilayer (symmetric and CH2 stretching), in the interfacial region (C=O stretching), and in the headgroup region (antisymmetric 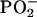 stretching).
stretching).
This infrared investigation is also focused on comparison of the orientation of the polar portions of POPA, POPU, and their synthetic precursor 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and on the correlations between their orientational differences and the capability of nucleolipids to interact by stacking and H-bonding. For a better understanding of the headgroup role in packing properties, a geometrical optimization of POPA and POPU molecules in a periodic box of water has also been performed to correlate, although qualitatively, neutron and infrared results.
MATERIALS
POPC was purchased from Avanti Polar Lipids (Alabaster, AL) and its purity checked by thin-layer chromatography. The lecithin was used as received since no oxidation or lyso products could be detected. Adenosine, uridine, HCl, CHCl3, MeOH, and NH3 (33% aqueous solution) used in the synthesis were purchased from Fluka (Buchs, Switzerland). Phospholipase D from Streptomyces sp. AA586 was a generous gift from Asahi Chemical Industry (Tokyo, Japan). Deuterium oxide (>99.5%) for neutron diffraction measurements was provided by Euriso-Top (Saclay, Gif sur Yvette, France).
POPU and POPA (whose molecular structures are sketched in Fig. 1) were synthesized starting from the corresponding phosphatidylcholine in a two-phase system (14) according to a modification of the method proposed by Shuto and coworkers (15,16), and obtained as an ammonium salt. Separation from the by-products was achieved by silica-gel flash chromatography. Purity was checked by thin-layer chromatography, 1H NMR, and elementary analysis.
FIGURE 1.

Schematic drawing of the molecular structure of 1-palmitoyl-2-oleoyl-sn-glycero-3-uridine (A) and 1-palmitoyl-2-oleoyl-sn-glycero-3-adenosine (B).
Sample preparation
Neutron diffraction
Lipid lyophilized powder, 20 mg, was dissolved in methanol/chloroform solution (4:1 v/v). The solution was deposited on a quartz microscope slide (75 × 25 mm) using an artist airbrush. The slides were placed in a vacuum desiccator for 12 h to remove all traces of the solvent before rehydration for 12 h at 37°C at a relative humidity (RH) of 98%.
Fourier transform infrared spectroscopy (FTIR)
Samples of ∼10 mg of pure lipids were dissolved in 2 ml chloroform/methanol (4:1 v/v). The solution was vortexed for a few seconds, deposited drop by drop on a CaF2 (41 × 23 × 6 mm) window, allowed to dry for 12 h in a dust-free environment, and placed in a vacuum to remove any traces of solvent. Samples were rehydrated for 12 h in a K2SO4 saturated atmosphere (98% RH).
Assuming an average area per molecule of ∼60 Å2, 700 bilayers were deposited for FTIR measurements and ∼650 bilayers for neutron diffraction measurements.
The hydration degree after equilibration was experimentally determined through gravimetric methods. POPA samples contain ∼15% (w/w) water, corresponding to an average number of water molecules per lipid, nw = 9 ± 3, whereas POPU samples result in a water content of ∼26% (nw = 17 ± 3).
METHODS
Neutron diffraction data acquisition and data analysis
Neutron diffraction measurements were performed on the membrane diffractometer V1 at the Berlin Neutron Scattering Center of the Hahn Meitner Institute. The samples were placed vertically in temperature-controlled aluminum containers (T = 37 ± 0.1°C for all samples) where the humidity was controlled by aqueous saturated solutions of K2SO4 in Teflon water baths at the base of the chamber. Contrast variation was achieved by adjusting the atmosphere in the sample container using three different D2O/H2O compositions (i.e., 100:0, 50:50, 0:100). Samples were equilibrated for 24 h after each solution change.
Diffraction patterns were measured with rocking scans, rocking the sample around the expected Bragg position θ by θ ± 2°. The duration of the scans ranged between 30 min and 4 h, depending on the intensities of the reflections. Diffraction reflections of POPU phase are good until the fifth order, whereas for POPA stacked bilayers only the first two orders are visible. For this latter sample, θ − 2θ scans were therefore performed. The lamellar spacing d of each sample was calculated by least-square fitting of the observed 2θ values to the Bragg equation nλ = 2d × sinθ, where n is the diffraction order and λ is the selected neutron wavelength (4.82 Å). Integrated intensities were calculated with Gaussian fits to the experimental Bragg reflections. Absorption and Lorentz corrections were applied and the intensities square rooted to produce structure factor amplitudes F(h).
The scattering density profile ρ(z) is given by
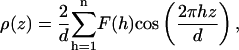 |
(1) |
where F is in units of scattering lengths, f(h) are the scaled structure factors, and the sum describes the distribution in scattering lengths across the bilayer.
The phase assignment was obtained with the isomorphous replacement method, using D2O/H2O exchange, since the structure factors are a linear function of the mole fraction of D2O/H2O, as shown in Fig. 2 (17); for data treatment, only the structure factors determined at 100% H2O were used, since coherent water scattering is in this case negligible with respect to the bilayer. At 100% and 50% D2O, the large coherent scattering of the heavy water layer partially hides that of the membrane.
FIGURE 2.

Structure factors and their phase assignment using D2O:H2O exchange of POPU stacked multilayers.
FTIR data acquisition and data analysis
Infrared spectra were collected with a Nexus 870 spectrophotometer (Thermo Nicolet, Paris, France) equipped with a liquid-nitrogen-cooled mercury cadmium telluride detector. All spectra were performed at room temperature with 4 cm−1 resolution and averaging 1500 scans. Vibrational linear dichroism, measurements were achieved by using a static linear polarizer (Graseby Specac, Waltham, MA) or a photoelastic modulator (Hinds Instruments PEM90, Hillsboro, OR).
Linear dichroism (LD), i.e., the different absorption of light polarized parallel and perpendicular with respect to an orientation direction, is related to the oscillatory strength of a transition and to the polarization of the transition with respect to the orientation axis. The LD signal (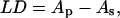 where Ap and As are the absorbance in the parallel and perpendicular directions, respectively) is nonzero when the sample has a nonrandom orientation, as in the case of membrane layers (18).
where Ap and As are the absorbance in the parallel and perpendicular directions, respectively) is nonzero when the sample has a nonrandom orientation, as in the case of membrane layers (18).
The sample orientation was varied with respect to the incoming beam. When the polarization is perpendicular, the angle between the incident light and the bilayer is 90° and the absorbance spectrum is As. When the sample is rotated at an angle ω with respect to parallel-polarized light, the angle between the incident beam and the bilayer normal is ω.
If the z axis of the molecular frame is chosen as coincident with the direction of a transition dipole, for a particular vibrational transition and uniaxial distribution, a relation between the dichroic ratio D = Ap/As and the order parameter S holds (18–20):
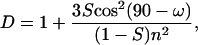 |
(2) |
where n is the refractive index of the lamellar liquid crystalline phase (n = 1.4) (18).
RESULTS AND DISCUSSION
The phospholipids investigated in this study differ from their synthetic precursor, POPC, in the nature of their polar headgroups. The enzymatic exchange could have a major impact on phase behavior: a net negative charge is introduced in POPA and POPU with respect to the zwitterionic POPC, and the bulkiness of the polar group is increased (VPOPC = 1256 Å3, VPOPU = 1349 Å3, and VPOPA = 1383 Å3) (21). A comparison of molecular properties of POPU and POPA highlights a slightly higher molecular volume (2.5%) for the purine derivative. However, from a functional point of view, the base moieties can give rise to selective intermolecular interactions when arranged on a supramolecular array. Therefore, since acyl chains are the same, we can attribute any deviations from the equilibrium structures of POPC multilamellar bilayers observed for POPA and POPU to different steric hindrance (either electrostatic or excluded volume) and interactions at the interface region of the membranes. Moreover, any differences encountered between POPA and POPU should be highlighted in view of possibly different “attractive” interaction patterns.
Our choice was directed to unsaturated derivatives since it is known from thermal studies that the incorporation of cis double bonds into a saturated lipid chain drastically lowers the gel to liquid crystalline transition temperature, that is, −5°C for fully hydrated POPC lamellae (22,23). This transition is strongly dependent both upon polar head kind and hydration degree, being generally increased in the low water content regime.
Since water uptake from the equilibrating atmosphere (98% RH) is modulated by the polar head nature (24) and results in a different extent of hydration for POPA (nw = 9 ± 3) and POPU (nw = 17 ± 3), as reported in the experimental section, the occurrence of different mesophases cannot in principle be ruled out. Differential scanning calorimetry measurements performed on samples prepared in the same way as for neutron diffraction have confirmed that both phospholiponucleosides are above the main transition temperature, which is −2°C for POPA and −7°C for POPU in these experimental conditions (S. Milani, F. Baldelli Bombelli, F. Ridi, D. Berti, and P. Baglioni, unpublished data). The different hydration degree is unexpected when one considers that the derivatives have a like-charged phosphate group, but can be explained by the fact that adenine rings have higher stacking constants and the stacked conformation could lead to water exclusion in the interfacial layer.
Although it is very difficult and even misleading to attribute different water-binding capacities to a single physicochemical property of the polar head (size, charge polarity, etc.), it is generally accepted that smaller and interacting polar heads, such as phosphatidylethanolamine (PE), have a lower water affinity, whereas a net negative charge should favor water binding. Therefore, we should expect an increased water affinity with respect to POPC (nw = 10), which actually occurs for POPU.
Fig. 3, A-C, shows the neutron diffraction patterns for POPU lamellar phases obtained by lipid hydration from the vapor phase at 37°C. POPU samples, at the three different H2O/D2O contrasts, show up to five lamellar orders displaying a well-ordered array of stacked bilayers (mosaicity >0.2°, comparable to the 0.5° obtained for POPC in the same experimental conditions) (25), with a lamellar repeat distance of 51 Å.
FIGURE 3.

Diffraction patterns of POPU bilayers. (A) 100% D2O. (B) 50% D2O. (C) 100% H2O.
The effective bilayer thickness, dL, can be obtained by multiplying the actual surfactant volume fraction, φL, for the smectic period. Once dL is determined (38.5 A°), we can evaluate the averaged surface area per lipid molecule, AL, occupied by polar headgroup through
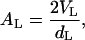 |
(3) |
where VL is the lipid molecular volume, obtained from phosphatidylcholine submolecular fragment volumes determined by Armen et al. through molecular dynamics simulations (21). The calculated value for POPU surface area equals 72 Å2, higher than that for POPC in the lamellar phase (≈58 Å2) (25), as expected for the increased bulkiness of nucleosidic polar heads both in terms of steric hindrance and Coulombic repulsion (26).
The acquisition of five lamellar orders allows us to obtain a POPU scattering-length density profile along the normal to the membrane plane through Fourier synthesis from the structure factors at 0% D2O (see Methods). This profile, reported in Fig. 4 C, represents the elementary cell of the oriented samples composed of one bilayer with its hydration shell. The water content is centered at the two edges of the diagram at d = 0 Å and d = 51 Å, respectively.
FIGURE 4.

(A) Chemical structure of POPU in which we labeled each submolecualr fragment that contributes to the scattering-length profile. (B) Calculated scattering-length profile evaluated by considering the contributions of different submolecular fragments as separated by sharp interfaces. (C) Experimental scattering-length density profiles at 0% D2O in the direction normal to the membrane plane of the POPU sample.
The minimum positioned at 25.5 Å represents the terminal methyl groups of the phospholipids and its appearance rules out any interdigitation in the lipid bilayer. The two maxima in the profiles at ±17 Å from the terminal methyl represent the carbonyl groups near the glycerol backbone, which have a scattering-length density higher than the hydrocarbon region and the uridine groups. For a better comprehension of the profile, Fig. 4 B shows the correlation of the scattering-length density profile with the chemical structure of the phospholipids, shown in Fig. 4 A, evaluated by considering the contributions of different submolecular fragments as separated by sharp interfaces.
POPA bilayers, equilibrated with the same procedure, reveal not only a less ordered lamellar phase, but even a slower equilibration process required to obtain monophasic samples (24 h, see Fig. 5). For the sample equilibrated for 12 h, the most likely interpretation suggests the presence of two lamellar phases, characterized by different smectic periods. After 24 h, the diffraction spectrum shows a single lamellar phase, with only two clearly visible Bragg orders. Concerning the behavior after 12 h, although the peak located at lower scattering vectors can be considered the precursor of the “equilibrium” lamellar phase, as the comparison of the two scattering profiles highlights, the exact nature of the second Bragg reflection is ambiguous due to the lack of higher reflection orders. However, it can be tentatively attributed to less hydrated lamellar stacks that eventually merge in the final lamellar phase. The lamellar phase observed for POPA after one day of equilibration is stable during the experimental time window and persists after isotopic exchange with H2O/D2O and H2O. A one-week exposure to the equilibrating atmosphere does not produce any scattering shift or evolution of the peaks, nor does it improve mosaicity.
FIGURE 5.

Diffraction patterns of POPA bilayers. (Top) 100% D2O after 12 and 24 h of equilibration at 37°C and 98% RH. (Bottom) 100% H2O after 24 h of equilibration at 37°C and 98% RH.
Two different aspects must be stressed: the equilibrium water uptake is lower and slower for POPA than for POPU; and both kinetic and equilibrium effects can be ascribed to apparently subtle chemical differences that are amplified in the supramolecular arrangement. A slower hydration process suggests the presence of a consistent rearrangement concerning polar head conformation induced by water adsorption.
Different equilibrium water uptakes and different imbibition kinetics are familiar for phospholipids with differing polar heads, and the most striking example of this emerges from the comparison of PC with PE lipids. Extensive investigations of both their thermotropic and lyotropic behavior has highlighted the fact that PE polar heads (smaller in size than POPC and with an H-donor group for possible interactions with the negatively charged neighboring phosphate group) are considerably less hydrated (27).
In our case, the similar anionic charge and the comparatively similar steric hindrance of the polar heads should be the dominant factor in both swelling equilibrium and kinetics. As a matter of fact, the different phase behavior is without doubt ascribable to subtler polar headgroup features, adenine being slightly different from uracil not only in size but also in its pronounced stacking efficiency.
The thermodynamic parameters for purine and pyrimidine nucleosides in aqueous solution indicate that association stacking constants are characteristic of weak interactions (the Gibbs free energy for the dimerization through stacking of nucleosides in aqueous solution is negative for adenosine (−1 Kcal/mol) and slightly positive for uridine (0.29 Kcal/mol)) and NMR experiments show that stacking interactions between purine and pyrimidine bases follow the trend purine-purine > purine-pyrimidine > pyrimidine-pyrimidine (28,29). Evidence for base-base stacking and H-bonding triggered by supramolecular arrangement has been highlighted for phospholiponucleosides (1,2,8,30,31), and the observed trend is the same as outlined previously (32). Thus, we can conclude that for POPA, stacking prevails over hydration energy for nw > 9 and precludes the uptake of further water molecules with respect to POPU.
The formation of a long-range well ordered lamellar phase like that shown by POPU is precluded by nucleobase-nucleobase interactions that alter packing requirements, rather than by steric factors. This interaction pattern affecting mesoscopic ordering is probably coupled to a preferred orientation of the sugar-phosphate-base group at the water/hydrocarbon interface; a comparison with the same moieties of POPU is therefore interesting and can confirm the above hypothesis.
On the basis of diffraction results, we used LD-FTIR to investigate the local arrangement of lipid molecules; this technique provides information both on the orientation of a given transition dipole moment and on the mesoscopic ordering through the evaluation of the order parameters. Such measurements were performed for the POPC, POPA, and POPU lyotropic phases, equilibrated at 98% RH. Although for POPU the results for the order parameters can be compared with POPC, given the similar mosaicity of the samples, the powder-like nature of POPA precludes such an analysis and gives more qualitative results.
Fig. 6 shows the absorption spectra of unpolarized infrared radiation for anhydrous lamellar stacks of POPC, POPA, and POPU, obtained by evaporation of a CHCl3/MeOH solution layered onto CaF2 windows. (Once dried, the samples were not equilibrated in a controlled RH atmosphere and therefore are “nominally” anhydrous, their water content deriving only from hydration of the lipid/powder and/or adsorption during sample preparation. However, as we can see from the first spectrum, that of POPC, the broad absorption centered at 3400 cm−1 is indicative of the presence of water. A parallel titration with thermogravimetric methods reveals a residual water content around 3% w/w, corresponding to 1.6 water molecules per lipid. For POPA and POPU, in the same region, the sugar OH stretching also gives a contribution, as the more complex band shape reveals. However this region was not taken into account for further analysis in this study.) The presence of nucleobases attached to a phospholipid skeleton affects mainly the region below 1800 cm−1, as a comparison of the spectra reveals.
FIGURE 6.

Infrared adsorption spectra of (A) POPC, (B) POPA, and (C) POPU bilayers.
The spectral assignment to characteristic vibrations of molecular groups was obtained from comparison with data reported in the relevant literature for lipids and nucleosides (18,33–37) and is shown in Table 1 for samples equilibrated in a 98% RH atmosphere, as done for neutron diffraction.
TABLE 1.
Assignments of selected absorption bands of POPC, POPA, and POPU lamellar phases
| Vibration modes | Wavenumber (cm−1) | S |
|---|---|---|
| POPC | ||
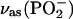
|
1235 | −0.318 |
| ν(C=O) | 1735 | −0.365 |
| ν(CH2) | 2854 | −0.365 |
| POPU | ||
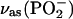
|
1214 | −0.443 |
| ν(N1C6) | 1275 | −0.330 |
| ν(C=O) | 1738 | −0.283 |
| ν(CH2) | 2853 | −0.335 |
| POPA | ||
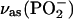
|
1208 | −0.140 |
| ν(ring) | 1600 | −0.189 |
| δ(NH2) | 1675 | −0.180 |
| ν(C=O) | 1730 | −0.200 |
| ν(CH2) | 2851 | −0.197 |
ν, symmetric and antisymmetric stretching vibration mode; δ, bending vibration mode.
The LD spectra of POPA and POPU lamellar stacks hydrated in a 98% RH atmosphere are shown in Fig. 7 for the 1800–1000 cm−1 region. Let us first emphasize the common features of these spectra: a negative linear dichroism is displayed for each band in the region ranging from 3000 to 1200 cm−1, which comprises the methylene stretching and bending regions and the ester CO stretching. In the 1200–1000 cm−1 region, the symmetric 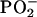 stretching vibrations give rise to a positive LD for both derivatives, as noted by us for POPC (data not shown) and obtained for DOPC by Holmgren (31) and by Akutsu for other lipid membranes (18,38). A comparison of Fig. 7 allows us to distinguish the carboxyl C=O stretching (∼1735 cm−1) from the scissoring of the adenine amino group and the stretching of uridine ring carbonyls.
stretching vibrations give rise to a positive LD for both derivatives, as noted by us for POPC (data not shown) and obtained for DOPC by Holmgren (31) and by Akutsu for other lipid membranes (18,38). A comparison of Fig. 7 allows us to distinguish the carboxyl C=O stretching (∼1735 cm−1) from the scissoring of the adenine amino group and the stretching of uridine ring carbonyls.
FIGURE 7.

LD spectrum (Ap – As) of POPA and POPU for an incident angle (90° − ω) equal to 40° in the region 1800–1110 cm−1.
The lower LD effect for POPA highlights a worse long-range ordering and is completely expected on the basis of neutron diffraction. Moreover some differences between nucleolipids arranged in lamellar stacks arise: besides the symmetric 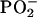 stretching, POPU shows in the low wavenumber limit several bands with positive LD, such as those occurring at 1140 cm−1 (C1′O4′) and 1175 cm−1. This latter band, which shows instead a negative LD for POPA, was also recognizable in the transmission spectrum of the hydrated samples as a shoulder of the
stretching, POPU shows in the low wavenumber limit several bands with positive LD, such as those occurring at 1140 cm−1 (C1′O4′) and 1175 cm−1. This latter band, which shows instead a negative LD for POPA, was also recognizable in the transmission spectrum of the hydrated samples as a shoulder of the 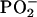 asymmetric stretching, particularly evident for POPA. In nucleic acids this band is considered a signature of the A form (C3′-endo sugar puckering mode with an anti conformation of the base about the ribose), and it has been attributed to sugar vibrations, including predominantly the C3′-O stretching (39). An evaluation of the order parameter is precluded in the phospholiponucleoside case by the fact that this absorption is also due to the C-O symmetrical stretching vibration of the ester group. A comparison with DOPC bilayers (Holmgren) and our data for POPC reveals that for conventional phospholipids the vibration of the ester C-O has a negative linear dichroism; this consideration might indicate that the positive LD found for POPU in the same spectral region and the fact that POPA shows instead a negative LD can be reasonably ascribed to a different orientation of the ribose rings with respect to the bilayer normal that overwhelms the negative contribution.
asymmetric stretching, particularly evident for POPA. In nucleic acids this band is considered a signature of the A form (C3′-endo sugar puckering mode with an anti conformation of the base about the ribose), and it has been attributed to sugar vibrations, including predominantly the C3′-O stretching (39). An evaluation of the order parameter is precluded in the phospholiponucleoside case by the fact that this absorption is also due to the C-O symmetrical stretching vibration of the ester group. A comparison with DOPC bilayers (Holmgren) and our data for POPC reveals that for conventional phospholipids the vibration of the ester C-O has a negative linear dichroism; this consideration might indicate that the positive LD found for POPU in the same spectral region and the fact that POPA shows instead a negative LD can be reasonably ascribed to a different orientation of the ribose rings with respect to the bilayer normal that overwhelms the negative contribution.
Another noteworthy feature is represented by the negative linear dichroism at 1335 cm−1 for POPA lamellar phases. This band, well visible also in the transmission spectrum, is considered the C3′ endo/anti marker band (40), which indicates the orientation of the glycosidic bond with respect to the sugar. A negative measured LD is indicative of the fact that the base is oriented preferentially parallel to the bilayer surface. In Fig. 7, an important LD signal at ∼1275 cm−1 for POPU lamellar phase arises. Following the attribution of Ivanov (36), we assign this peak to the stretching N1C6.
In Table 1, we report the order parameter arising for dichroic signals representative of the alkyl, interfacial, and base regions. The S values confirm the trend observed in diffraction experiments: POPA order parameters are considerably lower than those of POPU, whose S values are in turn lower than those of POPC with the exception of the phosphate group. The negative sign of the order parameters, determined for the CH2 symmetric stretching mode, indicates that the C2 axis of the chain is on average normal to the lipid bilayer surface.
Following the relation (41)
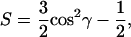 |
(4) |
we can also determine γ, the mean angle between the stretching transition moments (oriented along the bisector of the H-C-H angle) and the surface normal. This angle is 72° for POPC and 71° for POPU. The value found for POPA (62°) is not reliable due to the powder-like nature of the sample.
As far as the polar head region is concerned, the negative sign of the order parameter of the fatty ester C=O stretching supports that its direction lies mostly parallel to the bilayer interface. This result is in close agreement with some previous studies, in which it was shown that the ester group of the fatty acid in lipid membranes exhibits a planar configuration of the C-COO-C frame (18,42). The same observation holds for phosphate antisymmetric stretching modes, whose negative sign for all of the three derivatives indicates that the bisector of 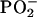 lies along the normal.
lies along the normal.
A comparison of POPU with POPC reveals an undramatic effect of the exchange choline-nucleoside concerning the hydrocarbon core of the bilayer, except that the lower absolute value of the order parameter indicates a more fluid-like phase, which can be expected on the basis of the higher cross-sectional area per lipid molecule and the lower thickness of the apolar core of the bilayer. More complex is the case of POPA: the low absolute values of the order parameters must be expected considering the unsatisfactory alignment of the smectic phase, and thus one cannot for instance ascribe the lower S value found for the symmetric stretching mode to a more fluid-like hydrocarbon phase.
The comparison with POPU indicates that the reason for this behavior resides in headgroup/headgroup interactions. Experimental evidence gathered in the past for liposomes (43) can help in the interpretation of these results. Ultraviolet hypochromism and circular dichroism spectra indicate stacking excess for POPA when locally arranged in zero-curvature aggregates. Mixtures of POPA and POPU 1:1 also show nonideal behavior, indicating that specific interactions similar to those found in nucleic acids are operating. These spectroscopic features are lost when the lipids are “diluted” with conventional surfactants, such as sodium dodecyl sulfate or octylglucoside (43). The same observations, corroborated by NMR results, have been reported by us for short-chain derivative micelles (globular or wormlike), and seem connected to the presence of an adenosine headgroup (either alone or in a 1:1 mixture with the uridine homolog), rather than to the interfacial curvature (30).
On the basis of previous results, we should expect a definite orientation pattern in the polar head region for POPA whose observed linear dichroism would be the result of the orientations of a given transition dipole moment that can be different in neighboring molecules due to the onset of interactions.
Further supporting our results, a simple geometrical optimization of POPA and POPU molecules has been performed with Hyperchem 5.1, using AMBER as a force field (44). These geometrical optimizations highlight meaningful differences in the headgroup conformation of two liponucleosides, as is visible in Fig. 8 A. In particular, the pyrimidine ring lies on average perpendicular to the bilayer plane, whereas the purine ring is oriented parallel to the membrane surface. According to Fig. 8 B, the connecting line of two nonesterified oxygens of the phosphate group of POPU is parallel to the bilayer plane, in agreement with FTIR results, whereas for POPA the alignment does not occur.
FIGURE 8.

(A) POPA and POPU geometrical optimizations performed with Hyperchem 5.1 using AMBER force field. (B) This particular of the headgroup region highlights the transition moments considered above and their orientation with respect to the bilayer surface.
When this energy minimization is run on a linear array of five or six molecules, as reported in Fig. 9, A and B, POPU molecules retain the same alignment for phosphate groups, whereas the POPA orientation (as far as this vector is concerned) varies for neighboring molecules. The geometric minimization in Fig. 9 indicates that uracyl bases are more or less oriented parallel to each other, with a slight alteration of the original conformation optimized for an isolated molecule. POPA, on the other side, arranges preferentially in pairs, varying highly its initial orientation. This behavior can be related to stronger stacking attitudes of the purine bases with respect to pyrimidines.
FIGURE 9.

Geometrical minimization for a group of POPU (A) and POPA (B) molecules performed with Hyperchem 5.1 using AMBER force field. The orientation of purinic rings is strongly altered by the interaction between bases, whereas the pyrimidinic bases keep, more or less, their original conformation.
To gain experimental support for the indications on base arrangement inferred from calculations, we have to look for vibration modes of molecular groups located on the bases. Unfortunately, the characteristic vibrations of uracyl (C=O, C-N, C=C, N-H) fall in frequency ranges where they overlap with other vibrational modes, so that it is very hard to perform an unambiguous assignment of the marker bands. The only distinctive uracyl band for which we can determine the order parameter (S = −0.33) is N1C6 stretching (1275 cm−1), whose dipolar moment results in an orientation almost parallel to the bilayer surface, in agreement with the simulated structure (see Fig. 8 B). For adenine, the peak at 1675 cm−1, δ(NH) of NH2 shows a negative linear dichroism, where the bisector of the NH2 group is on average parallel to the bilayer plane (38). This observation is strengthened by the attribution of the LD negative peak at 1335 cm−1 to C3′endo/anti conformation. Both these indications are in agreement with the geometrical arrangement of adenine bases reported in Fig. 9, therefore validating the proposed arrangement.
CONCLUSIONS
We performed neutron diffraction on bilayer membranes of POPA and POPU hydrated at controlled humidity. Our results support that the uridine derivative gives well ordered lamellar stacks under the same conditions in which POPC does. POPA hydrates more slowly and to a lesser extent: the final equilibrium phase is rather powder-like, and only two diffraction orders are collectable.
In the search for a closer correlation between molecular structure and functional properties of the supramolecular organization, we performed an FTIR and LD-FTIR investigation on the same samples. The combination of structural investigation of lyotropic phases and FTIR linear dichroism provides a wealth of information that is intimately correlable. For lecithin headgroup phospholipids much is known; more sophisticated derivatives with more conformational degrees of freedom can present simultaneously inside the bilayer and on the polar head different levels of molecular ordering.
POPA and POPU have the same glycerol-acyl chain set and the same charged group. What differentiates them is tiny from a structural point of view, but induces major differences in the lyotropic equilibria, as neutron data highlight.
LD-FTIR supports the diffraction results, indicating that these changes can be traced back to different orientations of the base moieties with respect to the bilayer normal. We interpret these differences as stemming from an orientation pattern for POPA. This conclusion is confirmed by simple geometrical optimization of a linear array of molecules of POPU and POPA, for which intermolecular interactions between neighboring molecules clearly emerge.
Acknowledgments
The authors are deeply grateful to Prof. Michael F. Brown (University of Tucson).
We acknowledge European Union financing for the neutron diffraction measurements (HPRI-CT-2001-00138 414; RII3-CT-2003-505925 1063). S.M., F.B.B., D.B., and P.B. thank the Consorzio Interuniversitario per lo sviluppo dei Sistemi a Grande Interfase and the Ministero dell‘Istruzione, dell’Università e della Ricerca for funding the research.
References
- 1.Berti, D., F. Baldelli Bombelli, M. Almgren, and P. Baglioni. 2003. Micellar aggregates formed by dilauroylphosphatidylnucleosides. In Self-Assembly. B. H. Robinson, editor. IOS Press, Amsterdam, The Netherlands.
- 2.Berti, D., U. Keiderling, and P. Baglioni. 2002. Supramolecular structures formed by phospholiponuclosides: aggregational properties and molecular recognition. Prog. Colloid Polym. Sci. 120:64–73. [Google Scholar]
- 3.Berti, D., L. Franchi, P. Baglioni, and P. L. Luisi. 1997. Molecular recognition in monolayers. Complementary base pairing in dioleoylphosphatidyl derivatives of adenosine, uridine, and cytidine. Langmuir. 13:3438–3444. [Google Scholar]
- 4.Fuhrhop, J.-H., and J. Koning. 1994. Membranes and Molecular Assemblies: The Synkinetic Approach. The Royal Society of Chemistry, London, UK.
- 5.Gosse, C., A. Boutorine, I. Aujard, M. Chami, A. Kononov, E. Cogné-Laage, J.-F. Allemand, J. Li, and L. Jullien. 2004. Micelles of lipid-oligonucleotide conjugates: implications for membrane anchoring and base pairing. J. Phys. Chem. B. 108:6485–6497. [DOI] [PubMed] [Google Scholar]
- 6.Kurihara, K., T. Abe, and N. Nakashima. 1996. Direct demonstration of attraction for a complementary pair of apposed nucleic acid base monolayers. Langmuir. 12:4053–4056. [Google Scholar]
- 7.Moreau, L., P. Barthelemy, M. El Maataoui, and M. W. Grinstaff. 2004. Supramolecular assemblies of nucleoside phosphocholine amphiphiles. J. Am. Chem. Soc. 126:7533–7539. [DOI] [PubMed] [Google Scholar]
- 8.Baglioni, P., and D. Berti. 2003. Self assembly in micelles combining stacking and H-bonding. Curr. Opin. Colloid Interface Sci. 8:55–61. [Google Scholar]
- 9.Mirkin, C. A. 2000. Programming the assembly of two- and three-dimensional architectures with DNA and nanoscale inorganic building blocks. Inorg. Chem. 39:2258–2272. [DOI] [PubMed] [Google Scholar]
- 10.Mirkin, C. A., R. L. Letsinger, R. C. Mucic, and J. J. Storhoff. 1996. A DNA-based method for rationally organizing nanoparticles into macroscopic materials. Nature. 382:607–609. [DOI] [PubMed] [Google Scholar]
- 11.Niemeyer, C. M. 2001. Nanoparticles, proteins, and nucleic acids: biotechnology meets materials science. Angew. Chem. Int. Ed. Engl. 40:4128–4158. [DOI] [PubMed] [Google Scholar]
- 12.Niemeyer, C. M. 2002. The developments of semisynthetic DNA-protein conjugates. Trends Biotechnol. 20:395–401. [DOI] [PubMed] [Google Scholar]
- 13.Berti, D., P. L. Luisi, and P. Baglioni. 2000. A SANS investigation on micelles from short-chain phospholiponucleosides. Colloids Surf. A. 167:95–103. [Google Scholar]
- 14.Berti, D. 1996. Reactivity and molecular recognition in organized system. PhD thesis. University of Florence, Florence, Italy.
- 15.Shuto, S., S. Ueda, S. Imamura, K. Fukukawa, A. Matsuda, and T. Ueda. 1987. A facile one-step synthesis of 5′-phosphatidylnucleosides by an enzymatic two-phase reaction. Tetrahedron Lett. 28:199–202. [Google Scholar]
- 16.Shuto, S., Itoh, H., S. Ueda, S. Imnamura, K. Fukukawa, A. Matsuda, M. Tsujino, and T. Ueda. 1988. A facile enzymatic synthesis of 5′-(3-sn-phosphatidyl)nucleosides and their antileukemic activities. Chem. Pharm. Bull. 36:209–217. [DOI] [PubMed] [Google Scholar]
- 17.Franks, N. P., and W. R. Lieb. 1979. The structure of lipid bilayers and the effects of general anaesthetics: An X-ray and neutron diffraction study. J. Mol. Biol. 133:469–500. [DOI] [PubMed] [Google Scholar]
- 18.Holmgren, A., L. B. Å. Johansson, and G. Lindblom. 1987. An FTIR linear dichroism study of lipid membranes. J. Phys. Chem. 91:5298–5301. [Google Scholar]
- 19.Johansson, L. B. Å., and G. Lindblom. 1980. Orientation and mobility of molecules in membranes studied by polarized light spectroscopy. Q. Rev. Biophys. 13:63–118 [DOI] [PubMed] [Google Scholar]
- 20.Nillson, A., A. Holmgren, and G. Lindblom. 1994. An FTIR study of the hydration and molecular ordering at phase transitions in the monooleoylglycerol/water system. Chem. Phys. Lipids. 71:119–131. [Google Scholar]
- 21.Armen, R. S., O. D. Uitto, and S. E. Feller. 1998. Phospholipid component volumes: determination and application to bilayer structure calculations. Biophys. J. 75:734–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Kruyff, B., R. A. Demel, A. J. Slotboom, L. L. M. van Deenen, and R. F. Rosenthal. 1973. The effect of the polar headgroup on the lipid-cholesterol interaction: a monolayer and differential scanning calorimetry study. Biochim. Biophys. Acta. 307:1–19. [DOI] [PubMed] [Google Scholar]
- 23.Seelig, A., and J. Seelig. 1977. Effect of a single cis double bond on the structure of a phospholipid bilayer. Biochemistry. 16:45–50. [DOI] [PubMed] [Google Scholar]
- 24.Rand, R. P., N. Fuller, V. A. Parsegian, and D. C. Rau. 1988. Variation in hydration forces between neutral phospholipid bilayers: evidence for hydration attraction. Biochemistry. 27:7711–7722. [DOI] [PubMed] [Google Scholar]
- 25.Dante, S., T. Hauss, and N. A. Dencher. 2002. β-Amyloid 25 to 35 is intercalated in anionic and zwitterionic lipid membranes to different extents. Biophys. J. 83:2610–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binder, H., and K. Gawrisch. 2001. Effect of unsaturated lipid chains on dimensions. Molecular order and hydration of membranes. J. Phys. Chem. B. 105:12378–12390. [Google Scholar]
- 27.Cevc, G. 1993. Phospholipids Handbook. Marcel Dekker, New York.
- 28.Mitchell, P. R., and H. Sigel. 1978. A proton nuclear-magnetic-resonance study of self-stacking in purine and pyrimidine nucleosides and nucleotides. Eur. J. Biochem. 88:149–154. [DOI] [PubMed] [Google Scholar]
- 29.Solie, T. N., and J. A. Schellman. 1968. The interaction of nucleosides in aqueous solution. J. Mol. Biol. 33:61–77. [DOI] [PubMed] [Google Scholar]
- 30.Berti, D., F. Pini, J. Teixeira, and P. Baglioni. 1999. Micellar aggregates from short-chain phospholiponucleosides: a SANS study. J. Phys. Chem. B. 103:1738–1745. [Google Scholar]
- 31.Berti, D., P. L. Barbaro, I. Bucci, and P. Baglioni. 1999. Molecular recognition through H-bonding in micelles formed by dioctylphosphatidyl nucleosides. J. Phys. Chem. B. 103:4916–4922. [Google Scholar]
- 32.Saenger, W. 1984. Principles of Nucleic Acid Structure. Springer Verlag, New York.
- 33.Banyay, M., M. Sarkar, and A. Graslund. 2003. A library of IR bands of nucleic acids in solution. Biophys. Chem. 104:477–488. [DOI] [PubMed] [Google Scholar]
- 34.Fringeli, U. P., and H. H. Gunthard. 1981. Infrared membrane spectroscopy. In Membrane Spectroscopy. E. Grell, editor. Springer-Verlag, Berlin, Germany. 270–332. [DOI] [PubMed]
- 35.Bellamy, L. J. 1958. The Infrared Spectra of Complex Molecules, Chapman and Hall, London.
- 36.Ivanov, A. Y., S. A. Krasnokutski, G. Sheina, and Y. P. Blagoi. 2003. Conformational structures and vibrational spectra of isolated pyrimidine nucleosides: Fourier transform infrared matrix isolation study of 2-deoxyuridine. Spectrochim. Acta A. 59:1959–1973. [DOI] [PubMed] [Google Scholar]
- 37.Yohimasa, K., R. C. Lord, and A. Rich. 1967. An infrared study of hydrogen bonding between adenine and uracil derivatives in chloroform solution. J. Am. Chem. Soc. 89:496–504. [DOI] [PubMed] [Google Scholar]
- 38.Akutsu, H., Y. K. H. Nakahara, and K. Fukuda. 1975. Conformational analysis of phosphatidylethanolamine in multilayers by infrared dichroism. Chem. Phys. Lipids. 15:222–242. [DOI] [PubMed] [Google Scholar]
- 39.Pohle, W., and H. Fritzsche. 1980. A new conformation-specific infrared band of A-DNA in films. Nucleic Acids Res. 8:2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liquier, J., A. Akhebat, E. Taillandier, F. Ceolin, T. Huynh-Dinh, and J. Igolen. 1991. Characterization by FTIR spectroscopy of oligoribonucleotide duplexes r(A-U)6 and r(A-U)8. Spectrochim. Acta. 47A:177–186. [Google Scholar]
- 41.Saupe, A. 1964. Nuclear resonances in crystalline liquids and in liquid crystalline solutions. Z. Naturforschg. 19a:161–171. [Google Scholar]
- 42.Fringeli, U. P. 1977. The structure of lipids and proteins studied by attenuated total reflection (ATR) infrared spectroscopy. II. Oriented layers of a homologous series: phosphatidylethanolamine to phosphatidylcholine. Z. Naturforsch. 32:20–45. [DOI] [PubMed] [Google Scholar]
- 43.Berti, D., S. Bonaccio, G. Barsacchi-Bo, P. L. Luisi, and P. Baglioni. 1998. Base complementarity and nucleoside recognition in phosphatidylnucleoside vesicles. J. Phys. Chem. B. 102:303–308. [Google Scholar]
- 44.Weiner, S. J., P. A. Kollman, D. T. Nguyen, and D. A. Case. 1986. An all atom force field for simulation of proteins and nucleic acids. J. Comput. Chem. 7:230–252. [DOI] [PubMed] [Google Scholar]