

Abstract
Interaction of the human antimicrobial peptide LL-37 with lipid monolayers has been investigated by a range of complementary techniques including pressure-area isotherms, insertion assay, epifluorescence microscopy, and synchrotron x-ray scattering, to analyze its mechanism of action. Lipid monolayers were formed at the air-liquid interface to mimic the surface of the bacterial cell wall and the outer leaflet of erythrocyte cell membrane by using phosphatidylglycerol (DPPG), phosphatidylcholine (DPPC), and phosphatidylethanolamine (DPPE) lipids. LL-37 is found to readily insert into DPPG monolayers, disrupting their structure and thus indicating bactericidal action. In contrast, DPPC and DPPE monolayers remained virtually unaffected by LL-37, demonstrating its nonhemolytic activity and lipid discrimination. Specular x-ray reflectivity data yielded considerable differences in layer thickness and electron-density profile after addition of the peptide to DPPG monolayers, but little change was seen after peptide injection when probing monolayers composed of DPPC and DPPE. Grazing incidence x-ray diffraction demonstrated significant peptide insertion and lateral packing order disruption of the DPPG monolayer by LL-37 insertion. Epifluorescence microscopy data support these findings.
INTRODUCTION
A worldwide increase in multidrug-resistant bacteria (1–4) has made it imperative to develop new classes of antibiotics. Certain antimicrobial peptides of the innate immune system can distinguish membranes having different lipid compositions (5–9). This property allows them to lyse bacterial membranes while leaving eukaryotic plasma membranes unaltered. A better understanding of this ability at a molecular level could enhance the design and development of antimicrobial peptides as alternatives to the conventional antibiotics used today.
During the last decade it has been established that antimicrobial peptides act by permeating cell membranes (7,10–12). Two main mechanisms of action of membrane perturbation by antimicrobial peptides have been previously proposed: the barrel stave model (13–17) and the carpet model (11,12,18–22).
In the barrel stave mechanism, peptide monomers bind to the lipid membrane and then assemble together to form bundles, in which the hydrophobic surfaces interact with the acyl chain part of the membrane. The hydrophilic regions align together to form a pore which may be increased in size by additional peptide monomers.
In the carpet model, peptides bind to the outside of the membrane by adsorption to the headgroup region of the lipid membrane, carpeting the surface of the membrane and orienting parallel to it. Membrane disruption may occur due to the formation of pores in the membrane or via membrane micellization. When a critical peptide/lipid ratio is reached, the peptides are thought to change orientation to a direction perpendicular to the membrane (23). It is these reorientated peptides that can then form the toroidal pores (7,22–24). The formation of toroidal pores may then be followed by the complete collapse of the membrane.
It has been proposed that human antimicrobial peptide LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES-NH2) acts by perturbing membranes via the carpet mechanism of action (19,25,26). Oren et al. (19) suggest that LL-37 carpets the surface of both zwitterionic phosphatidylcholine (PC) and negatively charged PC/phosphatidylserine (PS) vesicles. They suggest that LL-37 oligomerizes in solution, and although it is self-associated when bound to zwitterionic phospholipid vesicles, it dissociates into monomers upon binding to negatively charged vesicles. Henzler-Wildman et al. (25,26) also demonstrate that LL-37 first carpets lipid bilayers before membrane perturbation by using solid-state nuclear magnetic resonance and differential scanning calorimetry experiments. Their results support the carpet mechanism of action of lipid bilayer disruption by LL-37.
One of the most important stages of peptide-membrane interaction is an initial contact of the peptide with the outer leaflet of the plasma membrane. To investigate what defines the propensity of peptides to interact with specific membrane lipids, methods that allow the membrane to be modeled in a fluid environment are needed. One such approach is the use of a Langmuir monolayer to mimic the external leaflet of the cell membrane, coupled with the introduction of peptides into the subphase of a Langmuir trough to represent the extracellular fluid and thus the approach of the peptide toward the cell surface. Different lipid compositions can be used to represent the membranes of different cell types, and changes in membrane structure resulting from its interaction with peptides allow us to suggest a possible mechanism of peptide action.
The outer leaflet of mammalian cell membranes mainly comprises PC, phosphatidylethanolamine (PE), sphingomyelin, and cholesterol, which are charge-neutral at physiological pH.
In this work, we used dipalmitoylphosphatidylcholine (DPPC) and dipalmitoylphosphatidylethanolamine (DPPE) to study the interaction of LL-37 with the human red blood cell membrane. Nouri-Sorkhabi et al. (27) used P-31 NMR spectra of human erythrocyte lysates to find that PE accounts for 33.3% and PC makes up ∼30.3% of human erythrocyte membrane mass. Independently, Keller et al. (28) found that human red blood cell membrane contains on average ∼34% PE and 35% PC, although their distribution among the leaflets is asymmetrical: the majority of PC (∼28% total lipid content) is at the outer leaflet, whereas the majority of PE (∼28% total lipid content) is within the inner leaflet.
The surfaces of both Gram-negative and Gram-positive bacterial cell walls contain large amounts of negatively charged lipids (29,30). The outer layer of Gram-positive bacteria cell wall is composed of acidic polysaccharides (teichoic acids) and phosphatidylglycerol (PG) (31), whereas the outer leaflet of the outer membrane bilayer of the Gram-negative bacteria is predominantly composed of lipopolysaccharide (LPS), a polyanionic molecule (29,30,32–34).
In this article, we look into the interaction of LL-37 with DPPG lipid. In a consequent article (F. Neville, C. S. Hodges, C. Liu, O. Konovalov, and D. Gidalevitz, unpublished), we use lipid A, the main lipid component of lipopolysaccharides, to study Gram-negative membrane lysis by LL-37, protegrin-1, and SMAP-29 antimicrobial peptides.
A similar approach has been successfully used in conjunction with x-ray surface scattering and epifluorescence techniques to study interactions of phospholipid monolayers with the porcine antimicrobial peptide protegrin-1 (35). Surface x-ray scattering techniques yield information about the packing arrangement of the lipid layer before and after introduction of antimicrobial peptide into the system, whereas epifluorescence microscopy allows direct observation of the changes in lipid monolayer morphology caused by peptides. Langmuir monolayer techniques and surface x-ray scattering have also been successfully applied to study interactions of an amphibian antimicrobial peptide, PGLa (9). More recently, preliminary studies on the human antimicrobial peptide LL-37 using electrochemical techniques and epifluorescence microscopy have been carried out (36).
In this article, the interaction of LL-37 with phospholipid monolayers is further studied using a combination of pressure area isotherms, specular x-ray reflectivity (XR), grazing incidence x-ray diffraction (GIXD), and epifluorescence microscopy (EFM) in a complementary manner, with the purpose of probing the question of antimicrobial peptide selectivity between prokaryotic and eukaryotic cells and its possible mechanism of action. Discrimination of LL-37 between monolayers modeling outer layers of red blood cells and bacterial membranes has been demonstrated. Although no interactions of LL-37 with DPPC or DPPE monolayers were observed, significant insertion of LL-37 into DPPG monolayers was documented with each of the experimental techniques used. The mechanism of LL-37 insertion into DPPG monolayers is consistent with a “carpet” mechanism proposed earlier.
METHODS
Lipid monolayers
To study peptide-membrane interactions, Langmuir monolayers composed of lipids representative of membranes were studied. This approach has been utilized repeatedly over the years (37–43). The planar monolayer system allows versatile adjustments of parameters such as monolayer composition, surface pressure, packing density, temperature, and aqueous subphase condition to model the conditions under which peptides approach the outer surface of the membrane from the extracellular medium. The cell membranes of different organisms have characteristic lipid compositions. However, to develop a full understanding of membrane interactions, each component of a membrane should be studied separately to ascertain the contribution of each membrane component to the overall interaction of the peptide with the membrane. DPPC, DPPE, and dipalmitoylphosphatidylglycerol (DPPG) were used to form Langmuir monolayers. All lipids were purchased from Avanti Polar Lipids (Alabaster, AL) and were used without further purification.
Pressure-area compression isotherms
All experiments were performed using Dulbecco's phosphate buffered saline (DPBS) (Invitrogen, Carlsbad, CA) without calcium and magnesium ions. Monolayers of DPPC and DPPE were deposited from chloroform (high-performance liquid chromatography grade, Fisher Scientific, Pittsburgh, PA) solution, whereas DPPG was deposited from 9:1 v/v % chloroform/methanol solution (high-performance liquid chromatography grade, Fisher Scientific). Upon spreading, the lipid film was left undisturbed for 15 min to allow for solvent evaporation. At this point, barrier compression was initiated and the increase in surface pressure of the monolayer was monitored. This gives rise to a surface pressure (mN/m) versus area per lipid molecule (Å2/molecule) isotherm, which can be utilized to deduce the phases and phase transitions associated with the monolayer as a function of lateral lipid packing density.
Insertion experiments
Insertion experiments were carried out to quantify the interaction of LL-37 with the lipid monolayer. Initially, the lipid monolayer was deposited and equilibrated, followed by compression to the surface pressure corresponding to the liquid-condensed phase of lipids (30–40 mN/m), and equivalent to the packing density of the cell membrane (44,45). The surface pressure was kept constant via a built-in proportional-integral-derivative control feedback system by adjusting the surface area. The LL-37 solution (10 μg/ml LL-37 in 0.01% glacial acetic acid w/v) was then evenly injected underneath the monolayer using a microsyringe with an L-shaped needle (VDRL needle; Hamilton, Reno, NV) to make up the final concentration of 0.04 μg/ml or 0.1 μg/ml. The surface pressure immediately increased as a result of peptide incorporation into the lipid monolayer. Injected peptides interact with the lipid monolayer and result in an increase in the surface pressure. To keep the surface pressure constant, the surface area would have to increase. The resulting relative change in area per molecule, ΔA/A, was monitored throughout the experiment to compare the degree of LL-37 insertion into DPPC, DPPE, and DPPG monolayers. Epifluorescence microscopy was used concurrently with insertion experiments to monitor the surface morphology of the monolayers on insertion of LL-37; this involved the incorporation of a small amount of fluorescent dye into the different lipid-spreading solutions.
LL-37 at the interface
LL-37 (90–95% pure) was supplied by Pepceuticals (Nottingham, UK). LL-37 peptide was provided as a solid powder and made up to a working solution (10 μg/ml LL-37 in 0.01% glacial acetic acid w/v). Acetic acid was used to maintain the peptide structure while in solution. LL-37 is soluble in water, but, being amphipathic (46), is expected to be adsorbed at the air-liquid interface. LL-37 was injected into the pure subphase without a lipid monolayer present. The properties of pure LL-37 monolayer were then investigated using pressure-area isotherms in conjunction with x-ray reflectivity and grazing incidence x-ray diffraction.
Langmuir troughs
Pressure-area compression isotherms were performed using a twin-barrier rectangular Teflon micro Langmuir-Blodgett trough equipped with a Wilhelmy plate (Nima Technology, Coventry, UK). Insertion experiment data presented here were obtained using a custom-built trough (35,47) at the University of Chicago. X-ray scattering measurements were taken at the European Synchrotron Radiation Facility (ESRF) (9) and Advanced Photon Source (APS) synchrotrons, both of which utilizing custom-built Langmuir troughs equipped with a single moveable barrier. The subphase temperature was maintained at 22 ± 1°C for lateral compression and x-ray scattering experiments, and at 30 ± 1°C for insertion assays.
Epifluorescence microscopy
The Langmuir trough used for insertion experiments was equipped with an epifluorescence microscope mounted to observe the phase morphology of the lipid monolayer. The epifluorescence microscopy techniques were carried out as previously described (36,47–50). A resistively heated indium tin-oxide-coated glass plate was placed over the trough to minimize contamination and condensation on the microscope objective lens and to minimize. Excitation between 530 and 590 nm and emission between 610 and 690 nm was gathered through the use of an HYQ Texas Red filter cube. Lipid-linked Texas Red dye ((TR-DHPE) Molecular Probes, Eugene, OR; 0.5 mol %) was incorporated into the spreading phospholipid solutions. Due to steric hindrance, the dye partitions into the disordered phase, rendering it bright and the ordered phase dark. This assembly permits the monolayer morphology to be observed over a large lateral area while isotherm data are obtained concurrently.
GIXD and XR
Surface x-ray scattering experiments were carried out at the ID10B (Troïka II) beam line at the ESRF (Grenoble, France), and at the 9-ID (CMC-CAT) beam line at the APS, Argonne National Laboratory (Argonne, IL). The oxygen level was monitored as the vessel was filled with water-saturated helium and was allowed to reach a sufficiently low level before measurements were commenced (<0.1%). Water-saturated helium was used to reduce evaporation and scattering from the air. Control measurements of pure lipid monolayers were followed by subsequent injection of the desired amount of peptide into the subphase under the monolayer-covered area of the surface at 30 mN/m.
GIXD (51) was used to obtain in-plane information concerning the molecular structure of surfaces (52). For GIXD measurements, the angle of incidence (αi) was set to strike the air-aqueous interface at 0.8 αc, where αc is the critical angle for total external reflection. A linear position-sensitive detector that detected the diffracted beam recorded the intensity profile as a function of scattering angle. A Soller collimator with horizontal plates was placed after the detector. Such a configuration enables the incident wave to be reflected, whereas the refractive wave propagates along the surface, making this technique very sensitive to changes at the air-aqueous interface. The incident wavelength used at the ESRF was 1.54 Å, and the wavelength at APS was 0.92 Å.
Specular x-ray reflection measurements reveal information on the electron-density distribution along the surface normal and may be used to determine the density and thickness of thin layers (41,51,53). When specular x-ray reflection occurs, the scattering vector qz may be calculated from qz = 4π sin α/λ, where α is the grazing angle of the incident beam and λ the wavelength of the x-ray beam. When the reflectivity is measured as a function of the scattering vector qz, the reflectivity curve contains information regarding the gradient of the electron-density profile in the direction normal to the surface (54,55). XR measurements were carried out over a range of angles corresponding to qz values of ∼0–0.65 Å−1. A position-sensitive detector was used for detection and the reflected beam intensity was measured as a function of the incident angle.
RESULTS AND DISCUSSION
Pressure-area compression isotherms
Pressure-area (π-A) isotherms are a traditional method for the investigation of Langmuir monolayer phase behavior. As the area decreases at constant temperature, any increase in pressure on compression may be recorded.
Fig. 1 A shows typical π-A isotherms of DPPC, DPPE, and DPPG that exhibit trends similar to those described in earlier publications (35,50,56), as well as the π-A isotherm of LL-37. As found from the Langmuir isotherm, the area per DPPG molecule in a monolayer compressed at 30 mN/m is 45.6Å2 and that for DPPC is 46.4Å2 at the same pressure. These values will be compared with area per molecule values found from grazing incidence x-ray diffraction data.
FIGURE 1.

Pressure-area isotherms of pure lipids DPPC (solid line), DPPE (dashed line), and DPPG (dash-dotted line), and pure LL-37 peptide (dotted line). LL-37 isotherm has been presented at a scale of one-fifth of its real area per molecule; pressure values are those actually recorded.
Fig. 1 shows the surface pressure-area isotherm of an LL-37 peptide. Contrary to insoluble amphiphilic molecules like phospholipids, which are spread on an aqueous surface from a chloroform solution, LL-37 is water-soluble and a peptide surface film is created via its absorption from the bulk subphase at the interface after peptide injection into the DPBS buffer. Since an increase in the surface pressure can be seen with a decreased surface area, it is likely that the LL-37 aligns at the air-aqueous interface and is compressed slightly to increase the surface pressure before collapse at ∼30 mN/m. This is very similar to previous results for the PGLa peptide (9,57), which is reported to adapt α-helical conformation. Assuming that the LL-37 molecule is linear and α-helical in shape (19,58,59), with the approximate helix diameter of the molecule being ∼10 Å and a length of 1.5 Å per amino acid residue (60), the molecular area of an LL-37 peptide may be calculated to be in the region of A = ∼550 Å2 (assuming that helix volume is based approximately on a cylindrical shape).
Constant-pressure insertion isotherms
Constant-pressure isotherm experiments involve compression of the monolayer to the desired pressure followed by peptide injection into the subphase. Any increase or decrease in the surface pressure from the setpoint after peptide insertion will result in the compression or expansion of the barrier(s) to advert the change. Increase in the surface pressure value after injection is due to peptide insertion into the lipid layer and will therefore result in barrier expansion.
Constant-pressure insertion isotherms show little or no LL-37 insertion into DPPC and DPPE monolayers (Fig. 2, A and B), whereas a substantial increase in DPPG area per molecule indicates incorporation of peptide molecules into the monolayer structure (Fig. 2, A and B). However, when DPPG surface pressure is increased to 40 mN/m and peptide is injected to reach a subphase concentration of 0.1 μg/ml (Fig. 2 C), a critical destabilization of the monolayer takes place.
FIGURE 2.

Insertion isotherms of DPPC and DPPG showing percentage change in area per molecule after injection of LL-37. (A) Systems at concentration 0.04 μg/ml LL-37: DPPG + LL-37 at 30 mN/m (solid line); DPPC + LL-37 at 30 mN/m (dashed line); DPPE + LL-37 at 30 mN/m (dotted line); DPPG + LL-37 at 40 mN/m (dash-dotted line). (B) Systems at concentration 0.1 μg/ml LL-37: DPPG +LL-37 at 30 mN/m (solid line); DPPG + LL-37 at 40 mN/m (dashed line); DPPC + LL-37 at 30 mN/m (dotted line). (C) Critical destabilization of DPPG at 40 mN/m after injection of 0.1 μg/ml LL-37 (dashed line). (Concentration values refer to final concentrations of peptide in solution.)
Fig. 2 A illustrates the comparison of insertion results using the lower concentration of LL-37 (0.04 μg/ml) at constant pressures of 30 mN/m and 40 mN/m for DPPC, DPPE, and DPPG monolayers, whereas Fig. 2 B contains data for the higher concentration of LL-37 used (0.1 μg/ml). The DPPG insertion isotherms (Fig. 2, A and B), indicate that at 30 mN/m, increasing the concentration of the LL-37 by 2.5 times results in a subsequent fourfold increase in area per DPPG molecule (Fig. 2 B). On the other hand, an increase of LL-37 concentration in the subphase at the same pressure does not result in any substantial area per molecule increase in the DPPC monolayer (Fig. 2 B). From Fig. 2 A, it also appears that LL-37 inserts into the DPPG monolayer to the same final extent at both 30 and 40 mN/m at the lower concentration of LL-37, although the initial rate of insertion is greater at 30 mN/m (Fig. 2 A).
When the peptide concentration reaches 0.1 μg/ml (Fig. 2 B), a pronounced difference is seen between the insertion isotherms taken at 30 mN/m and those taken at 40 mN/m. A very large increase in area (∼180%) was seen on injection of 0.1 μg/ml LL-37 under the DPPG monolayer at 30 mN/m. However, when the pressure of the system was kept at 40 mN/m, injection of the peptide resulted in insertion during the first 5–10 min and then a gradual decrease in area to a value below that seen before the introduction of the peptide (Fig. 2 C). This effect is very similar to that of introducing the porcine PG-1 peptide into a lipid A system at 35 mN/m (35). It is likely that at this higher concentration and pressure, a critical threshold has been reached, resulting in a critical destabilization of the DPPG monolayer. This result is consistent with permeabilization of the bacterial membrane by LL-37 via the “carpet” route, which involves a threshold concentration of peptide to carpet the membrane before any pore formation can occur.
Epifluorescence
EFM measurements were performed simultaneously with the insertion isotherms to monitor the effect of peptide binding on the morphology of the monolayer. Fluorescence image contrast arises due to different phase densities and partitioning characteristics of the dye molecules in coexisting phases. Therefore, it is possible to gain insight into the structure of the lipid layer by imaging its lateral fluorescence distribution.
EFM images of the pure lipid monolayers at both 30 and 40 mN/m display an array of very densely packed “dark gray” domains of condensed phase with narrow, “light gray” liquid-phase borders between them. Little or no change in DPPE and DPPC morphology was observed after injection of LL-37 at either 0.04- or 0.1-μg/ml concentration (Fig. 3, A and C). A very slight increase in the amount of bright disordered phase of DPPC at the higher peptide concentration was observed after 19 min (Fig. 3 C3).
FIGURE 3.

Epifluorescence images of systems at 30 mN/m. (A1) DPPE monolayer at 30 mN/m before injection. (A2) DPPE monolayer 1 min after 0.04 μg/ml LL-37 injection. (A3) DPPE monolayer 2 min after 0.04 μg/ml LL-37 injection. (B1) DPPC monolayer at 30 mN/m before injection. (B2) DPPC monolayer 1 min after 0.04 μg/ml LL-37 injection. (B3) DPPC monolayer 6 min after 0.04 μg/ml LL-37 injection. (C1) DPPC monolayer at 30 mN/m before injection. (C2) DPPC monolayer 18 min after 0.1 μg/ml LL-37 injection. (C3) DPPC monolayer 19 min after 0.1 μg/ml LL-37 injection. (Concentration values 0.04 μg/ml and 0.1 μg/ml refer to final concentrations of peptide in solution.)
In contrast, DPPG displays profound changes at both pressures and concentrations of LL-37 used (Fig. 4). At 30 mN/m, with injection of the lower concentration of LL-37 (Fig. 4 A), a significant increase in the area of bright disordered phase and a decrease in the dark condensed-phase area can be seen, indicating substantial insertion of the peptide into the disordered phase of the monolayer. After only 3 min (Fig. 4 A3), large regions of disordered lipid phase appeared, and this quickly developed into a structure in which the majority of the lipid domains present were disordered and the remaining condensed phase dissipates into a “web-like” structure (Fig. 4 A5). When LL-37 was injected under the DPPG monolayer at 30 mN/m to give a final concentration of 0.1 μg/ml (Fig. 4 B), there ws an immediately noticeable difference in the epifluorescence images. The image taken before LL-37 injection (Fig. 4 B1) shows clear separation of the condensed and disordered phases. Immediately after introduction of LL-37 (Fig. 4 B2), it is clear that the domain edge becomes fuzzy, most likely due to the high speed of insertion of the LL-37 into the monolayer and the use of the domain edge as the preferred location for insertion.
FIGURE 4.

(A1) DPPG monolayer at 30 mN/m before injection and at 2 min (A2), 3 min (A3), 4 min (A4), 10 min (A5), and 20 min (A6) after LL-37 injection. (B1) DPPG monolayer at 30 mN/m before injection and at 4 min (B2) and 7 min (B3) after 0.1 μg/ml LL-37 injection. (Concentration values refer to final concentrations of peptide in solution.)
When the DPPG system was studied at the higher constant pressure of 40 mN/m, quite a different pattern of insertion was observed. The images before injection of LL-37 at either concentration of LL-37 demonstrate what seems to be a more closely packed layer, with clearly defined regions of condensed and disordered phase domains, due to the fact the monolayer is further compressed at 40 mN/m. After injection of 0.04 μg/ml LL-37, regions of bright disordered phase quickly appear (Fig. 5 A2). After 10 min (Fig. 5 A5), the monolayer is already mostly disordered by the LL-37 insertion and condensed-phase dark regions are indistinct.
FIGURE 5.

Epifluorescence images of DPPG + LL-37 at 40 mN/m. (A1) DPPG monolayer at 40 mN/m before injection, and at 3 min (A2), 6 min (A3),7 min (A4), 10 min (A5), and 13 min (A6) after 0.04 μg/ml LL-37 injection. (B1) DPPG monolayer at 40 mN/m before injection and at 8 min (B2), 14 min (B3), 31 min (B4), 63 min (B5), and 92 min (B6) after 0.1 μg/ml LL-37 injection. (Concentration values 0.04 μg/ml and 0.1 μg/ml refer to final concentrations of peptide in solution.)
When the higher concentration of LL-37 was injected under the DPPG monolayer at 40 mN/m (Fig. 5 B), the disordered phase grew larger whereas the size of the condensed-phase domains decreased. Nevertheless, distinct condensed-phase domains were clearly visible even half an hour after peptide injection (Fig. 5 B4). At the next stage of the film evolution, solid domains shrunk further with the phase separation border becoming clearly blurred (Fig. 5 B5), not unlike Fig. 5 A6. At this stage (see insertion isotherm at Fig. 2 B), the film was slowly disintegrating and at 1.5 h after peptide insertion not only had the condensed phase disappeared, but new, spontaneously-formed, small bright domains were visible at places formerly occupied by solid domains (Fig. 5 B6). It is possible that the peptide insertion causes the lipid to form some kind of self-assembled structure that combines both the lipid and the peptide. This suggests that the DPPG monolayer is destabilized due to the fact that it has been saturated with LL-37, which agrees with the “carpet” mechanism of LL-37 interaction with DPPG at this concentration and surface pressure.
Grazing incidence x-ray diffraction
Grazing incidence x-ray diffraction measurements are made with the x-ray momentum transfer in or close to the plane of the air-aqueous interface. The reflections of the Bragg peaks observed with this geometry can be indexed by two Miller indices, hk. Their angular positions 2θhk, corresponding to qhk = (4π/λ)sinθhk, yield the repeat distances dhk = 2π/qhk for the two-dimensional lattice structure (40,55).
GIXD was performed in conjunction with insertion isotherms (not shown here but similar to those taken with fluorescence experiments) to monitor the effect of peptide insertion on the molecular packing of the lipid monolayers of different compositions. Bragg peak profiles (intensity against qxy) were fitted with Gaussians and the peak position values were used to obtain unit-cell dimensions of the lipid lattices, whereas full-width half-maximum values of the peaks were used to determine the coherence length from the Scherrer formula, L = 0.9 × 2π/full-width half-maximum (qxy) (61,62). Bragg rod profiles were measured at Bragg peak positions. Bragg rod profiles were analyzed to determine the tilt of the hydrocarbon chains (52).
The observation of two Bragg peaks in the diffraction pattern of an amphiphilic monolayer is indicative of a distorted hexagonal unit cell (which can be viewed as centered rectangular). Therefore, all unit-cell dimensions in this study were calculated using the centered rectangular unit-cell approximation.
GIXD—DPPC
Pure DPPC monolayer data (Fig. 6 A1) show two Bragg peaks corresponding to d-spacings of 4.60 Å and 4.26 Å. This translates to a centered rectangular unit cell with dimensions of a = 5.47 Å and b = 8.53 Å and an area per DPPC molecule of A = 46.6 Å2, as each unit cell contains two hydrocarbon chain components of the phospholipid molecule. This area per molecule value agrees well with that obtained from the pressure-area isotherm (Fig. 1) and those obtained in previous studies (9,54,57,63–65). The coherence lengths, calculated with the Scherrer formula (52,62), give values of ∼126 Å and 1061 Å for the {1,1} (L11) and {0,2} (L02) reflections, respectively. Analysis of Bragg rod profile yields a molecular tilt of 30° for the lipid molecules in the condensed phase. These values are again very close to those obtained by other authors (9,54,57,63–65). The area per molecule obtained from GIXD data is valid only for the ordered part of the monolayer. The fact that the area per molecule from the π-A isotherm corresponds to values obtained from GIXD indicates that the monolayer is mostly ordered, corroborating our EFM observations.
FIGURE 6.

Bragg peak/rod combination plot of scattering vectors qxy and qz as a function of their respective intensities. (A) DPPC monolayer at 30 mN/m. (B) DPPC monolayer at 30 mN/m after 0.04 μg/ml LL-37 injection into the subphase. (C) DPPG monolayer at 30 mN/m. (D) DPPG monolayer at 30 mN/m after 0.04 μg/ml LL-37 injection into the subphase. The initial Bragg peaks from scattering of ordered structure of the monolayer vanish upon LL-37 insertion into the DPPG monolayer, whereas the DPPC monolayer was left practically unchanged. (Concentration values 0.04 μg/ml and 0.1 μg/ml refer to final concentrations of peptide in solution.)
Fig. 6 A2 shows a slight change in DPPC packing after LL-37 injection at a constant pressure of 30 mN/m. Here, the second peak position shifts only very slightly,yielding d-spacings of 4.64 and 4.27 Å. This computes to unit-cell dimensions of a = 5.52 Å and b = 8.54 Å and an area per molecule of A = 47.1 Å2, showing little interaction of LL-37 with the phosphatidylcholine headgroup. The coherence lengths changed only slightly from the DPPC monolayer alone, to L11 = 128 Å and L02 = 1193 Å. The calculated molecular tilt value did not change and was 30° after the injection of LL-37. This again is corroborated by the insertion isotherms and epifluorescence data (Figs. 2–5), which also show little change in lipid packing.
GIXD—DPPG
Pure DPPG at 30 mN/m (Fig. 6 B1) yields two peaks corresponding to d-spacings of 4.51 Å and 4.25 Å, centered-rectangular unit-cell dimensions of a = 5.32 Å and b = 8.51 Å, and an area per molecule of A = 45.2 Å2. This value is again in good agreement with that obtained using the area-pressure compression isotherm and with previously published GIXD results (9,35,57). The coherence lengths for a DPPG monolayer at 30 mN/m are calculated to be L11 = 75 Å and L02 = 448 Å. The molecular tilt is found to be 27° for the DPPG monolayer.
After LL-37 insertion, no Bragg peaks or rods were observed, indicating that the ordered structure of the DPPG monolayer has been totally disrupted by LL-37 (Fig. 6 B2). This is again confirmed by insertion isotherms (Fig. 2) and epifluorescence data (Fig. 4), which show increases in area per molecule and an increased area fraction of the disordered bright phase, respectively, demonstrating disruption of the membrane by the peptide. For easy comparison of the GIXD data, results for both the DPPC and DPPC lipid systems are shown in Table 1.
TABLE 1.
Grazing incidence x-ray diffraction data for systems at 30 mN/m, 22°C
| Coherence length (Å)
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Experiment | d11 (Å) | d02 (Å) | a (Å) | b (Å) | Area per molecule (Å2) | Tilt angle (°°) | L11 | L02 |
| DPPC | 4.60 | 4.26 | 5.47 | 8.53 | 46.6 | 30 | ∼126 | ∼1061 |
| DPPC + 0.04 μg/ml LL-37 | 4.64 | 4.27 | 5.52 | 8.54 | 47.1 | 30 | ∼128 | ∼1193 |
| DPPG | 4.51 | 4.25 | 5.32 | 8.51 | 45.2 | 27 | ∼75 | ∼448 |
| DPPG + 0.04 μg/ml LL-37 | No peaks visible | NA | NA | |||||
“11” and “02” are used to denote hk for a set of Bragg rods with equal in-plane components, which cannot be resolved from the GIXD data. In this case, data are calculated using the rectangular unit cell, thus {11} lattice means {(11), (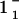 ), (
), (  ), (
), (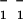 )} and {02} means {(02), (
)} and {02} means {(02), (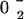 )}.
)}.
X-ray reflectivity
Specular x-ray reflection measurements record the intensity of those x-rays scattered from the air-aqueous interface with a momentum transfer strictly perpendicular to the interface. The structure factor associated with the XR profile is determined by the Fourier transform of the gradient of the electron density perpendicular to the interface. The data cannot in general be uniquely inverted to yield the electron-density profile (66). It is a common practice for XR data to be fitted to a model of the interface consisting of a stack of uniform slabs, each with a different electron density ρi and thickness Li (40,66–69). For example, each lipid molecule may be modeled with two slabs, corresponding to the head and tail regions of the molecule, with different electron densities and thicknesses. The effect of capillary waves on the density distribution in the interface is modeled by a Gaussian roughness, σ. The XR data were analyzed with RFit2000 (70–74), which uses elements of global minimization (75) to calculate the reflectivity based on the Parrat method (76).
XR—DPPC
Fig. 7 A shows the reflectivity curve normalized to the Fresnel reflectivity of a planar interface of the subphase (R/RF) against the scattering vector (q) in the z direction (qz) for the DPPC monolayer at 30 mN/m before and after injection of LL-37 into the subphase. It shows the measured experimental data obtained and the calculated least-square fitted model. Successful fits to the data demonstrated that the DPPC monolayer may be modeled as two slabs with different thicknesses and electron densities. The thickness of the tail layer was found to be 15.2 Å and the headgroup layer thickness 8.8 Å, with tail- and head-layer electron-density values (normalized to the electron density of the subphase) of 0.91 and 1.33, respectively. The roughness of the layers was found to be 3.3 Å. These values agree well with previously published values (54,63,68).
FIGURE 7.

Reflectivity data and corresponding fits normalized by Fresnel reflectivity plotted against scattering vector (qz). (A) DPPC monolayer reflectivity data (square) and fit (solid line), DPPC after 0.04 μg/ml LL-37 injection, and data (circle) and fit (dashed line) 0.1 min after 0.04 μg/ml LL-37 injection; (A4) 7 min after 0.04 μg/ml LL-37 injection; and (A5) 10 min after 0.04 μg/ml LL-37 injection. (A6) DPPG monolayer 13 min after 0.04 μg/ml LL-37 injection. (B1) DPPG monolayer at 40 mN/m before injection and at 8 min (B2), 14 min (B3), 31 min (B4), 63 min (B5), and 92 min (B6) after 0.1 μg/ml LL-37 injection. (Concentration values 0.04 μg/ml and 0.1 μg/ml refer to final concentrations of peptide in solution.)
In addition to the insertion, epifluorescence, and GIXD results, the XR data also show little difference in the DPPC monolayer electron-density profile after LL-37 injection. The data of the DPPC/LL-37 system was again modeled with two slabs. Two different concentrations of LL-37 were used in these experiments: 0.04 μg/ml and 0.1 μg/ml. The DPPC + 0.04 μg/ml LL-37 data yielded a tail thickness of 16.2 Å and a head-layer thickness of 8.2 Å. The normalized tail and head electron-density values were 0.95 and 1.38, respectively, and a molecular roughness between layers was determined to be 3.5 Å. The fit to the data of the system where 0.1 μg/ml LL-37 was injected under the DPPC monolayer gave values similar to those presented above. The tail layer was found to be 16.5 Å thick, with a normalized electron density of 0.95, and the head layer was 6.9 Å thick, with a density of 1.39. Both layers had a roughness value of 3.5 Å.
XR—DPPG
The R/RF plot in Fig. 7 B shows the DPPG data obtained at 30 mN/m. The best fit was again obtained using two slabs, as with DPPC, yielding 18.1 Å for the tail layer thickness and 5.9 Å for the headgroup layer thickness. The normalized electron-density values for the tail and headgroup layers were 0.98 and 1.55, respectively, with a roughness value of 3.7 Å for both. These results are in line with previous results (35,77), with small differences most likely attributable to the use of different subphases.
Fig. 7 B shows substantial change in the reflectivity profile upon injection of LL-37 under the DPPG monolayer at 30 mN/m. Since the minima that are observable in R/RF plots are due to interference of x-ray waves reflected from the interfaces of the slabs in the surface layer, the minima are characteristic of the thickness of the lipid monolayer. After injection of 0.04 μg/ml LL-37, the XR profile is very different from that of the DPPG monolayer alone, with the minimum of the curve being shifted to a greater qz value by ∼0.11 Å−1. This indicates a decrease in monolayer thickness, similar to that observed by Huang et al. (78). Although the amplitude of the second peak was reduced, it was possible to fit the data using a two-slab model (Fig. 8). The top slab has been modeled as the lipid tailgroups, with peptide inserted slightly into the hydrocarbon tail region, and the second slab has been modeled as a mix of headgroups and peptide. The least-square fit parameters gave 7.4 Å for the thickness of the top layer (the layer closest to the air), with a normalized electron density of 1.23, and 8.1 Å for the thickness of the second layer, with a density of 1.33. The roughness obtained was 5.6 Å. The results of the fitting suggest that the peptide has penetrated the headgroup region, as the electron density has decreased by ∼17%. It is clearly seen that the peptide has also partially penetrated into the tail region, destabilizing it, as its electron density has increased by ∼26%. All the results of the XR data fitting are presented in Table 2 for clarity.
FIGURE 8.

Cartoon schematic of possible interactions of LL-37 with bacterial membranes. (A) DPPG. (B) DPPG after insertion of LL-37. Cartoon based on x-ray data (GIXD and XR).
TABLE 2.
X-ray reflectivity fitting data for systems at 30 mN/m, 22°C
| Experiment | L1 (Å) | ρ1/ρDPBS | L2 (Å) | ρ2/ρDPBS | Roughness σ (Å) |
|---|---|---|---|---|---|
| DPPC | 15.2 | 0.91 | 8.8 | 1.33 | 3.3 |
| DPPC + 0.04 μg/ml LL-37 | 16.2 | 0.95 | 8.2 | 1.38 | 3.5 |
| DPPC + 0.1 μg/ml LL-37 | 16.5 | 0.95 | 6.9 | 1.39 | 3.5 |
| DPPG | 18.1 | 0.98 | 5.9 | 1.55 | 3.7 |
| DPPG + 0.04 μg/ml LL-37 | 7.4 | 1.23 | 8.1 | 1.33 | 5.6 |
L1 refers to the slab modeled to be between the air and the L2 slab, and L2 refers to the layer modeled between the top layer (L1) and the aqueous subphase. ρ1 and ρ2 are the respective electron densities of these layers. Electron-density values are normalized by the electron-density value of the DPBS subphase (0.337 e−/Å3). Data show a comparison of systems before and after the addition of LL-37. Very little difference was seen upon addition of LL-37 to the DPPC system, even with increasing concentration, whereas a significant difference was seen when LL-37 was injected under the DPPG monolayer, even at the lower of the two concentrations used with DPPC.
Complementary data from insertion experiments, epifluorescence measurements, and x-ray scattering allow us to speculate as to how the LL-37 peptide differentiates between different model membranes. The data analysis suggests that there is little interaction of LL-37 with DPPC and DPPE monolayers, but distinct interaction with DPPG monolayers. Fig. 8 shows a schematic cartoon of the proposed mode of interaction of LL-37 with DPPG monolayers at 30 mN/m based on results from the EFM and x-ray data. The DPPG monolayer has a tightly packed ordered structure at 30 mN/m, which, after injection of LL-37 into the subphase, is disturbed as the peptide penetrates the DPPG monolayer. Some regions of ordered lipid structure may remain, but the magnitude of peptide interaction and possible movement of the lipid molecules to accommodate the peptide in the monolayer have a great effect, resulting in the overall thinning of the film. The change in monolayer morphology after peptide insertion was monitored with epifluorescence (Fig. 4), whereas the change in film thickness was obtained from XR data modeling. Our results suggest that it is highly likely that the peptide penetrates the lipid monolayer, lying parallel to the surface. Thus the inserted peptides carpet the lipid monolayer, in agreement with Y. Shai and colleagues' “carpet” mechanism of action of antimicrobial peptides (11,22). Since our study uses monolayers, it is not possible to know whether this could later lead to the formation of toroidal pores in bilayer systems. Therefore, this work is presented as an important but complementary approach to studies involving live cells, vesicles, and bilayers.
XR—LL-37
X-ray reflectivity data for LL-37 (Fig. 7 C) show slight differences in the structure of the film when the pressure was increased from 0 to 28 mN/m. A small increase in layer thickness was observed between these two pressures, suggesting that the LL-37 molecules become more tightly packed after the film compression and thus the molecular rearrangement produces a thickening of the peptide layer. Moreover, XR analysis indicates a second partial layer of LL-37 forming underneath the top layer. XR data at 0 mN/m was modeled as a one-slab model with an electron density normalized to the electron density of the subphase of 1.18 and a thickness of L = 7.7 Å. This suggests that LL-37 aligns at the air-liquid interface with the molecules oriented parallel to the surface. XR data at π = 28 mN/m can no longer be fitted with a single slab and it was necessary to use a two-slab model. It is most likely that at 28 mN/m there is not enough space for every LL-37 molecule to align at the surface in the same way as at 0 mN/m, so some molecules would get forced out from a single layer to make a partial second layer. The fitting of the data using a two-slab model returned values of L1 = 8.5 Å and L2 = 4.3 Å for the two layers (going from the air-liquid interface toward the subphase). Corresponding normalized density values for the layers were 1.13 and 0.96, which could correspond to an upper layer and a partial second layer consisting of some peptide molecules that might be partly in the upper layer, with the more hydrophilic regions extending into the subphase. The fact that the LL-37 XR data do not show defined structural features, such as those seen for lipids like DPPC and DPPG, suggests that the LL-37 peptide layer is somewhat amorphous, as is seen with other protein structures at the air-liquid interface (9,79). This is corroborated by GIXD data (not shown), which show no Bragg peaks at either pressure (0 or 28 mN/m) and thus demonstrate that LL-37 does not show long-range lateral two-dimensional structure at the air-aqueous interface.
CONCLUSIONS
This is the first study we know of in which x-ray reflectivity and grazing incidence x-ray diffraction were used to observe the effects of the antimicrobial peptide LL-37 on phospholipid monolayers at the air-liquid interface. Moreover, synchrotron surface x-ray scattering has been combined with epifluorescence microscopy to give a further insight into the interactions of LL-37 with lipid monolayers.
GIXD experiments show that there is very little change in the phosphatidylcholine lipid packing upon addition of LL-37 to the subphase. This indicates that there is no detrimental interaction between the human antimicrobial peptide and the DPPC monolayer at the concentrations used here, contradicting the results of Oren et al. (19) and Henzler-Wildman et al. (26), who observed LL-37 lytic activity toward human red blood cell membrane mimics. However, our working peptide concentrations of 0.04 μg/ml (9 nM) and 0.1 μg/ml (22 nM) were significantly lower, and it is entirely possible that with an increase of LL-37 concentration to MIC levels (1–10 μM) we would observe peptide insertion into DPPC. This is in stark contrast to the DPPG monolayer, whose structure is completely destroyed after injection of LL-37. These results agree well with insertion assay and epifluorescence data, which show LL-37 insertion into DPPG, but not into DPPC, films.
The GIXD data obtained from the LL-37 peptide alone reveal that it does not form an ordered two-dimensional crystalline structure at the air-aqueous interface although it is able to form a film at the surface, as demonstrated by the pressure-area isotherms carried out with the peptide. XR data of the peptide alone corroborate the GIXD findings, suggesting that the peptide molecules self-assemble in-plane with the surface, forming layers of approximately the thickness expected for an α-helical peptide. These results prove very useful in suggesting a proposed model for the lipid-peptide interactions of the bacterial membrane with LL-37.
As expected, the x-ray reflectivity model fits suggest that the DPPC monolayers show little change after addition of the LL-37 to the subphase. Even increasing the concentration of the peptide by 2.5 times the original concentration makes little difference in the results obtained. However, the XR data of the DPPG/LL-37 system suggest that LL-37 molecules penetrate in-plane with the monolayer and insert predominantly into the phospholipid headgroup region, partitioning the peptide partly into the hydrocarbon tail region. The peptide/lipid ratio at the interface can be calculated analyzing insertion isotherms and x-ray reflectivity data. The insertion isotherm yields an area per lipid molecule increase caused by peptide absorption at the interface, whereas x-ray reflectivity yields the thickness of the peptide layer, and, thus, the peptide insertion angle. For DPPG at 30 mN/m this gives us a lipid/peptide ratio of 28:1 for peptide concentrations of 0.04 μg/ml and 6:1 for those of 0.1 μg/ml.
This is in agreement with recent work using other techniques (25,26,36), which proposes that the peptide carpets bacterial membranes. The work presented here also suggests that the LL-37 carpets the outside of the membrane before complete membrane disruption.
In summary, the research presented here used Langmuir-trough-generated lipid monolayers to investigate the lipid discrimination of LL-37 and its mechanism of action. By coupling synchrotron x-ray scattering and epifluorescence microscopy techniques with the use of Langmuir films, the interactions of LL-37 with phospholipid monolayers can be directly examined. Our results suggest that LL-37 can differentiate between eukaryotic and bacterial cell membrane lipid types. This work also supports previous work suggesting that the human antimicrobial peptide LL-37 acts against bacterial membranes via the “carpet” mechanism of membrane perturbation. The knowledge of the discrimination of LL-37 for different cell types and its mechanism of action bodes well for the production of future pharmaceutical therapeutic agents in the ongoing battle against antibiotic-resistant bacterial disease.
Acknowledgments
We thank Jeff Keen and Deidre Devine for providing the peptide. We acknowledge the European Synchrotron Radiation Facility for providing synchrotron radiation facilities at the ID10B beam line.
Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-ENG-38. This project is sponsored by the Engineering and Physical Sciences Research Council, Swindon, UK. Y.I. and K.Y.C.L. are grateful for the support of the Packard Foundation (99-1465).
References
- 1.Tonks, A. 1994. Drug resistance is a worldwide threat, warns report. Brit. Med. J. 309:1109–1110.7987100 [Google Scholar]
- 2.Hart, C. A., and S. Kariuki. 1998. Antimicrobial resistance in developing countries. BMJ. 317:647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies, J. 1996. Bacteria on the rampage. Nature. 383:219–220. [DOI] [PubMed] [Google Scholar]
- 4.Reacher, M. H., A. Shah, D. M. Livermore, M. C. J. Wale, C. Graham, A. P. Johnson, H. Heine, M. A. Monnickendam, K. F. Barker, D. James, and R. C. George. 2000. Bacteraemia and antibiotic resistance of its pathogens reported in England and Wales between 1990 and 1998: trend analysis. Brit. Med. J. 320:213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andreu, D., and L. Rivas. 1998. Animal antimicrobial peptides: an overview. Biopolymers. 47:415–433. [DOI] [PubMed] [Google Scholar]
- 6.Matsuzaki, K., K. Sugishita, N. Fujii, and K. Miyajima. 1995. Molecular basis for membrane selectivity of an antimicrobial peptide, magainin 2. Biochemistry. 34:3423–3429. [DOI] [PubMed] [Google Scholar]
- 7.Matsuzaki, K. 1999. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta. 1462:1–10. [DOI] [PubMed] [Google Scholar]
- 8.Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature. 415:389–395. [DOI] [PubMed] [Google Scholar]
- 9.Konovalov, O., I. Myagkov, B. Struth, and K. Lohner. 2002. Lipid discrimination in phospholipid monolayers by the antimicrobial frog skin peptide PGLa. A synchrotron X-ray grazing incidence and reflectivity study. Eur. Biophys. J. 31:428–437. [DOI] [PubMed] [Google Scholar]
- 10.Matsuzaki, K. 1998. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta. 1376:391–400. [DOI] [PubMed] [Google Scholar]
- 11.Oren, Z., and Y. Shai. 1998. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers. 47:451–463. [DOI] [PubMed] [Google Scholar]
- 12.Shai, Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta. 1462:55–70. match the journal [DOI] [PubMed] [Google Scholar]
- 13.Ehrenstein, G., and H. Lecar. 1977. Electrically gated ionic channels in lipid bilayers. Q. Rev. Biophys. 10:1–34. [DOI] [PubMed] [Google Scholar]
- 14.Westerhoff, H. V., D. Juretic, R. W. Hendler, and M. Zasloff. 1989. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. USA. 86:6597–6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuzaki K., M. Harada, S. Funakoshi, N. Fujii and K. Miyajima. 1991. Physicochemical determinants for the interactions of magainins-1 and magainins-2 with acidic lipid bilayers. Biochim. Biophys. Acta. 1063:162–170. [DOI] [PubMed] [Google Scholar]
- 16.Rapaport, D., and Y. Shai. 1991. Interaction of fluorescently labeled pardaxin and its analogs with lipid bilayers J. Biol. Chem. 266:23769–23775. [PubMed] [Google Scholar]
- 17.Shai, Y. 1994. Pardaxin-channel formation by a shark repellant peptide from fish. Toxicology. 87:109–129. [DOI] [PubMed] [Google Scholar]
- 18.Pouny, Y., D. Rapaport, A. Mor, P. Nicolas, and Y. Shai. 1992. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry. 31:12416–12423. [DOI] [PubMed] [Google Scholar]
- 19.Oren, Z., J. C. Lerman, G. H. Gudmundsson, B. Agerberth, and Y. Shai. 1999. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 341:501–513. [PMC free article] [PubMed] [Google Scholar]
- 20.Wu, M. H., E. Maier, R. Benz, and R. E. W. Hancock. 1999. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry. 38:7235–7242. [DOI] [PubMed] [Google Scholar]
- 21.Shai, Y., and Z. Oren. 2001. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides. 22:1629–1641. [DOI] [PubMed] [Google Scholar]
- 22.Shai, Y. 2002. Mode of action of membrane active antimicrobial peptides. Biopolymers. 66:236–248. [DOI] [PubMed] [Google Scholar]
- 23.Yang, L., T. A. Harroun, T. M. Weiss, L. Ding, and H. W. Huang. 2001. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 81:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, F. Y., M. T. Lee, and H. W. Huang. 2003. Evidence for membrane thinning effect as the mechanism for peptide induced pore formation. Biophys. J. 84:3751–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henzler Wildman, K. A., D. K. Lee, and A. Ramamoorthy. 2003. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry. 42:6545–6558. [DOI] [PubMed] [Google Scholar]
- 26.Henzler-Wildman, K. A., G. V. Martinez, M. F. Brown, and A. Ramamoorthy. 2004. Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37. Biochemistry. 43:8459–8469. [DOI] [PubMed] [Google Scholar]
- 27.Nouri-Sorkhabi, M. H., L. C. Wright, D. R. Sullivan, and P. W. Kuchel. 1996. Quantitative 31P nuclear magnetic resonance analysis of the phospholipids of erythrocyte membranes using detergent. Lipids. 31:765–770. [DOI] [PubMed] [Google Scholar]
- 28.Keller, S. L., W. H. Pitcher, W. H. Huestis, and H. M. Mcconnell. 1998. Red blood cell lipids form immiscible liquids. Phys. Rev. Lett. 81:5019–5022. [Google Scholar]
- 29.Ratledge, C., and S. G. Wilkinson. 1988. Microbial Lipids. Academic Press, London.
- 30.Graham, J. M. 1997. Membrane Analysis. Springer, New York.
- 31.Brock, T. D. 1974. Biology of Microorganisms. Prentice-Hall, Englewood Cliffs, NJ.
- 32.Goldfine, H. 1972. Comparative aspects of bacterial lipids. Adv. Microb. Physiol. 8:1–58. [DOI] [PubMed] [Google Scholar]
- 33.Op Den Kamp, J. A. F. 1979. Lipid asymmetry in membranes. Annu. Rev. Biochem. 79:47–71. [DOI] [PubMed] [Google Scholar]
- 34.Basanez, G., A. E. Shinnar, and J. Zimmerberg. 2002. Interaction of hagfish cathelicidin antimicrobial peptides with model lipid membranes. FEBS Lett. 532:115–120. [DOI] [PubMed] [Google Scholar]
- 35.Gidalevitz, D., Y. J. Ishitsuka, A. S. Muresan, O. Konovalov, A. J. Waring, R. I. Lehrer, and K. Y. C. Lee. 2003. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA. 100:6302–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neville, F., M. Cahuzac, A. Nelson, and D. Gidalevitz. 2004. The interaction of antimicrobial peptide LL-37 with artificial biomembranes: epifluorescence and impedance spectroscopy approach. J. Phys. Condens. Matter. 16:S2413–S2420. [Google Scholar]
- 37.Maget-Dana, R. 1999. The monolayer technique: a potent tool for studying the interfacial properties of antimicrobial and membrane-lytic peptides and their interactions with lipid membranes. Biochim. Biophys. Acta. 1462:109–140. [DOI] [PubMed] [Google Scholar]
- 38.Brockman, H. 1999. Lipid monolayers: why use half a membrane to characterize protein-membrane interactions? Curr. Opin. Struct. Biol. 9:438–443. [DOI] [PubMed] [Google Scholar]
- 39.Castano, S., B. Desbat, and J. Dufourcq. 2000. Ideally amphipathic beta-sheeted peptides at interfaces: structure, orientation, affinities for lipids and hemolytic activity of (KL)(m)K peptides. Biochim. Biophys. Acta. 1463:65–80. [DOI] [PubMed] [Google Scholar]
- 40.Jensen, T. R., K. Balashev, T. Bjornholm, and K. Kjaer. 2001. Novel methods for studying lipids and lipases and their mutual interaction at interfaces. Part II. Surface sensitive synchrotron X-ray scattering. Biochimie. 83:399–408. [DOI] [PubMed] [Google Scholar]
- 41.Losche, M. 2002. Surface-sensitive X-ray and neutron scattering characterization of planar lipid model membranes and lipid/peptide interactions. Curr. Top. Membr. 52:117–161. [Google Scholar]
- 42.Sun, F. 2002. Constant normal pressure, constant surface tension, and constant temperature molecular dynamics simulation of hydrated 1,2-dilignoceroylphosphatidylcholine monolayer. Biophys. J. 82:2511–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ambroggio, E. E., F. Separovic, J. Bowie, and G. D. Fidelio. 2004. Surface behaviour and peptide-lipid interactions of the antibiotic peptides, Maculatin and Citropin. Biochim. Biophys. Acta. 1664:31–37. [DOI] [PubMed] [Google Scholar]
- 44.Demel, R. A., W. S. M. Geurts Van Kessel, R. F. A. Zwaal, B. Roelofsen, and L. L. M. Van Deenen. 1975. Relation between various phospholipase actions on human red cell membranes and the interfacial phospholipid pressure in monolayers. Biochim. Biophys. Acta. 406:97–107. [DOI] [PubMed] [Google Scholar]
- 45.Blume, A. 1979. A comparative study of the phase transitions of phospholipid bilayers and monolayers. Biochim. Biophys. Acta. 557:32–44. [DOI] [PubMed] [Google Scholar]
- 46.Gennaro, R., and M. Zanetti. 2000. Structural features and biological activities of the cathelicidin-derived antimicrobial peptides. Biopolymers. 55:31–49. [DOI] [PubMed] [Google Scholar]
- 47.Gopal, A., and K. Y. C. Lee. 2001. Morphology and collapse transitions in binary phospholipid monolayers. J. Phys. Chem. B. 105:10348–10354. [Google Scholar]
- 48.Maskarinec, S. A., J. Hannig, R. C. Lee, and K. Y. C. Lee. 2002. Direct observation of poloxamer 188 insertion into lipid monolayers. Biophys. J. 82:1453–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maskarinec, S. A., and K. Y. C. Lee. 2003. Comparative study of polaxmer insertion into lipid monolayers. Langmuir. 19:1809–1815. [Google Scholar]
- 50.Ege, C., and K. Y. C. Lee. 2004. Insertion of Alzheimer's Aß40 Peptide into lipid monolayers. Biophys. J. 87:1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Als-Nielsen, J., and D. McMorrow. 2001. Elements of Modern X-Ray Physics. John Wiley & Sons, Chichester, UK.
- 52.Jensen, T. R., and K. Kjaer. 2001. Structural properties and interactions of thin films at the air-liquid interface explored by synchrotron X-ray scattering. In Novel Methods to Study Interfacial Layers. D. Möbius and R. Miller, editors. Elsevier, Amsterdam. 205–254.
- 53.Schalke, M., and M. Losche. 2000. Structural models of lipid surface monolayers from X-ray and neutron reflectivity measurements. Adv. Colloid Interface Sci. 88:243–274. [DOI] [PubMed] [Google Scholar]
- 54.Helm, C. A., H. Mohwald, K. Kjaer, and J. Als-Nielsen. 1987. Phospholipid monolayer density distribution perpendicular to the water-surface: a synchrotron x-ray reflectivity study. Europhys. Lett. 4:697–703. [Google Scholar]
- 55.Als-Nielsen, J., D. Jacquemain, K. Kjaer, F. Leveiller, M. Lahav, and L. Leiserowitz. 1994. Principles and applications of grazing incidence X-ray and neutron scattering from ordered molecular monolayers at the air-water interface. Phys. Rep. 246:251–313. [Google Scholar]
- 56.Dubreil, L., V. Vie, S. Beaufils, D. Marion, and A. Renault. 2003. Aggregation of puroindoline in phospholipid monolayers spread at the air-liquid interface. Biophys. J. 85:2650–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Konovalov O., S. M. O'Flaherty, E. Saint-Martin, G. Deutsch, E. Sevcsik, and K. Lohner. 2005. The bending rigidity of phospholipid monolayers in presence of an antimicrobial frog peptide studied by X-ray grazing incidence diffraction. Physica B. 357:185–189. [Google Scholar]
- 58.Agerberth, B., H. Gunne, J. Odeberg, P. Kogner, H. G. Boman, and G. H. Gudmundsson. 1995. Fall-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone-marrow and testis. Proc. Natl. Acad. Sci. USA. 92:195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johansson, J., G. H. Gudmundsson, M. E. Rottenberg, K. D. Berndt, and B. Agerberth. 1998. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 273:3718–3724. [DOI] [PubMed] [Google Scholar]
- 60.Stryer, L. 1995. Biochemistry. W. H. Freeman, New York.
- 61.Guinier, A. 1963. X-ray diffraction. In Crystals, Imperfect Crystals, and Amorphous Bodies. H. M. Foley and M. A. Ruderman, editors. W. H. Freeman, San Francisco and London. 142–143
- 62.Leveiller, F., D. Jacquemain, L. Leiserowitz, K. Kjaer, and J. Als-Nielsen. 1992. Toward a determination at near atomic resolution of 2-dimensional crystal structures of amphiphilic molecules on the water-surface: a study based on grazing-incidence synchrotron x-ray-diffraction and lattice energy calculations. J. Phys. Chem. 96:10380–10389. [Google Scholar]
- 63.Brezesinski, G., A. Dietrich, B. Struth, C. Bohm, W. G. Bouwman, K. Kjaer, and H. Mohwald. 1995. Influence of ether linkages on the structure of double-chain phospholipid monolayers. Chem. Phys. Lipids. 76:145–157. [Google Scholar]
- 64.Lee, K. Y. C., A. Gopal, A. Von Nahmen, J. A. Zasadzinski, J. Majewski, G. S. Smith, P. B. Howes, and K. Kjaer. 2002. Influence of palmitic acid and hexadecanol on the phase transition temperature and molecular packing of dipalmitoylphosphatidyl-choline monolayers at the air-water interface. J. Chem. Phys. 116:774–783. [Google Scholar]
- 65.Wu, G., J. Majewski, C. Ege, K. Kjaer, M. J. Weygand, and K. Y. C. Lee. 2004. Lipid corralling and poloxamer squeeze-out in membranes. Phys. Rev. Lett. 93:028101. [DOI] [PubMed] [Google Scholar]
- 66.Kjaer, K., J. Als-Nielsen, C. A. Helm, P. Tippmankrayer, and H. Mohwald. 1989. Synchrotron X-ray-diffraction and reflection studies of arachidic acid monolayers at the air-water interface. J. Phys. Chem. 93:3200–3206. [Google Scholar]
- 67.Pedersen, J. S., and I. W. Hamley. 1994. Analysis of neutron and X-ray reflectivity data by constrained least-squares methods. Physica B (Amsterdam). 198:16–23. [Google Scholar]
- 68.Thoma, M., M. Schwendler, H. Baltes, C. A. Helm, T. Pfohl, H. Riegler, and H. Möhwald. 1996. Ellipsometry and X-ray reflectivity studies on monolayers of phosphatidylethanolamine and phosphatidylcholine in contact with n-dodecane, n-hexadecane, and bicyclohexyl. Langmuir. 12:1722–1728. [Google Scholar]
- 69.Vaknin D., P. Kruger, and M. Losche. 2003. Anomalous x-ray reflectivity characterization of ion distribution at biomimetic membranes. Phys. Rev. Lett. 90:178102. [DOI] [PubMed] [Google Scholar]
- 70.Konovalov, O., L. A. Feigin, and B. M. Shchedrin. 1996. Allowance for apparatus distortions in modeling the structure of Langmuir-Blodgett films from reflectivity data. Crystallogr. Rep. 41:592–597. [Google Scholar]
- 71.Konovalov, O., L. A. Feigin, and B. M. Shchedrin. 1996. Statistical evaluation of the accuracy of structure parameter determination from x-ray and neutron reflectivity data. Crystallogr. Rep. 41:603–606. [Google Scholar]
- 72.Konovalov, O., I. I. Samoilenko, L. A. Feigin, and B. M. Shchedrin. 1996. Application of a normalizing function in the simulation of x-ray and neutron reflectivity data. Crystallogr. Rep. 41:598–602. [Google Scholar]
- 73.Konovalov, O. V., I. I. Samoilenko, L. A. Feigin, B. M. Shchedrin, and L. G. Yanusova. 1999. Statistical substantiation of parametrization of a film model in reflectometry. Crystallogr. Rep. 44:319–323. [Google Scholar]
- 74.Samoilenko, I. I., O. V. Konovalov, L. A. Feigin, B. M. Shchedrin, and L. G. Yanusova. 1999. Processing of experimental reflectivity data within the REFLAN software package. Crystallogr. Rep. 44:310–318. [Google Scholar]
- 75.Samoilenko, I. I., B. M. Shchedrin, and L. A. Feigin. 1996. Global minimization in reconstruction of the scattering-length density profile by x-ray reflectivity data. Physica B (Amsterdam). 221:542–546. [Google Scholar]
- 76.Parrat, L. G. 1954. Surface studies of solids by total reflection of x-rays. Phys. Rev. 95:359–369. [Google Scholar]
- 77.Kruger, P., M. Schalke, J. Linderholm, and M. Losche. 2001. Multipurpose x-ray reflectometer optimized for the characterization of organic surface films on aqueous subphases. Rev. Sci. Instrum. 72:184–192. [Google Scholar]
- 78.Huang H. W., F. Y. Chen, and M. T. Lee. 2004. Molecular mechanism of peptide-induced pores in membranes. Phys. Rev. Lett. 92:198304. [DOI] [PubMed] [Google Scholar]
- 79.Gidalevitz, D., Z. Q. Huang, and S. A. Rice. 1999. Protein folding at the air-water interface studied with x-ray reflectivity. Proc. Natl. Acad. Sci. USA. 96:2608–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]