

Abstract
To monitor binding of tropomyosin to yeast actin, we mutated S235 to C and labeled the actin with pyrene maleimide at both C235 and the normally reactive C374. Saturating cardiac tropomyosin (cTM) caused about a 20% increase in pyrene fluorescence of the doubly labeled F-actin but no change in WT actin C374 probe fluorescence. Skeletal muscle tropomyosin caused only a 7% fluorescence increase, suggesting differential binding modes for the two tropomyosins. The increased cTM-induced fluorescence was proportional to the extent of tropomyosin binding. Yeast tropomyosin (TPM1) produced less increase in fluorescence than did cTM, whereas that caused by yeast TPM2 was greater than either TPM1 or cTM. Cardiac troponin largely reversed the cTM-induced fluorescence increase, and subsequent addition of calcium resulted in a small fluorescence recovery. An A230Y mutation, which causes a Ca+2-dependent hypercontractile response of regulated thin filaments, did not change probe235 fluorescence of actin alone or with tropomyosin ± troponin. However, addition of calcium resulted in twice the fluorescence recovery observed with WT actin. Our results demonstrate isoform-specific binding of different tropomyosins to actin and suggest allosteric regulation of the tropomyosin/actin interaction across the actin interdomain cleft.
INTRODUCTION
The tropomyosins (TMs) are a family of conserved eukaryotic F-actin binding proteins with molecular masses from 19 to 40 KDa (1,2). TM has a dimeric α-helical coiled-coil structure and contains four to seven quasirepeat regions, depending on the TM isoform. Each quasirepeat contacts a separate actin monomer when the protein binds to the actin filament, but the contribution of different pseudorepeats to the binding of TM to actin is not equivalent (3,4). The overlapping interaction of termini on neighboring TMs is important for the binding of TM to the actin filament (5,6).
Nonmuscle TM is believed to play an important role in maintaining the integrity of the actin cytoskeleton (7). Previous studies have demonstrated that TM can restore polymerizability to a polymerization-defective yeast actin mutant V266G, L267G (8). Liu et al. found that deletion of yeast TM1 (TPM1), which comprises ∼85% of the total TM in the cell, led to disruption of actin cables and resulted in a temperature-sensitive phenotype (9,10). Overexpression of a second minor yeast TM, TPM2, in the mutant cells largely rescued the phenotype described above (11). Overexpression of TPM1 in yeast cells also rescued the defect in actin cable formation caused by mutations in other genes like mdm20, which catalyzes the N-acetylation of TM (12). The interface in the actin/TM complex has not yet been characterized, and it has been proposed that there are no specific contacts between the two proteins. Instead there may be merely a loose interaction between complementary charged surfaces (13).
In muscle cell contraction, the actin thin filament is the transducer of the force exerted by the myosin head. This process is highly controlled at the level of the thin filament by the calcium-regulated TM/troponin complex (14). Troponin (TN) is a TM-associated protein composed of three subunits, TNC, I, and T. The efficiency of contraction is dictated in large part by the contacts at the actin/myosin interface and may also depend on the conformational changes in the actin exerted by its interaction with myosin and the two regulatory proteins (15).
Calcium regulation of myosin activity in muscle cells, dependent on the intermediacy of the TM/TN complex, can be described by a three-state model (16). In this model, in the absence of Ca2+ binding to the low affinity site(s) on TNC, the TM/TN complex sits on the actin thin filament in a manner that sterically blocks the ability of the myosin head to interact with the actin (blocked or B-state). Upon the binding of Ca2+ to the TNC subunit, a conformational change in the complex occurs, resulting in its translation over the actin surface to a more inner position on the filament (closed or C-state). These states are depicted in Fig. 1. This movement now allows partial accessibility of the myosin head to binding sites on the actin. This actin/myosin interaction now forces the TM/TN complex even further toward the filament center, allowing the strong actin/myosin complex to form, which ultimately results in the generation of contractile force (open or M state).
FIGURE 1.

Actin structural models. (A) Depiction of actin/TM binding states and the actin mutations used in this study. (Left) Shown are the approximate binding regions for TM on actin in the blocked (B), closed (C), and open (M) states. The binding envelopes are based on work by Lehman and colleagues (17,18). Roman numerals I–IV denote the four subdomains of actin. Highlighted are amino acid residues E224 (red), A230 (green), S235 (blue), M305 (orange), and C374 (purple). An ATP/ADP binding site is located at the bottom of the central cleft. (Right) Side view (subdomains III and IV face forward) of the actin monomer. The G-actin structure was constructed based on the yeast actin atomic coordinates (PDB, 1YAG (27)). (B) The distance between the C374 and the E224 in the actin filament. The F-actin model is constructed based on the Holmes model with the hydrophobic 3/4 loop extended (52). The mutated residues and C374 in each actin monomer are highlighted with the same color scheme as described for panel A. The distance between the sulfur of C374 in Unit 3 of the actin monomer and the γ-carbon of E224 in Unit 2 of the actin monomer is ∼21 Å. Not shown in the figure are the following intraresidue differences in which 374 is in Unit 3 and the remaining residues are in Unit 2: 374–230, 19.6 Å; 374–235, 28.7 Å; 374–305, 39 Å. Residue 230 is pointing in the opposite direction from 374 and thus probes attached to these two could not interact. The 235 and 305 residues are on the opposite face of actin from 374, and attached probes would thus be blocked by intervening mass from interacting with a pyrene at 374.
Results of recent high resolution electron microscopy studies using both full length and shortened TMs are generally consistent with this three-state model (17). Subsequent work revealed that the position of binding of TM to actin depended both on the nature of the actin isoform and the nature of the TM. Electron microscopy (EM) studies showed that skeletal muscle TM (skTM) binds in the C-state to muscle actin, bridging the cleft borders of subdomains 2 and 4 of the actin. However, it binds in the B-state on a yeast actin filament, running through the middle of subdomains 1 and 2 (18). It has also been demonstrated that with skeletal muscle actin, cardiac and smooth muscle TMs are in the B-state instead of the C-state where skTM binds (18). The position of binding of bovine cardiac TM (cTM) to yeast actin has not been assessed by EM.
A number of studies have used fluorescently labeled TMs to assess the behavior of TM on the actin filament in solution. Ishii and Lehrer (19) used the excimer formed by pyrene-labeled TM to study the kinetics of the on-off change of TM resulting from the displacement of myosin S1 from the actin filament. Graceffa (20) used fluorescence energy transfer between a donor probe on TM and an acceptor probe on C374 of actin to assess the movement of smooth muscle TM caused by myosin heads. Chandy et al. (21) used the polarized phosphorescence of erythrosine-labeled TM to measure the microsecond rotational dynamics of cardiac versus skTMs on the actin surface. They concluded that the cTM was more mobile than its skeletal muscle counterpart. Finally, Bacchiocchi and Lehrer (22,23) have employed multisite fluorescence resonance energy transfer to study the problem. They used a naphthalene derivative attached to TM as a donor and rhodamine phalloidin attached to actin as an acceptor. They then applied a sophisticated mathematical analysis to the data they acquired. With this approach, they demonstrated first that TM can roll over the actin surface. They also used the system to demonstrate myosin-induced movement of different smooth muscle TMs on actin. The ability to easily measure such movement is important to appreciate the molecular basis of the regulation of muscle contractility, but the technology involved in these newer studies is not straightforward. Routine assessment of the motion of these proteins over actin, from actin's perspective, has been difficult in part because of a lack of convenient sites on actin to attach critically placed fluorescent probes for detection of changes in binding.
Yeast provides a convenient system for generating mutated actins in large enough quantities for biochemical studies (24). Mutations can be introduced which will either test the importance of specific residues in actin function or provide attachment sites for fluorescent probes. Yeast actin, 87% identical with muscle actin (25), is encoded by a single gene, and it can easily be replaced by a gene carrying the mutated site of choice (26). Its tertiary structure is almost identical to that of muscle actin (27). Yeast actin is a valid model for studying regulation of thin filament activity since previous studies have shown that its ability to activate muscle S1 ATPase activity can be controlled in vitro by TM/TN in much the same way as muscle F-actin (28).
In this study, we investigate the interaction between actin and TM and the effect TN exerts on this complex. Lorenz's model (13) and recent EM studies of the actin/TM interaction (17) suggested that two amino acids on subdomain 4, residues 235 and 305, might represent convenient attachment sites for probes to assess the interaction of TM with actin (Fig. 1 A). To test this hypothesis, we separately mutated yeast actin Ser235, Glu224, and Met305 to Cys so that we could label these two sites with pyrene maleimide. One can thus potentially investigate the effect of the interaction among actin, TM, and TN on these two sites by monitoring the change in pyrene fluorescence caused by the interaction of actin and TM, assuming the probes are in the correct position to report on such an interaction.
We have used this system to assess whether there are differences in the interaction of yeast TPM1, yeast TPM2, cTM, and skTM with actin and to assess the effect of TN on the actin/TM interaction. Finally, we have studied the effect of an A230Y mutation on the fluorescence behavior of this probe. This mutation, in the presence of TM/TN, causes a hypercontractile response to Ca2+. We have assessed whether or not the A230Y substitution might result in a change in the environment sampled by the pyrene235 in the presence of cTM and cardiac TN (cTN).
MATERIALS AND METHODS
Materials
The DNA primers used for site-directed mutagenesis were obtained from Integrated DNA Technologies (Coralville, IA). N-(1-pyrenyl)maleimide was purchased from Sigma-Aldrich (St. Louis, MO). The yeast cake used for WT actin purification was purchased locally. Bovine cardiac TM (cTM), which is ∼85% α-isoform and 15% β-isoform (29), yeast TPM1, and yeast TPM2 were purified from cardiac acetone powder and yeast cells, respectively, according to methods described previously (30,31). Bovine cTN, and skeletal muscle (skTM), which is an α,β TM mixture, were the generous gifts of Larry Tobacman and Emil Reisler, respectively.
Mutagenesis and plasmid construction
Plasmid purification and manipulation were performed according to methods described previously (32). Site-directed mutagenesis was performed using kits purchased from Stratagene (La Jolla, CA), according to the manufacturer's instructions. The template plasmid is the TRP1-marked centromeric plasmid pRS314-WN (33) in which the yeast ACT1 promoter and coding sequence had been cloned between the BamH I and EcoR I sites in the polylinker site of pRS314 (34). S235C, M305C, E224C, and A230Y/mutations were successfully constructed, and all plasmids were sequenced to confirm the correctness of the DNA sequences. Plasmids containing the mutant actin sequences were introduced into a ura3, trp1, leu2 haploid yeast cell in which the chromosomal actin gene had been disrupted by a LEU2 gene. In this cell, the WT actin was expressed from another centromeric plasmid marked with the URA3 gene. pRS314 transformed cells were selected for by growth in Trp-deficient medium. Plasmid shuffling produced viable haploid cells expressing only the mutant actins. The plasmids were reisolated from the transformed yeast and sequenced to confirm the presence of the desired actin mutation.
Actin purification and labeling of actin with pyrene maleimide
Actins were purified from the mutant cells by DNase I affinity chromatography and diethylaminoethyl cellulose chromatography as described previously (35,36), and purified actins were stored in G buffer (10 mM Tris-HCl, pH 7.5, containing 0.2 mM ATP, 0.2 mM CaCl2, and 0.1 mM DTT) at 4°C and used for experiments within 3 days. Pyrene labeling of actins was performed as described previously (37) with modifications as follows. Before pyrene labeling, actins were dialyzed against G buffer without dithiothreitol. The reaction concentrations of actin and pyrene maleimide were initially 40 μM. The reaction solution was G buffer with 2 mM MgCl2 and 50 mM KCl added after the probe to promote actin polymerization at room temperature for 30 min. Another equal aliquot of pyrene maleimide was added for an additional 30 min. The labeled actin was then pelleted in a Beckman TL-100 ultracentrifuge (Fullerton, CA). Actins were depolymerized by dialysis against G buffer and stored in G buffer (pH 7.5) at 4°C. The labeling ratio was determined from the absorbance of bound pyrene maleimide at 334 nm and was 1.6–1.9 mol of probe/mole of actin for S235C and E224C actins, indicating labeling of both C374 and the cysteine introduced by mutagenesis. For M305C actin, the labeling efficiency was ∼1.4 mol of dye/mol of actin, suggesting that the sulfur at 305 was more sequestered than the other residues.
Actin polymerization
Actin polymerization was carried out with 5 μM G-actin in G buffer in a total volume of 120 μl in a thermostatted fluorimeter cuvette. Polymerization was initiated by addition of MgCl2 and KCl to final concentrations of 2 and 50 mM, respectively, and formation of actin filaments was followed as a function of time by monitoring the increase in light scattering with excitation and emission wavelengths set at 360 nm. For these and all pyrene fluorescence measurements, we used a FluoroMax (Jobin Yvon, Edison, NJ) fluorescence spectrometer.
Fluorescence of pyrene maleimide-labeled actin
The fluorescence of pyrene-labeled actins was monitored with an excitation wavelength of 365 nm. Emission spectra were recorded between 370–550 nm, and the emission wavelength for single wavelength measurements was 385 nm.
To assess TM binding, varying amounts of TM in F-buffer were mixed with 5 μM pyrene maleimide-labeled actin filaments. To correct for dilution of the reaction solution by the addition of TM, an equal volume of the buffer was mixed with the labeled actin filaments, and the change in the pyrene fluorescence was measured. Comparisons of the relative effects of cTM and yeast TPM1 and TPM2 were performed using equal weights of the three proteins to account for the differences in their molecular masses. This approach normalized the data to the total number of quasirepeats present in a TM sample.
To assess the effect of adding TN to the actin/TM complex, the pyrene maleimide labeled actin/cTM complex was formed as described above. To the solution was added 0.3 mM EGTA, pH 7.5, to chelate Ca2+, and the change in pyrene fluorescence was measured. cTN, equimolar to the amount of TM present, was then added, and the change in the fluorescence was measured. −CaCl2, 0.3 mM was then added, which saturated the TN, and the change in the fluorescence was measured again. Corrections for change in fluorescence due to dilution from the added TN and Ca2+ aliquots were addressed as above.
Cosedimentation assay
Measurement of the binding of TM to F-actin using a centrifugation-based assay was performed according to methods described previously (38) with modification as follows. G-actin, 10 μM, in the presence of various concentrations of TM was induced to polymerize by the addition of 2 mM MgCl2 and 50 mM KCl. After incubation at room temperature for at least 30 min to ensure attainment of the steady state, the actin-TM solution was centrifuged at 25°C for 30 min in a Beckman TL-100 ultracentrifuge at 75,000 rpm. The pellet was dissolved in SDS sample buffer and was subjected to 12.5% SDS-PAGE analysis. Gels were stained with Coomassie blue, and the image of the gel was captured with an Epson Perfection 2450 photo scanner (Long Beach, CA). The intensities of the yeast actin and TM bands were further quantified using UN-SCAN-IT (Silk Scientific, Orem, UT) software. The ratio of the bound TPM1/pelleted actin band densities were calculated at each point and normalized to the maximum ratio at saturating TM concentrations. To check for extent of binding, at saturation, the staining density of the individual bands were corrected for differences in molecular weights. The TM band was further corrected to account for the fact that the native protein is composed of two polypeptide chains. The final corrected ratio of the bands showed that at saturation the molar ratio of bound TPM1/pelleted actin was ∼1:5 as expected.
Calculation of the apparent binding affinity of actin to TM
The apparent binding constants (Kapp) and Hill coefficents (αH) of cTM and TPM1 binding to pyrene235 F-actin were determined by fitting the data to the Hill equation as described in Cho et al. (39) using Microsoft Excel (Redmond, WA):
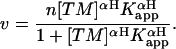 |
(1) |
The molar concentration of bound TM at each point was calculated as follows. The total fluorescence change at saturating TM was set at 100% and the fraction of this total change Δ was determined for each point assessed. For cTM, at saturation, seven actins are covered by a single TM, so that for 5 μM actin, a 100% fluorescence change would be caused by 0.71 μM TM. Thus, the concentration of bound cTM at each point is 0.71Δ. Free TM is the total TM – bound TM. For yeast TPM1, which only covers five actin monomers, the calculation is made the same way except that 100% of the fluorescence change at saturating TM is caused by 1 μM TPM1.
Molecular distance calculations
Molecular distances in our modeling figures were determine using Swiss PDB viewer, version 3.7 (http://www.expasy.ch/spdbv/mainpage.html) applied to the atomic coordinates for a muscle F-actin trimer based on the Holmes actin filament model (13).
RESULTS
To create a system to monitor TM binding from actin's perspective, we first mutated yeast actin E224, S235, and M305 individually to C as potential fluorescent probe attachment sites (Fig. 1). We selected these residues because they were on the surface of the protein, their side chains were facing away from the body of the actin monomer, and an attached pyrene should be located in a position that might be affected by bound TM (13). The mutation at 224 was unique in that a probe attached here would point away from, not toward, the bound TM and would thus serve as a directional control. All mutant actins were compatible with yeast viability and produced no observable defective phenotype, and the actins behaved normally in our standard purification protocol.
We reacted the mutant actins with at least a 2:1 molar ratio of pyrene maleimide/actin, since the reactive C374 was also present. The fluorescence signal of C374 pyrene-labeled wild-type (WT) actin does not change when it is mixed with TM (37). To eliminate the potential for problems of interpretation arising from the presence of multiple probes, we attempted to replace the reactive C374 with an A. However, this change caused purification problems, even though we had successfully used the same approach with similar mutant actins (37).
Polymerization of mutant actins
By light scattering, all three unlabeled mutant actins polymerized similarly to WT actin both in terms of rate and extent (data not shown). The polymerization kinetics of pyrene-labeled E224C and M305C actins were also similar to the kinetics of pyrene-labeled WT actin (data not shown). However, we observed increased light scattering with the labeled S235C actin compared with WT actin. Examination of polymerized pyrene-S235C actin by electron microscopy showed that the light scattering increase was probably caused by the formation of disordered filament bundles not observed with the unlabeled mutant actin (Fig. 2 A). Copolymerization of a 1:1 mixture of labeled and unlabeled mutant actins caused the bundles to disappear (Fig. 2 B). Addition of TM to pure labeled F-actin also caused the disappearance of the bundles (Fig. 2 C).
FIGURE 2.

Electron microscopic analysis of pyrene-labeled S235C actin filaments. An aliquot of 5 μM actin, after inducement of polymerization by addition of KCl and MgCl2, was examined by electron microscopy as described in Materials and Methods. (A) pyrene-labeled S235C actin alone, (B) a mixture of pyrene-labeled and unlabeled S235C actins at a 1:1 molar ratio, or (C) pyrene-labeled S235C actin with cTM at 2:1 molar ratio. Bar = 10 μm.
Fig. 3 A shows that a polymerization-dependent increase in fluorescence for the pyrene-labeled S235C and pyrene WT actins was roughly proportional to the extent of labeling of the actin in comparison to that produced by WT actin labeled at C374 alone. In the latter case, the labeling efficiency usually approaches 100%. Interestingly, the postpolymerization fluorescence spectrum of the C224-labeled actin (Fig. 3 B) showed the appearance of a new pyrene excimer band (37,40), centered at ∼490 nm, formed by the interaction of the pyrene at C224 on one monomer with a pyrene at C374 of another monomer on the opposing strand of the actin helix.
FIGURE 3.

The fluorescence emission spectrum of pyrene-labeled WT, S235C, and E224C F-actin. Each pyrene-labeled actin was triggered to polymerize until the steady state was reached, and the emission spectrum of each sample from 370 to 550 nm was recorded as described in Materials and Methods. Excitation wavelength was 365 nm. (A) 5 μM pyrene WT (line 1) and pyrene-S235C (line 2). (B) 2.5 μM pyrene-E224C before (line 1) and after (line 2) polymerization. The inset is the difference spectrum for these plots. The excimer peak at 490 nm is denoted by an arrow.
Effect of cTM on the fluorescence of pyrene-labeled E224C, S235C, and M305C actins
The addition of saturating amounts of cTM to pyrene-labeled S235C F-actin produced a significant increase in F-actin fluorescence (19%) at an emission wavelength of 385 nm (Fig. 4 A). We next determined whether the bundling described earlier affected the fluorescence change of the labeled C235 F-actin when mixed with cTM. Using a different preparation of labeled actin, we performed the same experiment with 50% pyrene-labeled (no bundling) and 100% pyrene-labeled (bundling) C235 F-actin. cTM caused similar increases in the fluorescence signals for 50% (17% increase) and 100% (22% increase) labeled C235 F-actin, suggesting that the bundling does not significantly interfere with the fluorescence change induced by cTM. We therefore used completely labeled actin for the remainder of our experiments. The different values for the 100% labeled actin samples from different preparations can be explained by slightly different degrees of attachment of pyrene to the actin. Since only one of the two labeled sites is affected by the TM, slight differences in the ratio of labeling of the two sites can cause a difference in the percentage change in fluorescence values which are based on total fluorescence.
FIGURE 4.

Change in fluorescence of pyrene-labeled S235C actin after addition of saturating amounts of cTM (A) or skTM (B). The experiment was performed as described in Materials and Methods. (Solid lines) F-actin only; (dashed lines) F-actin + TM.
Addition of cTM to either labeled M305C or E224C F-actin resulted in no significant increase in fluorescence (data not shown), in contrast to the case for labeled S235C F-actin. Also, in agreement with previous results (37), addition of TM to C374-labeled WT yeast F-actin produced no change in pyrene fluorescence. Cosedimentation experiments demonstrated that binding of the TM to the labeled actins was normal (data not shown). Thus, the TM-dependent change in fluorescence of the probe at C235 is extremely position specific.
Comparison of the effect of cTM, skTM, and yeast TPM1 and TPM2 on the fluorescence of pyrene-labeled S235C actin
Lehman et al. (18) previously showed that rabbit skTM, a mixture of the α- and β-isoforms, bound in the C-state to skeletal muscle actin and cTM (85% α) bound in the B-state. The skTM mixture is ∼60% α,α dimer and 40% α,β dimer; the α and β polypeptides differ from one another at 39 positions, only two of which involve charged residues (41). However, the same skTM mixture bound to yeast F-actin in the B-state, far away from the probes we engineered into the actin. Fig. 4 B shows that the addition of saturating amounts of the same skTM mixture to C235-pyrene actin caused a 6–7% increase in F-actin fluorescence in comparison with the 20% observed with cTM (Fig. 4 A). This result suggested that cTM and skTM might bind to different parts of the yeast actin filament but in the opposite order compared with that observed with muscle actin.
We next compared the abilities of yeast TPM1, TPM2, and cTM to alter the fluorescence of the pyrene-labeled S235C actin. Overall, yeast TPM1 and TPM2 are 64.5% identical (1); however, removal of a large insert present in TPM1 but not TPM2 raises the identity to 85%. For yeast TPM1 and TPM2, the molar ratios of TM/actin used were greater than that used for the binding of cTM to account for the fact the TPM1 is roughly 5/7 and TPM2 is 4/7 the length of cTM. The percentage increase caused by the yeast TPM1 at saturating concentrations was slightly but reproducibly less than that caused by cTM, whereas that caused by TPM2 was larger than that of cTM (Table 1). This result suggests that the two yeast TPM isoforms do not bind to the same sites on F-actin.
TABLE 1.
Summary of the effects of saturating amounts of different TMs on the fluorescence of pyrene-labeled S235C F-actin
| CTM | skTM | TPM1 | TPM2 | |
|---|---|---|---|---|
| F-actin | 21.5% | 7.25% | 15.5% | 24% |
| ±1.7% | ±2.6% | +/−1% | +/−2% | |
| (n = 4) | (n = 4) | (n = 2) | (n = 2) |
For these determinations, ± represents SD, whereas +/− represents the range after the averaging of two values.
Relationship of the extent of increased fluorescence to the extent of TM binding
We wished to know whether there was a stoichiometric relationship between TM binding and increased actin fluorescence or whether substoichiometric amounts of TM caused long-range changes in actin conformation. We thus assessed the increase in C235 probe fluorescence resulting from the addition of increasing amounts of cTM or yeast TPM1 to a constant amount of labeled F-actin. In both cases, a 1:4 mixture of TM/yeast actin produced saturable binding as previously reported (8).
Fig. 5 A shows that the fluorescence change with yeast TPM1 resembles a typical saturable cooperative TM binding curve. A centrifugation assay confirmed that the increase in fluorescence reflected an increase in TM binding. We further fitted the fluorescence data to the Hill equation (39). The results yielded a Kapp of 2.34 ± 0.70 ×106 M−1 (average of three individual experiments), similar to the result obtained by Strand et al. (42) of Kapp = 1.42 × 106 M−1 using muscle actin and recombinant TPM1. Fig. 5 B shows that the fluorescence change caused by the binding of cTM is also cooperative with a Kapp of 2.79 × 106 M−1 (averaging from two individual experiments), again, similar to the Kapp = 2.3 ± 0.6 × 106 M−1 from Korman and Tobacman (38) using yeast actin and cTM, demonstrating the validity of our approach.
FIGURE 5.

Comparison of the titration curves for the binding of yeast TPM1 and cTM to pyrene-labeled S235C F-actin using fluorescence and cosedimentation assays. (A) The pyrene fluorescence of 5 μM F-actin at 385 nm was recorded before and after the addition of increasing concentration of TPM1. The excitation wavelength was 365 nm. The maximum difference in fluorescence, shown on the left vertical axis, was set at 100%. Other values were normalized to this number and plotted against the concentration of TPM1 added to the sample. Two separate experiments (•, ♦) are shown. A cosedimentation analysis of the reaction, using SDS-PAGE analysis of the proteins in the pelleted fraction, was performed as described in Materials and Methods. The ratios of the intensity of the pelleted TPM1 bands versus the pelleted actin bands at each point were calculated, normalized to the maximum ratio at saturation, and plotted as % maximum TPM1 bound on the right vertical axis (▴). (B) Titration curve for the binding of cTM to pyrene-labeled mutant S235C F-actin. The experiment was performed as in panel A. The results of two separate experiments (▪, •) are shown. Cosedimentation analysis was not performed with this TM.
Effect of TN ± Ca2+ on TM-dependent increase in fluorescence of pyrene-labeled S235C F-actin
Previous models of the calcium-dependent regulation of muscle contraction predict a Ca2+-TN-dependent modulation of TM binding to the actin filament surface. We determined whether the fluorescence of labeled F-actin was sensitive to such modulation. Saturating cTM was added to F-actin followed by enough EGTA to complex the calcium. The resulting emission spectrum of the TM-actin complex showed no EGTA-dependent change. A molar amount of cTN, equal to the amount of cTM, was introduced, and the emission spectrum was again recorded. Finally, enough calcium was added to achieve a final concentration of 0.3 mM, and the emission spectrum was recorded again.
The results of this experiment are shown in Fig. 6 A. The addition of TN caused a decrease in the fluorescence of the TM-actin solution to near that obtained with actin alone. As a control, TN in the absence of TM had no effect on probe fluorescence (data not shown). Addition of calcium to the actin-TM-TN complex caused a subsequent increase in fluorescence but not back to the level caused by the presence of TM alone. If the difference in fluorescence between F-actin and actin + TM represents 100%, addition of Ca2+ caused a fluorescence gain of 22.2 ± 6.6% (n = 5) of this difference or 4.2% of the initial F-actin fluorescence. In each experiment, the fluorescence observed after addition of Ca2+ exceeded the fluorescence in the presence of TM and TN alone. The size of the standard deviation probably reflects the ratio of labeling of the 235–374 residues in different actin preparations as discussed above. We did not repeat this experiment with yeast TMs since they do not bind to TN.
FIGURE 6.

Effect of cTM, cTN, and Ca2+ on the fluorescence of pyrene-labeled A230Y/S235C actin. TM, 1.2 μ M, was added to 5 μM pyrene-labeled F-actin polymerized by the addition of 2 mM MgCl2 and 50 mM KCl. EGTA was added to remove calcium from the solution. cTN, 1.2 μM, was then added followed by the addition of enough calcium to produce a final concentration of 0.3 mM. Beginning with F-actin, the fluorescence spectrum was recorded after each addition using an excitation wavelength of 365 nm. Addition of EGTA caused no change in the fluorescence spectrum (data not shown). F-actin (solid line); after TM addition (solid line with □); after TN addition (dashed line with ⋄); after calcium addition (solid line with ×). (A) S235C actin; (B) A230Y/S235C actin.
Next, we determined if saturation of the TN-dependent fluorescence might occur at subsaturating TN concentrations. In other words, with our system, could we demonstrate cooperative effects of TN on the TM conformation reported by the pyrene at residue 235? We repeated the experiment shown in Fig. 6 A with different amounts of TN in the absence of calcium. As shown in Fig. 7, the effect of TN addition follows a linear relationship until a plateau is reached at a 1:7 ratio of TN/actin complex. This linear relationship between the effect of TN and the ratio of TN/actin/TM complex suggests that the binding of TN to one molecule of TM does not affect neighboring TMs in a cooperative fashion. This experiment was performed twice with the same result. This result agrees with a previous study by Hill et al. (43), who carried out an analysis of the effect of TN on TM binding via a cosedimentation assay and concluded the effect of TN on a particular TM did not extend to neighboring TMs.
FIGURE 7.

Effect of increasing TN concentration on the fluorescence of the complex of labeled S235C actin with cTM. TM, 1.2 μM, and 5 μM pyrene-labeled F-actin were combined, and the fluorescence at 385 nm recorded. EGTA was added to remove calcium, and increasing amounts of cTN were added as indicated. The magnitude of the fluorescence decrease caused by each addition was plotted against the ratio of [TN]/[actin]. The excitation wavelength was 365 nm. Shown is a representative sample of two different experiments.
Effect of an A230Y mutation on behavior of pyrene at residue 235
Saeki and Wakabayashi (44) previously showed that alteration of A230 to tyrosine (A230Y) did not affect the binding of TM to actin but resulted in a hyperresponsive Ca2+-dependent stimulation of actomyosin ATPase activity in the presence of TM and TN. This result suggested that the mutation in subdomain 4 caused a conformational change in the actin-myosin binding site involving subdomains 1 and 2 that led to this hyperactivation. This mutation might also affect the TM-dependent behavior of the pyrene at residue 235 if similar conformation changes were involved in the fluorescence enhancement.
We thus examined the fluorescence behavior of labeled A230Y/actin in the presence of cardiac-TM/TN + Ca2+. The A230Y mutation does not affect the starting actin fluorescence (compare Fig. 6, A and B), the TM-induced fluorescence increase, or the fluorescence decrease after addition of the TN. However, using the same analysis employed in Fig. 6 A, the A230Y/actin recovers about twice the fluorescence difference as does the S235C actin at this position, 52% with a range of ∼12% based on two determinations. This result indicates communication between the 230 helix on one hand and TM bound in the C-state on the other, probably via conformational changes involving these two separate areas of the actin.
DISCUSSION
We show that a pyrene at residue 235 of yeast actin provides a novel and sensitive fluorescence-based assay for assessing, in solution, the binding of TM to F-actin. This assay can detect differences in the interactions of specific TM isoforms with actin, and it can detect changes in TM positioning brought about by factors that promote TM movement.
The increase in actin fluorescence caused by TM binding could be theoretically due to either an interaction of the TM with the probe, per se, or a TM-induced change in conformation of the actin in the vicinity of the probe leading to an altered probe environment. The distance between the sulfur on C235 to the tip of the attached pyrene is ∼9 Å. The distance from the –SH (sulfhydryl group) to the edge of the TM envelope for a C-state bound molecule is ∼20 Å. The 11 Å gap between these measurements, together with the specificity of the probe response, suggests that the TM-dependent enhancement of pyrene235 fluorescence is due to a TM-induced conformational change in the actin and not a direct TM-probe interaction. If true, the fluorescence change would be brought about by allosteric interactions between the small outer domain of actin (subdomains 1 and 2) and the larger inner domain (subdomains 3 and 4) in which residue 235 resides. The effect of the subdomain 4 A230Y mutation on the interaction of actin with TM/TN and with myosin on the outside of the actin filament on subdomains 1 and 2 is consistent with such interactions.
Our work suggests that the differential enhancement in pyrene fluorescence caused by cTM versus skTM results from the binding of the two TMs to different sites on the yeast actin surface. A possible alternative explanation is that both TMs bind to the same site, but the differences in amino acid sequences of the two TMs result in different conformational changes in the area of the probe. However, this latter possibility seems less likely. EM (17,18) shows that these two TMs bind to different places on the surface of muscle actin. Second, skTM, which binds near the interdomain cleft of actin in muscle actin, binds near the outside of the filament on yeast actin. One might thus infer that with yeast actin, the relative binding positions of these two TMs have been reversed: cTM is away from the outer edge in the C-position. The TN-induced decrease in the fluorescence of the actin-cTM complex to that of actin alone supports our hypothesis since TN clamps TM on the filament at or near where skTM resides. Although the fluorescence changes for the mammalian TMs seem consistent with the structural work, we cannot exclude the possibility of sequence-specific effects at this time.
The small increase in pyrene fluorescence caused by skTM might reflect a minor portion of the protein binding in the C-position. Such binding would agree with Lorenz's proposal (13) that the actin-TM interaction can be approximated by a broad energy well, controlled by ionic interactions between the two proteins, in which substantial movement of the TM occurs. Helical averaging over long stretches of filament used originally for the EM reconstructions might have caused a smaller second TM population to be missed. Recent work from the same laboratory using single particle analysis comparing regulated muscle thin filaments in the presence and absence of Ca+2 shows two distinct populations of TM on the actin surface (45).
The effects of addition of Ca+2 on the fluorescence of the TN/TM-actin complex correlate with the predictions of the three-state model of Geeves and co-workers (16). The calcium-dependent increase in fluorescence of the cTM/TN/actin complex would be caused by the movement of the TM back toward the C-position. The failure to return to the original cTM/actin fluorescence level suggests that the movement of the TM in the presence of TN does not reach the same position the TM occupied in the absence of TN. This decreased fluorescence gain might involve an effect caused by the TN as part of a TN/TM complex, but it is likely not caused by the interaction of TN with the actin per se since TN had no effect on the probe 235 fluorescence in the absence of TM as described in Results.
An enhanced fluorescence recovery associated with the A230Y mutation when calcium is added to the TN/TM/actin complex was observed. This result suggests that this mutation causes an allosteric change that makes the C-state more accessible. It cannot be caused by the effect of the A230Y mutation on the probe per se, since cTM causes the same fluorescence change in the presence or absence of the A230Y mutation. An increased C-state accessibility would be consistent with the hypercontractility seen with this mutant actin in the presence of calcium, TM, and TN, since it would result in greater access of the myosin head to its binding site on the actin surface.
There is precedence for such a proposal since a mutation in the inner domain of actin has been shown to affect TM binding to the outer domain. Korman and Tobacman (38) demonstrated that mutations at residues 315/316 of actin resulted in decreased binding of TM to the actin filament. The actin structure provides a clue as to how such an allosteric change might occur. A potentially close contact exists between the two major actin domains at the top of the interdomain cleft around the positions of Arg62 and Thr203. Furthermore, Thr203 on the edge of the cleft is on a strand that runs directly to the 223–231 helix (27). Interestingly, the TM envelope in the C-state contacts these apposing residues, potentially forming a bridge between the secondary structural elements in which they reside (18).
Possible different modes of binding of related TMs to actin is not confined to mammalian TMs. Yeast TPM1 and TPM2 appear to exhibit an analogous difference (1). The smaller fluorescence difference observed with the yeast TMs suggests that they may be binding much closer to one another on the actin surface than do the two muscle TMs. Again, the structural work has not been performed that would allow confirmation of this postulate. Earlier experiments with these two proteins suggested they had overlapping but partially different functions in the yeast cell, and our results here provide physical evidence for differential binding that could lead to differential function (1,11,42). Nonmuscle TMs from higher cells also fall into two different molecular size classes (46). The larger ones, with seven pseudorepeats, are predominant in nonmalignant cells, whereas the smaller ones predominate in malignant cells (46). Upregulation of the larger TMs can cause reversal of the malignant phenotype, but nothing is known about the underlying biochemical mechanisms by which these changes are regulated (47). The system we have developed here may provide an avenue for increasing our understanding of the roles of the two TMs in yeast at the molecular level. Ultimately, it may also be useful in ascertaining the differences in the two classes of nonmuscle TMs from higher cells as well.
Finally, the observance of an excimer peak after polymerization of actin labeled at positions 374 and 224 with pyrene provides important insight into the structure of the actin filament. Excimer formation by pyrene maleimides attached to Cys spacially separated Cys residues requires a distance of ∼15–8 Å between the two sulfurs (40). This distance fits well with the structural constraints of the Holmes actin filament model, which predicts an intersulfur distance of ∼21 Å (Fig. 1 B). Considering the dynamic flexibility of the C-terminal actin peptide (48), this additional 6 Å space could easily be traversed. Within F-actin, the 224–224 and 374–374 distances are each >50 Å. This fact, combined with the presence of large amounts of protein mass directly between cysteines in each of these two pairs, would exclude these last two alternatives as explanations for the excimer. The structural constraint arising from our experiment can be added to those imposed from previous studies using either intermonomer cross-linking (49,50) or excimer fluorescence studies in defining a final model of the actin filament (37,51).
Acknowledgments
We thank Larry Tobacman for the bovine cardiac tropomyosin and troponin, and Emil Reisler for skeletal muscle tropomyosin. We also thank S. S. Lehrer and C. Bacchiocci for their suggestions regarding the placement of the pyrene.
This work was supported in part by grants from the National Institutes of Health (GM-33689) and the American Heart Association to P.A.R.
Weizu Chen and Kuo-Kuang Wen contributed equally to this work.
References
- 1.Drees, B., C. Brown, B. G. Barrell, and A. Bretscher. 1995. Tropomyosin is essential in yeast, yet the TPM1 and TPM2 products perform distinct functions. J. Cell Biol. 128:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huxley, H. E. 1973. Muscular contraction and cell motility. Nature. 243:445–449. [DOI] [PubMed] [Google Scholar]
- 3.Hitchcock-DeGregori, S. E., Y. Song, and J. Moraczewska. 2001. Importance of internal regions and the overall length of tropomyosin for actin binding and regulatory function. Biochemistry. 40:2104–2112. [DOI] [PubMed] [Google Scholar]
- 4.Landis, C. A., A. Bobkova, E. Homsher, and L. S. Tobacman. 1997. The active state of the thin filament is destabilized by an internal deletion in tropomyosin. J. Biol. Chem. 272:14051–14056. [DOI] [PubMed] [Google Scholar]
- 5.Butters, C. A., K. A. Willadsen, and L. S. Tobacman. 1993. Cooperative interactions between adjacent troponin-tropomyosin complexes may be transmitted through the actin filament. J. Biol. Chem. 268:15565–15570. [PubMed] [Google Scholar]
- 6.Urbancikova, M., and S. E. Hitchcock-DeGregori. 1994. Requirement of amino-terminal modification for striated muscle alpha-tropomyosin function. J. Biol. Chem. 269:24310–24315. [PubMed] [Google Scholar]
- 7.Gunning, P. W., G. Schevzov, A. J. Kee, and E. C. Hardeman. 2005. Tropomyosin isoforms: divining rods for actin cytoskeleton function. Trends Cell Biol. 15:333–341. [DOI] [PubMed] [Google Scholar]
- 8.Wen, K. K., B. Kuang, and P. A. Rubenstein. 2000. Tropomyosin-dependent filament formation by a polymerization-defective mutant yeast actin (V266G,L267G). J. Biol. Chem. 275:40594–40600. [DOI] [PubMed] [Google Scholar]
- 9.Liu, H. P., and A. Bretscher. 1989. Disruption of the single tropomyosin gene in yeast results in the disappearance of actin cables from the cytoskeleton. Cell. 57:233–242. [DOI] [PubMed] [Google Scholar]
- 10.Liu, H. P., and A. Bretscher. 1989. Purification of tropomyosin from Saccharomyces cerevisiae and identification of related proteins in Schizosaccharomyces and Physarum. Proc. Natl. Acad. Sci. USA. 86:90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho, J., and A. Bretscher. 2001. Ras regulates the polarity of the yeast actin cytoskeleton through the stress response pathway. Mol. Biol. Cell. 12:1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singer, J. M., G. J. Hermann, and J. M. Shaw. 2000. Suppressors of mdm20 in yeast identify new alleles of ACT1 and TPM1 predicted to enhance actin-tropomyosin interactions. Genetics. 156:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorenz, M., K. J. Poole, D. Popp, G. Rosenbaum, and K. C. Holmes. 1995. An atomic model of the unregulated thin filament obtained by x-ray fiber diffraction on oriented actin-tropomyosin gels. J. Mol. Biol. 246:108–119. [DOI] [PubMed] [Google Scholar]
- 14.Tobacman, L. S. 1996. Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 58:447–481. [DOI] [PubMed] [Google Scholar]
- 15.Cassell, M., and L. S. Tobacman. 1996. Opposite effects of myosin subfragment 1 on binding of cardiac troponin and tropomyosin to the thin filament. J. Biol. Chem. 271:12867–12872. [DOI] [PubMed] [Google Scholar]
- 16.McKillop, D. F., and M. A. Geeves. 1993. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 65:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu, C., R. Craig, L. Tobacman, R. Horowitz, and W. Lehman. 1999. Tropomyosin positions in regulated thin filaments revealed by cryoelectron microscopy. Biophys. J. 77:985–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehman, W., V. Hatch, V. Korman, M. Rosol, L. Thomas, R. Maytum, M. A. Geeves, J. E. Van Eyk, L. S. Tobacman, and R. Craig. 2000. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol. 302:593–606. [DOI] [PubMed] [Google Scholar]
- 19.Ishii, Y., and S. S. Lehrer. 1993. Kinetics of the “on-off” change in regulatory state of the muscle thin filament. Arch. Biochem. Biophys. 305:193–196. [DOI] [PubMed] [Google Scholar]
- 20.Graceffa, P. 1999. Movement of smooth muscle tropomyosin by myosin heads. Biochemistry. 38:11984–11992. [DOI] [PubMed] [Google Scholar]
- 21.Chandy, I. K., J. C. Lo, and R. D. Ludescher. 1999. Differential mobility of skeletal and cardiac tropomyosin on the surface of F-actin. Biochemistry. 38:9286–9294. [DOI] [PubMed] [Google Scholar]
- 22.Bacchiocchi, C., and S. S. Lehrer. 2002. Ca(2+)-induced movement of tropomyosin in skeletal muscle thin filaments observed by multi-site FRET. Biophys. J. 82:1524–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bacchiocchi, C., P. Graceffa, and S. S. Lehrer. 2004. Myosin-induced movement of αα, αβ, and ββ smooth muscle tropomyosin on actin observed by multisite FRET. Biophys. J. 86:2295–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cook, R. K., W. T. Blake, and P. A. Rubenstein. 1992. Removal of the amino-terminal acidic residues of yeast actin. Studies in vitro and in vivo. J. Biol. Chem. 267:9430–9436. [PubMed] [Google Scholar]
- 25.Ng, R., and J. Abelson. 1980. Isolation and sequence of the gene for actin in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 77:3912–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuang, B., and P. A. Rubenstein. 1997. Beryllium fluoride and phalloidin restore polymerizability of a mutant yeast actin (V266G,L267G) with severely decreased hydrophobicity in a subdomain 3/4 loop. J. Biol. Chem. 272:1237–1247. [DOI] [PubMed] [Google Scholar]
- 27.Vorobiev, S., B. Strokopytov, D. G. Drubin, C. Frieden, S. Ono, J. Condeelis, P. A. Rubenstein, and S. C. Almo. 2003. The structure of nonvertebrate actin: implications for the ATP hydrolytic mechanism. Proc. Natl. Acad. Sci. USA. 100:5760–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korman, V. L., V. Hatch, K. Y. Dixon, R. Craig, W. Lehman, and L. S. Tobacman. 2000. An actin subdomain 2 mutation that impairs thin filament regulation by troponin and tropomyosin. J. Biol. Chem. 275:22470–22478. [DOI] [PubMed] [Google Scholar]
- 29.Payne, M. R., and S. F. Rudnick. 1985. Tropomyosin, structure and functional diversity. In Cell and Muscle Motility. J. W. Shay, editor. Plenum Press, New York. 141–184. [PubMed]
- 30.Potter, J. D. 1982. Preparation of troponin and its subunits. Methods Enzymol. 85(Pt B):241–263. [DOI] [PubMed] [Google Scholar]
- 31.Tobacman, L. S., and R. S. Adelstein. 1986. Mechanism of regulation of cardiac actin-myosin subfragment 1 by troponin-tropomyosin. Biochemistry. 25:798–802. [DOI] [PubMed] [Google Scholar]
- 32.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. A. Smith, J. B. Seidman, and K. Struhl. 1987. Chapters 1 and 2. In Current Protocols in Molecular Biology. John Wiley and Sons, New York.
- 33.Chen, X., R. K. Cook, and P. A. Rubenstein. 1993. Yeast actin with a mutation in the “hydrophobic plug” between subdomains 3 and 4 (L266D) displays a cold-sensitive polymerization defect. J. Cell Biol. 123:1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christianson, T. W., R. S. Sikorski, M. Dante, J. H. Shero, and P. Hieter. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene. 110:119–122. [DOI] [PubMed] [Google Scholar]
- 35.Cook, R. K., and P. A. Rubenstein. 1992. The generation and isolation of mutant actins. In Practical Approaches in Cell Biology. K. Carraway and C. C. Carraway, editors. IRL Press, Oxford. 99–122.
- 36.Kron, S. J., D. G. Drubin, D. Botstein, and J. A. Spudich. 1992. Yeast actin filaments display ATP-dependent sliding movement over surfaces coated with rabbit muscle myosin. Proc. Natl. Acad. Sci. USA. 89:4466–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng, L., E. Kim, W. L. Lee, C. J. Miller, B. Kuang, E. Reisler, and P. A. Rubenstein. 1997. Fluorescence probing of yeast actin subdomain 3/4 hydrophobic loop 262–274. Actin-actin and actin-myosin interactions in actin filaments. J. Biol. Chem. 272:16829–16837. [DOI] [PubMed] [Google Scholar]
- 38.Korman, V. L., and L. S. Tobacman. 1999. Mutations in actin subdomain 3 that impair thin filament regulation by troponin and tropomyosin. J. Biol. Chem. 274:22191–22196. [DOI] [PubMed] [Google Scholar]
- 39.Cho, Y. J., and S. E. Hitchcock-DeGregori. 1991. Relationship between alternatively spliced exons and functional domains in tropomyosin. Proc. Natl. Acad. Sci. USA. 88:10153–10157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehrer, S. S. 1995. Pyrene excimer fluorescence as a probe of protein conformational change. Subcell. Biochem. 24:115–132. [DOI] [PubMed] [Google Scholar]
- 41.Mak, A. S., L. B. Smillie, and G. R. Stewart. 1980. A comparison of the amino acid sequences of rabbit skeletal muscle α- and β-tropomyosins. J. Biol. Chem. 255:3647–3655. [PubMed] [Google Scholar]
- 42.Strand, J., M. Nili, E. Homsher, and L. S. Tobacman. 2001. Modulation of myosin function by isoform-specific properties of Saccharomyces cerevisiae and muscle tropomyosins. J. Biol. Chem. 276:34832–34839. [DOI] [PubMed] [Google Scholar]
- 43.Hill, L. E., J. P. Mehegan, C. A. Butters, and L. S. Tobacman. 1992. Analysis of troponin-tropomyosin binding to actin. Troponin does not promote interactions between tropomyosin molecules. J. Biol. Chem. 267:16106–16113. [PubMed] [Google Scholar]
- 44.Saeki, K., and T. Wakabayashi. 2000. A230Y mutation of actin on subdomain 4 is sufficient for higher calcium activation of actin-activated myosin adenosinetriphosphatase in the presence of tropomyosin-troponin. Biochemistry. 39:1324–1329. [DOI] [PubMed] [Google Scholar]
- 45.Rosol, M., W. Lehman, R. Craig, C. Landis, C. Butters, and L. S. Tobacman. 2000. Three-dimensional reconstruction of thin filaments containing mutant tropomyosin. Biophys. J. 78:908–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin, J. J., K. S. Warren, D. D. Wamboldt, T. Wang, and J. L. Lin. 1997. Tropomyosin isoforms in nonmuscle cells. Int. Rev. Cytol. 170:1–38. [DOI] [PubMed] [Google Scholar]
- 47.Pawlak, G., T. W. McGarvey, T. B. Nguyen, J. E. Tomaszewski, R. Puthiyaveettil, S. B. Malkowicz, and D. M. Helfman. 2004. Alterations in tropomyosin isoform expression in human transitional cell carcinoma of the urinary bladder. Int. J. Cancer. 110:368–373. [DOI] [PubMed] [Google Scholar]
- 48.Holmes, K. C., D. Popp, W. Gebhard, and W. Kabsch. 1990. Atomic model of the actin filament. Nature. 347:44–49. [DOI] [PubMed] [Google Scholar]
- 49.Orlova, A., A. Shvetsov, V. E. Galkin, D. S. Kudryashov, P. A. Rubenstein, E. H. Egelman, and E. Reisler. 2004. Actin-destabilizing factors disrupt filaments by means of a time reversal of polymerization. Proc. Natl. Acad. Sci. USA. 101:17664–17668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galkin, V. E., A. Orlova, M. S. VanLoock, A. Shvetsov, E. Reisler, and E. H. Egelman. 2003. ADF/cofilin use an intrinsic mode of F-actin instability to disrupt actin filaments. J. Cell Biol. 163:1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bobkov, A. A., A. Muhlrad, A. Shvetsov, S. Benchaar, D. Scoville, S. C. Almo, and E. Reisler. 2004. Cofilin (ADF) affects lateral contacts in F-actin. J. Mol. Biol. 337:93–104. [DOI] [PubMed] [Google Scholar]
- 52.Kuang, B., and P. A. Rubenstein. 1997. The effects of severely decreased hydrophobicity in a subdomain 3/4 loop on the dynamics and stability of yeast G-actin. J. Biol. Chem. 272:4412–4418. [DOI] [PubMed] [Google Scholar]